
Characterization of the Interaction between Gallic Acid and Lysozyme by Molecular Dynamics Simulation and Optical Spectroscopy

Abstract
:
1. Introduction

2. Results and Discussion
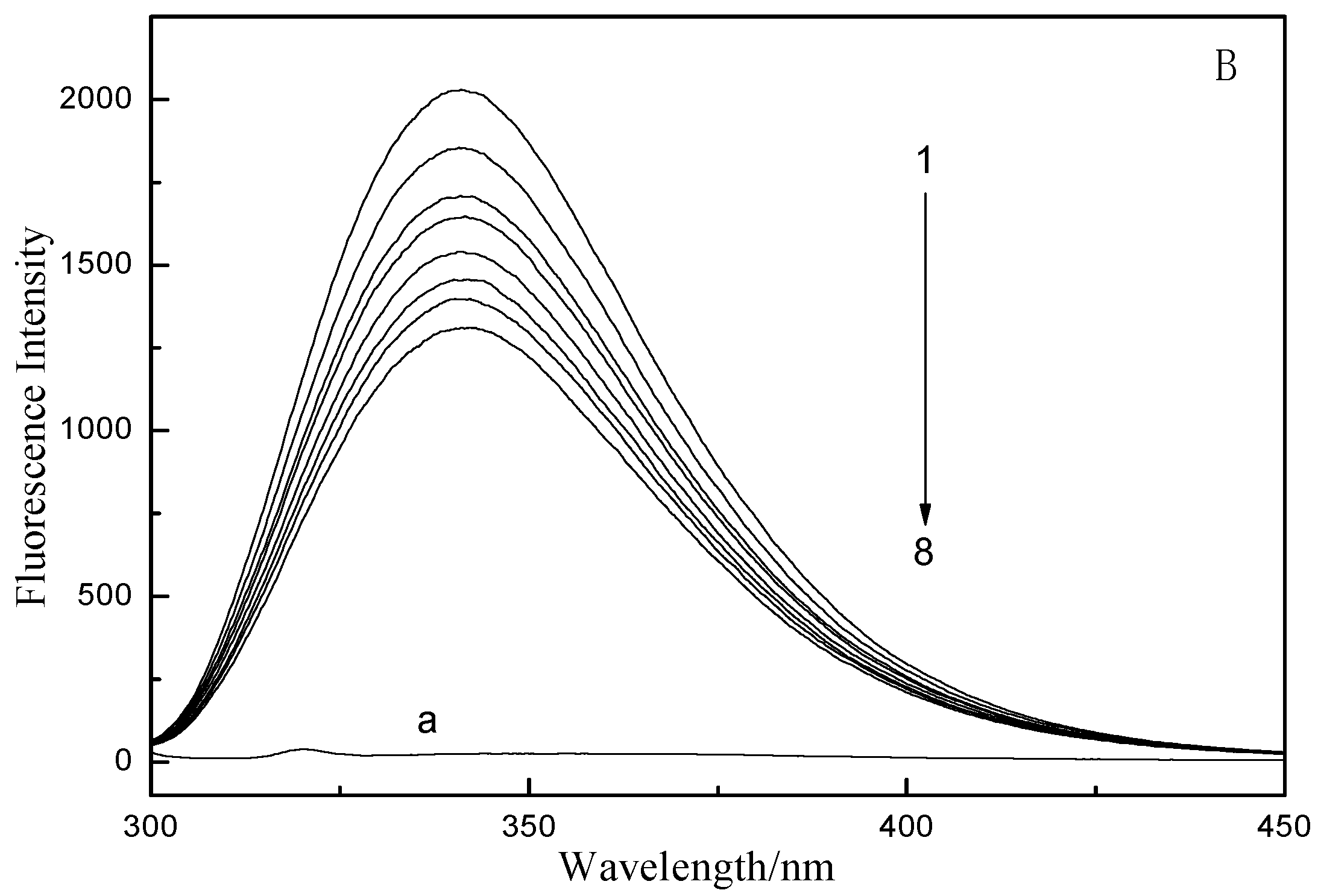
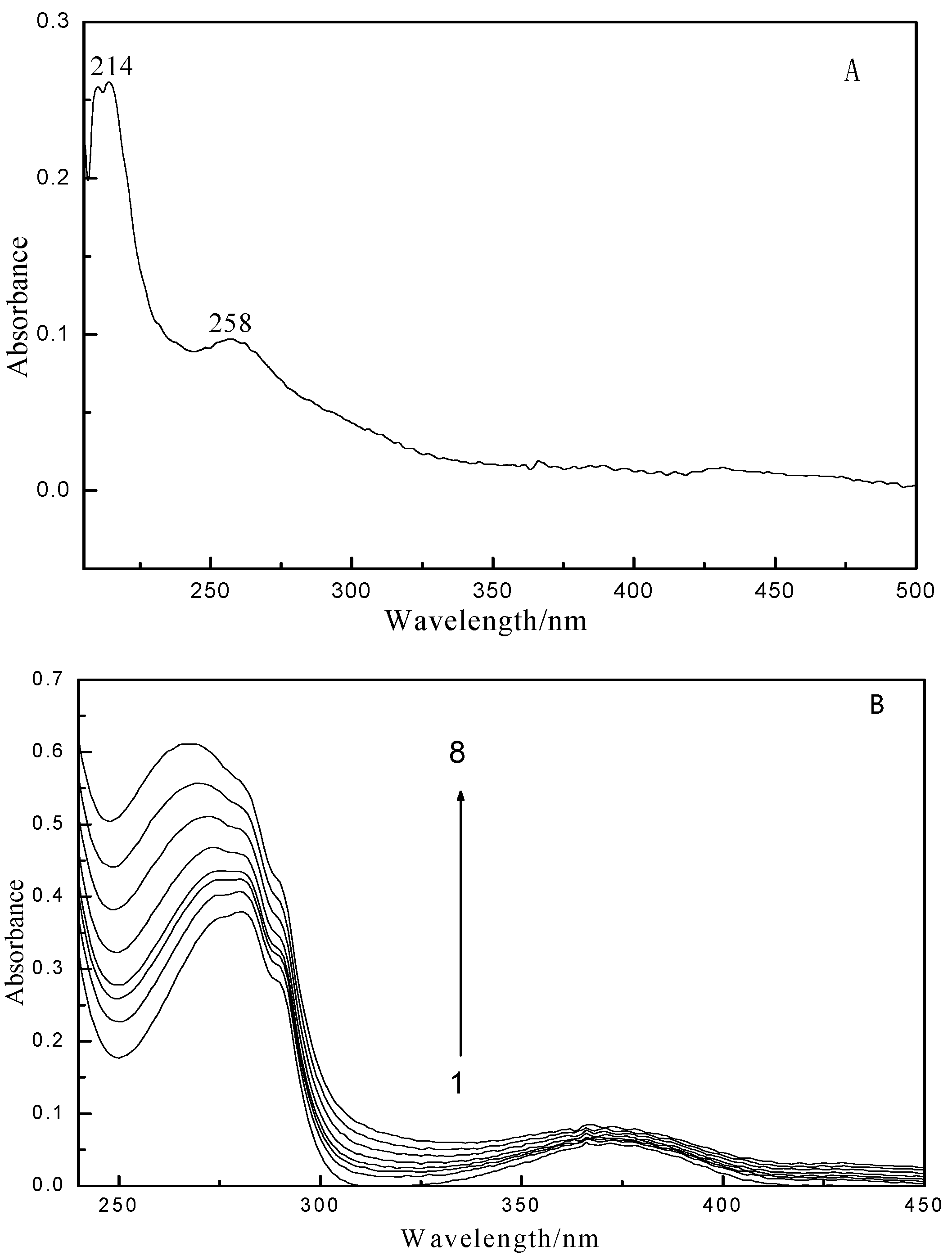
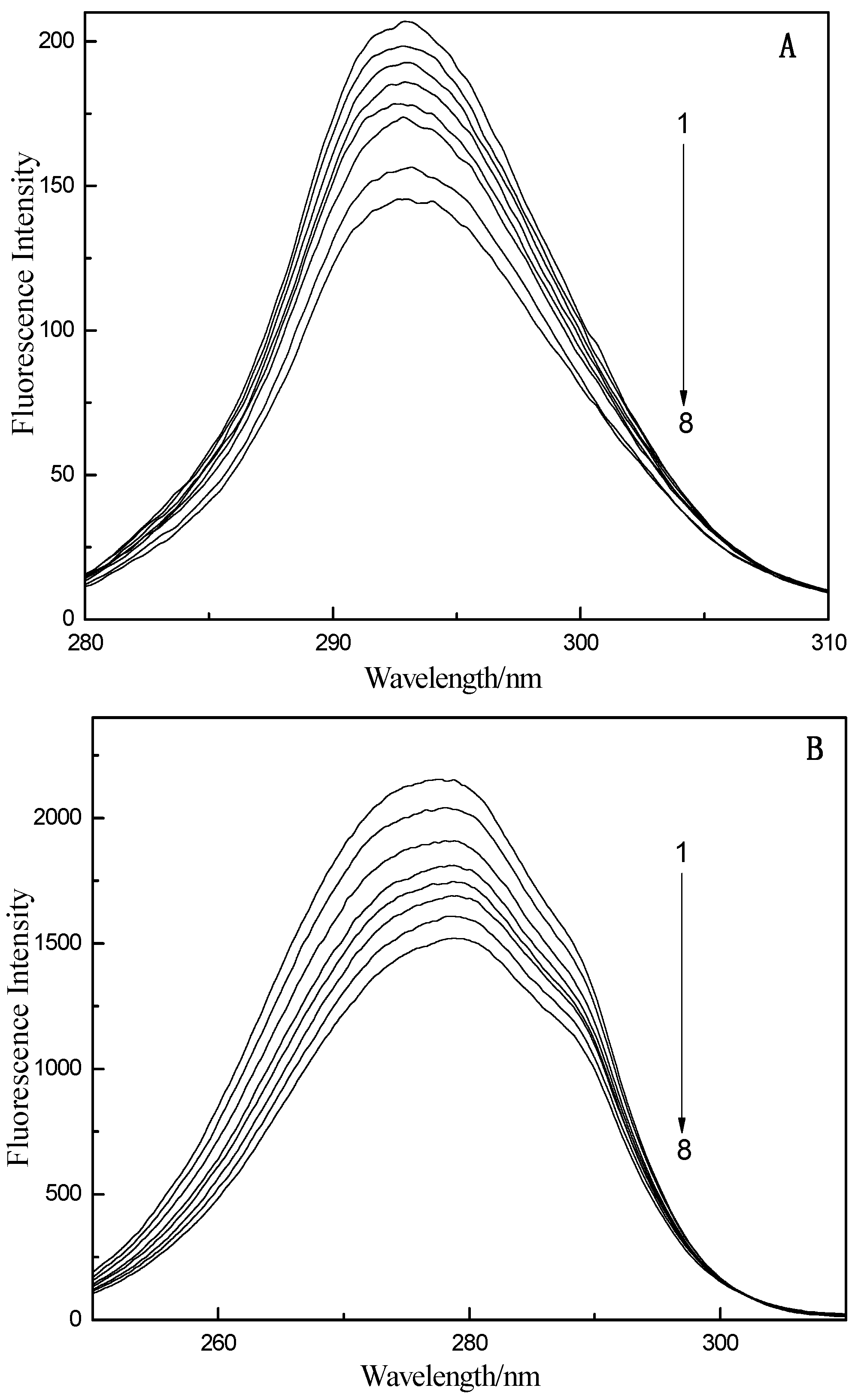
2.1. Fluorescence and UV–vis Spectroscopy


2.2. Fluorescence Quenching Mechanism



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T (K) | Ksv (L/mol) | Kq (L/(mol·s)) | R | SD | p |
|---|---|---|---|---|---|
| 298 K | 7.15 × 103 | 7.15 × 1010 | 0.9980 | 0.0066 | <0.0001 |
| 310 K | 1.17 × 104 | 1.17 × 1012 | 0.9829 | 0.0321 | <0.0001 |
2.3. Binding Mechanism and Thermodynamic Parameters
| T (K) | K (L/mol) | n | ΔG (kJ/mol) | ΔS (kJ/(mol·K)) | ΔH (kJ/mol) |
|---|---|---|---|---|---|
| 298 | 3.93 × 105 | 1.3985 | −31.92 | −1.20 | −388.81 |
| 310 | 9.04 × 102 | 0.7274 | −17.54 | −1.20 | −388.81 |
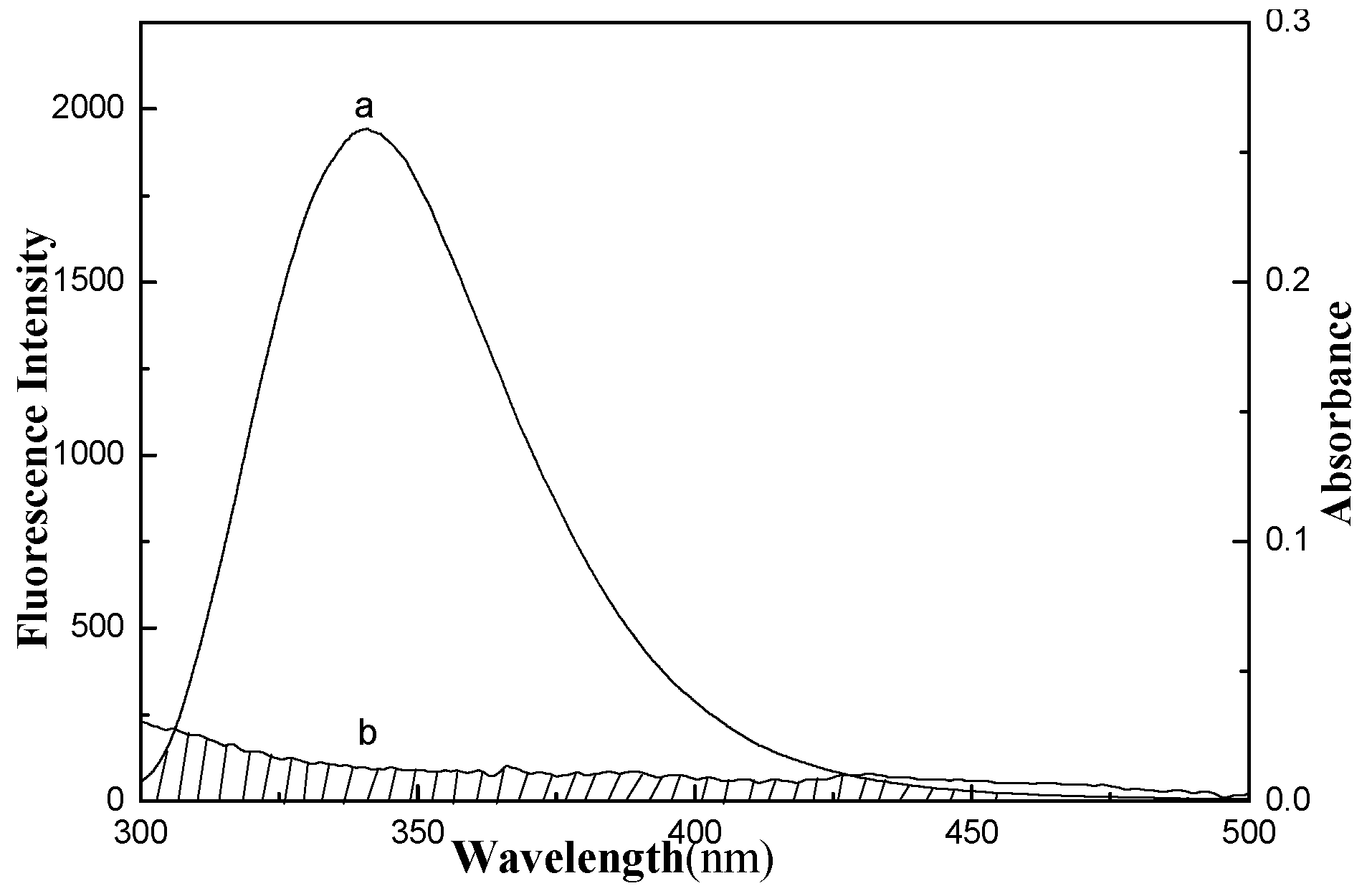
2.4. Energy Transfer from GA to LYS

2.5. Conformation Investigation

2.6. Molecular Dynamics Simulation Analysis






| Temperatures (T/K) | Donor | Acceptor | Duration (% of the Total Simulation Time Considered) | Mean Distance (nm) | Mean Angle Degrees (°) |
|---|---|---|---|---|---|
| 298 | O1(GA) | O(Trp63) | 7.20 | 2.808 | 18.63 |
| H5(GA) | O(Asp101) | 3.40 | 2.974 | 33.08 | |
| 310 | O5(GA) | NH2(Arg73) | 7.25 | 2.946 | 32.11 |
| H6(GA) | OD1(Asp101) | 0.77 | 2.924 | 29.85 |
2.7. MM-PBSA Free Energy
| T (K) | ΔGvdw | ΔGelec | ΔGpol-solv | ΔGnonpol-solv | ΔGsolv | ΔGgas | ΔG |
|---|---|---|---|---|---|---|---|
| 298 | −44.92 | −24.22 | 58.98 | −1.79 | 57.20 | −69.13 | −11.93 |
| 310 | −35.36 | −52.48 | 81.42 | −1.14 | 80.28 | −87.84 | −7.56 |
