
Homo-FRET Based Biosensors and Their Application to Multiplexed Imaging of Signalling Events in Live Cells

{kind=link}
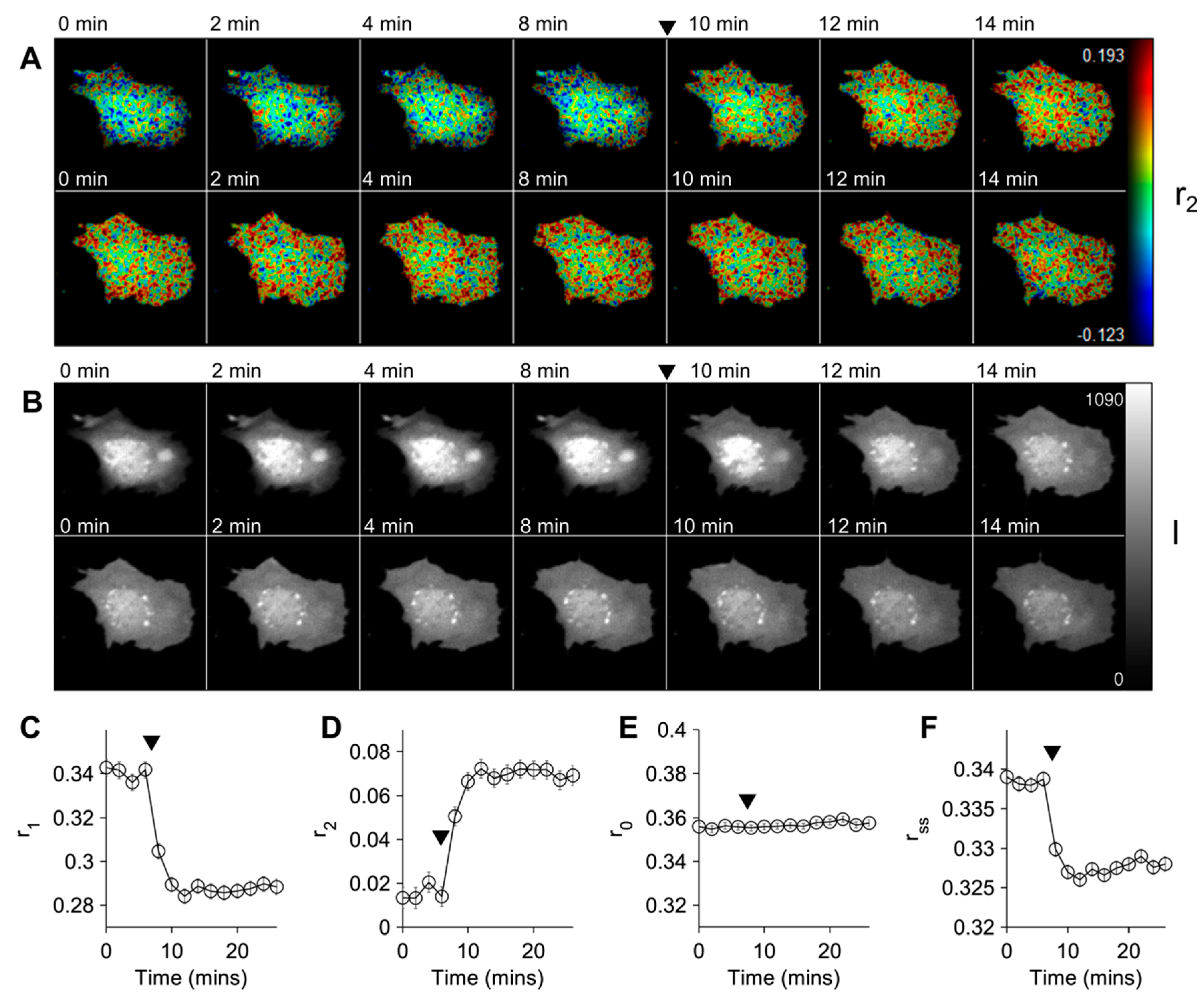{kind=link}
{kind=link}
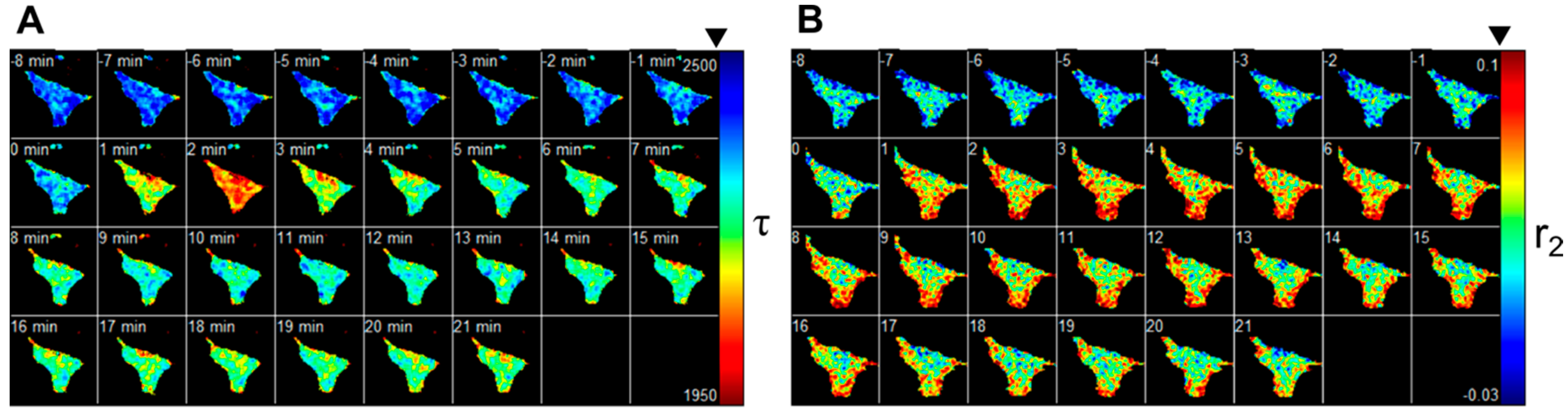{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Approaches to Multiplexed Förster Resonant Energy Transfer (FRET) Measurements
1.2. Fluorescence Anisotropy

1.3. Experimental Approaches to Homo-FRET Imaging
1.4. Quantifying Homo-FRET Aggregation Using Time Resolved Anisotropy
1.5. Phosphoinositide and Calcium Signalling
2. Results and Discussion
2.1. Homo-FRET between Aggregating mCherry-Akt-PH Molecules


2.2. Multiplexed mCherry-Akt-PH and TN-L15 Measurements


3. Experimental Section
3.1. Cells and Plasmids
3.2. Imaging

3.3. Instrument Response Function (IRF) Measurement and Polarisation Alignment
3.4. Analysis
3.4.1. Fitting of Anisotropy Data
3.4.2. Calibration of Calcium Measurements
3.4.3. Reconstruction and Fitting of Enhanced Cyan Fluorescent Protein (ECFP) Intensity Decays
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Niino, Y.; Hotta, K.; Oka, K. Simultaneous live cell imaging using dual FRET sensors with a single excitation light. PLoS ONE 2009, 4, e6036. [Google Scholar] [CrossRef] [PubMed]
- Piljic, A.; Schultz, C. Simultaneous recording of multiple cellular events by FRET. ACS Chem. Biol. 2008, 3, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Grant, D.M.; McGhee, E.J.; Zhang, W.; Bunney, T.D.; Talbot, C.B.; Kumar, S.; Munro, I.; Dunsby, C.; Neil, M.A.A.; Katan, M.; et al. Multiplexed FRET to image multiple signaling events in live cells. Biophys. J. 2008, 95, L69–L71. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.; Lin, M.; McKeown, M.; Steinbach, P. Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat. Methods 2008, 5, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Laine, R. Fluorescence Lifetime Spectroscopy and Imaging of FRET Probes for the Study of Cell Signalling. Ph.D. Thesis, Imperial College London, London, UK, 2013. [Google Scholar]
- Dean, K.M.; Lubbeck, J.L.; Binder, J.K.; Schwall, L.R.; Jimenez, R.; Palmer, A.E. Analysis of red-fluorescent proteins provides insight into dark-state conversion and photodegradation. Biophys. J. 2011, 101, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Chen, Y.; Müller, J.D. Fluorescence fluctuation spectroscopy of mCherry in living cells. Biophys. J. 2009, 96, 2391–2404. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, J.; Flors, C.; Dedecker, P.; Hofkens, J.; Engelborghs, Y. Dark states in monomeric red fluorescent proteins studied by fluorescence correlation and single molecule spectroscopy. Biophys. J. 2008, 94, 4103–4113. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy,, 2nd ed.; Kluwer Academic: AA Dordrecht, The Netherlands, 1999. [Google Scholar]
- Cross, A.J.; Fleming, G.R. Analysis of time-resolved fluorescence anisotropy decays. Biophys. J. 1984, 46, 45–56. [Google Scholar] [CrossRef]