3. Experimental Section
3.1. Materials and Apparatus
3.2. Absorption and Fluorescence Spectroscopy
3.3. Synchronous Fluorescence Spectroscopy and Fluorescence Phase Diagram
3.4. Molecular Docking and Molecular Dynamics Simulations
3.5. MM-PBSA Method
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sakurai, Y.; Kajimoto, K.; Hatakeyama, H.; Harashima, H. Advances in an active and passive targeting to tumor and adipose tissues. Expert Opin. Drug Deliv. 2015, 12, 41–52. [Google Scholar] [CrossRef]
- Vhora, I.; Patil, S.; Bhatt, P.; Gandhi, R.; Baradia, D.; Misra, A. Receptor-targeted drug delivery: Current perspective and challenges. Ther. Deliv. 2014, 5, 1007–1024. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Yang, F.; Lee, N.; Wu, X. HSA-based anti-inflammatory therapy: A new and improved approach. Future Med. Chem. 2014, 6, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhang, Y.; Liang, H. Interactive association of drugs binding to human serum albumin. Int. J. Mol. Sci. 2014, 15, 3580–3595. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.J.; Liu, R.; Jiang, X.H. Spectroscopic studies on the interaction between tetrandrine and two serum albumins by chemometrics methods. Spectrochim. Acta A 2013, 115, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Lu, Y.; Cao, X.; Huang, Y.; Liu, Y.; Tang, L.; Liao, S.G.; Wang, A.M.; Li, Y.J.; Lan, Y.Y.; et al. Evaluation of the impact of polygonum capitatum, a traditional Chinese herbal medicine, on rat hepatic cytochrome P450 enzymes by using a cocktail of probe drugs. J. Ethnopharmacol. 2014, 158, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Gao, Y.; Jiang, W.; Huang, M.; Xu, A.; Paxton, J.W. Interactions of herbs with cytochrome P450. Drug Metab. Rev. 2003, 35, 35–98. [Google Scholar] [CrossRef] [PubMed]
- Zientek, M.A.; Youdim, K. Reaction phenotyping: Advances in the experimental strategies used to characterize the contribution of drug-metabolizing enzymes. Drug Metab. Dispos. 2015, 43, 163–181. [Google Scholar] [CrossRef] [PubMed]
- Mateus, L.; Costa, L.; Silva, Y.J.; Pereira, C.; Almeida, A. Effect of lysozyme addition on the activity of phages against Vibrio parahaemolyticus. Aquaculture 2014, 432, 125–129. [Google Scholar] [CrossRef]
- Na, P.; Chen, B.H.; Wang, Y.F.; Wang, J.; Li, Y.N. Analysis and simulation of molecular dynamics of lysozyme in water cluster system. Trans. Tianjin Univ. 2012, 18, 1–7. [Google Scholar] [CrossRef]
- Eichenberger, A.P.; van Gunsteren, W.F.; Smith, L.J. Structure of hen egg-white lysozyme solvated in TFE/water: A molecular dynamics simulation study based on NMR data. J. Biomol. NMR 2013, 55, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Zhao, G.Y.; Huang, J.L.; Sun, Y.; Zhang, L. Fluorescence spectroscopic investigation of the interaction between chloramphenicol and lysozyme. Eur. J. Med. Chem. 2009, 44, 4083–4089. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Hao, F.; Liu, R.T. Interactions of lead (II) acetate with the enzyme lysozyme: A spectroscopic investigation. J. Lumin. 2013, 142, 144–149. [Google Scholar] [CrossRef]
- Piyali, M.; Mousumi, B.; Sampa, B.; Samita, B. Protein interactions of merocyanine 540: Spectroscopic and crystallographic studies with lysozyme as a model protein. J. Photochem. Photobiol. B 2013, 121, 46–56. [Google Scholar]