- Jovin, T.M.; Lidke, D.S.; Post, J.N. Dynamic and static fluorescence anisotropy in biological microscopy (rFLIM and emFRET). In Biomedical Optics 2004; Periasamy, A., So, P.T.C., Eds.; International Society for Optics and Photonics: Bellingham, WA, USA, 2004; pp. 1–12. [Google Scholar]
- Tramier, M.; Coppey-Moisan, M. Fluorescence anisotropy imaging microscopy for homo-FRET in living cells. Methods Cell Biol. 2008, 85, 395–414. [Google Scholar] [PubMed]
- Squire, A.; Verveer, P.J.; Rocks, O.; Bastiaens, P.I.H. Red-edge anisotropy microscopy enables dynamic imaging of homo-FRET between green fluorescent proteins in cells. J. Struct. Biol. 2004, 147, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Gautier, I.; Tramier, M.; Durieux, C.; Coppey, J.; Pansu, R.B.; Nicolas, J.C.; Kemnitz, K.; Coppey-Moisan, M. Homo-FRET microscopy in living cells to measure monomer-dimer transition of GFP-tagged proteins. Biophys. J. 2001, 80, 3000–3008. [Google Scholar] [CrossRef]
- Clayton, A.H.A.; Hanley, Q.S.; Arndt-Jovin, D.J.; Subramaniam, V.; Jovin, T.M. Dynamic fluorescence anisotropy imaging microscopy in the frequency domain (rFLIM). Biophys. J. 2002, 83, 1631–1649. [Google Scholar] [CrossRef]
- Clayton, A.H.A. The polarized AB plot for the frequency-domain analysis and representation of fluorophore rotation and resonance energy homotransfer. J. Microsc. 2008, 232, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Kozer, N.; Henderson, C.; Jackson, J.T.; Nice, E.C.; Burgess, A.W.; Clayton, A.H.A. Evidence for extended YFP-EGFR dimers in the absence of ligand on the surface of living cells. Phys. Biol. 2011, 8, 066002. [Google Scholar] [CrossRef] [PubMed]
- Bader, A.N.; Hofman, E.G.; van Bergen en Henegouwen, P.M.; Gerritsen, H.C. Imaging of protein cluster sizes by means of confocal time-gated fluorescence anisotropy microscopy. Opt. Express 2007, 15, 6934–6945. [Google Scholar] [CrossRef] [PubMed]
- Bader, A.N.; Hofman, E.G.; Voortman, J.; en Henegouwen, P.M.; Gerritsen, H.C. Homo-FRET imaging enables quantification of protein cluster sizes with subcellular resolution. Biophys. J. 2009, 97, 2613–2622. [Google Scholar] [CrossRef] [PubMed]
- Thaler, C.; Koushik, S.V.; Puhl, H.L.; Blank, P.S.; Vogel, S.S. Structural rearrangement of CaMKIIα catalytic domains encodes activation. Proc. Natl. Acad. Sci. USA 2009, 106, 6369–6374. [Google Scholar] [CrossRef] [PubMed]
- Runnels, L.W.; Scarlata, S.F. Theory and application of fluorescence homotransfer to melittin oligomerization. Biophys. J. 1995, 69, 1569–1583. [Google Scholar] [CrossRef]
- Agranovich, V.M.; Galanin, M.D. Electron-Excitation Energy Transfer in Condensed Media; Izdatel’stvo Nauka: Moscow, Russia, 1978. [Google Scholar]
- Swaminathan, R.; Hoang, C.P.; Verkman, A.S. Photobleaching recovery and anisotropy decay of green fluorescent protein GFP-S65T in solution and cells: Cytoplasmic viscosity probed by green fluorescent protein translational and rotational diffusion. Biophys. J. 1997, 72, 1900–1907. [Google Scholar] [CrossRef]
- Michell, R.H. Inositol derivatives: Evolution and functions. Nat. Rev. Mol. Cell Biol. 2008, 9, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Takasuga, S.; Sasaki, J.; Kofuji, S.; Eguchi, S.; Yamazaki, M.; Suzuki, A. Mammalian phosphoinositide kinases and phosphatases. Prog. Lipid Res. 2009, 48, 307–343. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.T.; Janetopoulos, C.; Lee, S.; Charest, P.G.; Takeda, K.; Sundheimer, L.W.; Meili, R.; Devreotes, P.N.; Firtel, R.A. G protein-independent Ras/PI3K/F-actin circuit regulates basic cell motility. J. Cell Biol. 2007, 178, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Kölsch, V.; Charest, P.G.; Firtel, R.A. The regulation of cell motility and chemotaxis by phospholipid signaling. J. Cell Sci. 2008, 121, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Servant, G. Polarization of chemoattractant receptor signaling during neutrophil chemotaxis. Science 2000, 287, 1037–1040. [Google Scholar] [CrossRef] [PubMed]
- Parent, C.A. A cell’s sense of direction. Science 1999, 284, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Funamoto, S.; Meili, R.; Lee, S.; Parry, L.; Firtel, R.A. Spatial and temporal regulation of 3-phosphoinositides by PI 3-kinase and PTEN mediates chemotaxis. Cell 2002, 109, 611–623. [Google Scholar] [CrossRef]
- Iijima, M.; Devreotes, P. Tumor suppressor PTEN mediates sensing of chemoattractant gradients. Cell 2002, 109, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Wang, F.; Glavas, S.; Ott, A.; Hofmann, F.; Aktories, K.; Kalman, D.; Bourne, H.R. Rac and Cdc42 play distinct roles in regulating PI(3,4,5)P3 and polarity during neutrophil chemotaxis. J. Cell Biol. 2003, 160, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.T.; Firtel, R. Regulation of chemotaxis by the orchestrated activation of Ras, PI3K, and TOR. Eur. J. Cell Biol. 2006, 85, 873–895. [Google Scholar] [CrossRef] [PubMed]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef] [PubMed]
- Escobedo, J.; Williams, L. Role of phosphatidylinositol kinase in PDGF receptor signal transduction. Science 1989, 243, 1191–1194. [Google Scholar]
- Kumjian, D.A.; Wahl, M.I.; Rhee, S.G.; Daniel, T.O. Platelet-derived growth factor (PDGF) binding promotes physical association of PDGF receptor with phospholipase C. Proc. Natl. Acad. Sci. USA 1989, 86, 8232–8236. [Google Scholar] [CrossRef] [PubMed]
- Mikoshiba, K. IP3 receptor/Ca2+ channel: From discovery to new signaling concepts. J. Neurochem. 2007, 102, 1426–1446. [Google Scholar] [CrossRef] [PubMed]