- Jang, A.; Srinivasan, P.; Lee, N.Y.; Song, H.P.; Lee, J.W.; Lee, M.; Jo, C. Comparison of hypolipidemic activity of synthetic gallic acid-linoleic acid ester with mixture of gallic acid and linoleic acid, gallic acid, and linoleic acid on high-fat diet induced obesity in C57BL/6 Cr Slc mice. Chem. Biol. Interact. 2008, 174, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, C.; Filippin-Monteiro, F.B.; Creczynski-Pasa, T.B. Alkyl esters of gallic acid as anticancer agents: A review. Eur. J. Med. Chem. 2013, 60, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Jayamani, J.; Shanmugam, G. Gallic acid, one of the components in many plant tissues, is a potential inhibitor for insulin amyloid fibril formation. Eur. J. Med. Chem. 2014, 85, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Zheng, W. Decrypting the structural, dynamic, and energetic basis of a monomeric kinesin interacting with a tubulin dimer in three ATPase states by all-atom molecular dynamics simulation. Biochemistry 2015, 54, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, E. Molecular dynamics simulations. Methods Mol. Biol. 2015, 1215, 3–26. [Google Scholar] [PubMed]
- Della-Longa, S.; Arcovito, A. Intermediate states in the binding process of folic acid to folate receptor α: Insights by molecular dynamics and metadynamics. J. Comput. Aided Mol. Des. 2015, 29, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Hsu, W.C.; Liu, A.L.; Hsu, C.J.; Sun, Y.C. Using thermodynamic integration MD simulation to compute relative protein–ligand binding free energy of a GSK3β kinase inhibitor and its analogs. J. Mol. Graph. Model. 