- Mouneimne, G.; Soon, L.; DesMarais, V.; Sidani, M.; Song, X.; Yip, S.-C.; Ghosh, M.; Eddy, R.; Backer, J.M.; Condeelis, J. Phospholipase C and cofilin are required for carcinoma cell directionality in response to EGF stimulation. J. Cell Biol. 2004, 166, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Kortholt, A.; King, J.S.; Keizer-Gunnink, I.; Harwood, A.J.; van Haastert, P.J.M. Phospholipase C regulation of phosphatidylinositol 3,4,5-trisphosphate-mediated chemotaxis. Mol. Biol. Cell 2007, 18, 4772–4779. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.; Johnell, M.; Siegbahn, A.; Rorsman, C.; Engström, U.; Wernstedt, C.; Heldin, C.H.; Rönnstrand, L. Mutation of a Src phosphorylation site in the PDGF β-receptor leads to increased PDGF-stimulated chemotaxis but decreased mitogenesis. EMBO J. 1996, 15, 5299–5313. [Google Scholar] [PubMed]
- Chen, L.; Iijima, M.; Tang, M.; Landree, M.A.; Huang, Y.E.; Xiong, Y.; Iglesias, P.A.; Devreotes, P.N. PLA2 and PI3K/PTEN pathways act in parallel to mediate chemotaxis. Dev. Cell 2007, 12, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Insall, R. The interaction between pseudopods and extracellular signalling during chemotaxis and directed migration. Curr. Opin. Cell Biol. 2013, 25, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Várnai, P.; Balla, T. Live cell imaging of phosphoinositide dynamics with fluorescent protein domains. Biochim. Biophys. Acta 2006, 1761, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Lasserre, R.; Guo, X.-J.; Conchonaud, F.; Hamon, Y.; Hawchar, O.; Bernard, A.-M.; Soudja, S.M.; Lenne, P.-F.; Rigneault, H.; Olive, D.; et al. Raft nanodomains contribute to Akt/PKB plasma membrane recruitment and activation. Nat. Chem. Biol. 2008, 4, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Lowry, P.R.; Zhou, X.; Depry, C.; Wei, Z.; Wong, G.W.; Zhang, J. PI3K/Akt signaling requires spatial compartmentalization in plasma membrane microdomains. Proc. Natl. Acad. Sci. USA 2011, 108, 14509–14514. [Google Scholar] [CrossRef] [PubMed]
- Van der Wal, J.; Habets, R.; Várnai, P.; Balla, T.; Jalink, K. Monitoring agonist-induced phospholipase C activation in live cells by fluorescence resonance energy transfer. J. Biol. Chem. 2001, 276, 15337–15344. [Google Scholar] [CrossRef] [PubMed]
- Warren, S.C.; Margineanu, A.; Alibhai, D.; Kelly, D.J.; Talbot, C.; Alexandrov, Y.; Munro, I.; Katan, M.; Dunsby, C.; French, P.M.W. Rapid global fitting of large fluorescence lifetime imaging microscopy datasets. PLoS ONE 2013, 8, e70687. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.S.; Nguyen, T.A.; van der Meer, B.W.; Blank, P.S. The impact of heterogeneity and dark acceptor states on FRET: Implications for using fluorescent protein donors and acceptors. PLoS ONE 2012, 7, e49593. [Google Scholar] [CrossRef] [PubMed]
- Laine, R.; Stuckey, D.W.; Manning, H.; Warren, S.C.; Kennedy, G.; Carling, D.; Dunsby, C.; Sardini, A.; French, P.M.W. Fluorescence lifetime readouts of troponin-C-based calcium FRET sensors: A quantitative comparison of CFP and mTFP1 as donor fluorophores. PLoS ONE 2012, 7, e49200. [Google Scholar] [CrossRef] [PubMed]
- Martins, M.; Warren, S.; Kimberley, C.; Margineanu, A.; Peschard, P.; McCarthy, A.; Yeo, M.; Marshall, C.J.; Dunsby, C.; French, P.M.W.; et al. Activity of phospholipase C epsilon contributes to chemotaxis of fibroblasts towards platelet-derived growth factor. J. Cell Sci. 2012, 125, 5758–5769. [Google Scholar] [CrossRef] [PubMed]
- Daddysman, M.K.; Fecko, C.J. DNA multiphoton absorption generates localized damage for studying repair dynamics in live cells. Biophys. J. 2011, 101, 2294–2303. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, W.; Arakawa, N.; Matsunaga, S.; Higashi, T.; Fukui, K.; Isobe, K.; Itoh, K. Femtosecond laser disruption of subcellular organelles in a living cell. Opt. Express 2004, 12, 4203–4213. [Google Scholar] [CrossRef] [PubMed]
- Edelstein, A.; Amodaj, N.; Hoover, K.; Vale, R.; Stuurman, N. Computer control of microscopes using µManager. Curr. Protoc. Mol. Biol. 2010. [Google Scholar] [CrossRef]
- Gesztelyi, R.; Zsuga, J.; Kemeny-Beke, A.; Varga, B.; Juhasz, B.; Tosaki, A. The Hill equation and the origin of quantitative pharmacology. Arch. Hist. Exact Sci. 2012, 66, 427–438. [Google Scholar] [CrossRef]
- Bajzer, Ž.; Therneau, T.M.; Sharp, J.C.; Prendergast, F.G. Maximum likelihood method for the analysis of time-resolved fluorescence decay curves. Eur. Biophys. J. 1991, 20, 247–262. [Google Scholar]
- Shaner, N.C.; Steinbach, P.A.; Tsien, R.Y. A guide to choosing fluorescent proteins. Nat. Methods 2005, 2, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Tsien, R.Y. Tsien Lab Website. Available online: http://www.tsienlab.ucsd.edu/ (accessed on 20 November 2014).
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Warren, S.C.; Margineanu, A.; Katan, M.; Dunsby, C.; French, P.M.W. Homo-FRET Based Biosensors and Their Application to Multiplexed Imaging of Signalling Events in Live Cells. Int. J. Mol. Sci. 2015, 16, 14695-14716. https://doi.org/10.3390/ijms160714695
Warren SC, Margineanu A, Katan M, Dunsby C, French PMW. Homo-FRET Based Biosensors and Their Application to Multiplexed Imaging of Signalling Events in Live Cells. International Journal of Molecular Sciences. 2015; 16(7):14695-14716. https://doi.org/10.3390/ijms160714695
Chicago/Turabian StyleWarren, Sean C., Anca Margineanu, Matilda Katan, Chris Dunsby, and Paul M. W. French. 2015. "Homo-FRET Based Biosensors and Their Application to Multiplexed Imaging of Signalling Events in Live Cells" International Journal of Molecular Sciences 16, no. 7: 14695-14716. https://doi.org/10.3390/ijms160714695
APA StyleWarren, S. C., Margineanu, A., Katan, M., Dunsby, C., & French, P. M. W. (2015). Homo-FRET Based Biosensors and Their Application to Multiplexed Imaging of Signalling Events in Live Cells. International Journal of Molecular Sciences, 16(7), 14695-14716. https://doi.org/10.3390/ijms160714695