2014, 51, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Adcock, S.A.; McCammon, J.A. Molecular dynamics: Survey of methods for simulating the activity of proteins. Chem. Rev. 2006, 106, 1589–1615. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Li, Y.; Shen, M.; Tian, S.; Xu, L.; Pan, P.; Guan, Y.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 5. Improved docking performance using high solute dielectric constant MM/GBSA and MM/PBSA rescoring. Phys. Chem. Chem. Phys. 2014, 16, 22035–22045. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Sun, H.; Li, Y.; Wang, J.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 3. The impact of force fields and ligand charge models. J. Phys. Chem. B 2013, 117, 8408–8421. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Li, Y.; Tian, S.; Xu, L.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys. Chem. Chem. Phys. 2014, 16, 16719–16729. [Google Scholar] [CrossRef] [PubMed]
- Decherchi, S.; Masetti, M.; Vyalov, I.; Rocchia, W. Implicit solvent methods for free energy estimation. Eur. J. Med. Chem. 2015, 91, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.L.; Beroza, P.; Artis, D.R. Including explicit water molecules as part of the protein structure in MM/PBSA calculations. J. Chem. Inf. Model. 2014, 54, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Jiang, F.; Jiang, H.; Wua, K.; Zheng, X.G.; Cai, Y.Z.; Katakowski, M.; Chopp, M.; Tony To, S.S. Gallic acid suppresses cell viability, proliferation, invasion and angiogenesis in human glioma cells. Eur. J. Pharmacol. 2010, 641, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Devendra, S.; Mohan, S.M.R.; Ajay, S.; Mona, S. Gallic acid-phospholipid complex: Drug incorporation and physicochemical characterization. Lett. Drug Des. Discov. 2011, 8, 284–291. [Google Scholar]
- Lakowicz, J.R.; Weber, G. Quenching of protein fluorescence by oxygen. Detection of structural fluctuations in proteins on the nanosecond time scale. Biochemistry 1973, 12, 4171–4179. [Google Scholar] [CrossRef] [PubMed]
- Birdsall, B.; King, R.W.; Wheeler, M.R. Correctionfor light absorption in fluorescence studies of protein-ligand interactions. Anal. Biochem. 1983, 132, 353–361. [Google Scholar] [CrossRef]
- Yang, J.; Qu, L.L.; Yang, W.Y.; Huang, Y.; Jiao, N.; Zhan, W.H.; Zhao, D.; Cui, L.J. Interaction of hyperoside with human serum albumin and effect of glucose on the binding. J. Spectrosc. 2014, 2014, 1–9. [Google Scholar] [CrossRef]
- Kumari, M.; Maurya, J.K.; Tasleem, M.; Singh, P.; Patel, R. Probing HSA-ionic liquid interactions by spectroscopic and molecular docking methods. J. Photochem. Photobiol. B 2014, 138, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Równicka-Zubik, J.R.; Sułkowski, L.; Toborek, M. Interactions of PCBs with human serum albumin: In vitro spectroscopic study. Spectrochim. Acta A 2014, 124, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.J.; Liu, Y.; Jiang, W.; Zhao, R.M.; Qu, S.S. Fluorometric investigation of the interaction of bovine serum albumin with surfactants and 6-mercaptopurine. J. Photochem. Photobiol. B 2005, 80, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Paramaguru, G.; Kathiravan, A.; Selvaraj, S.; Venuvanalingam, P.; Renganathan, R. Interaction of anthraquinone dyes with lysozyme: Evidences from spectroscopic and docking studies. J. Hazard. Mater. 2010, 175, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Zou, J.W.; Yi, P.G.; Shang, Z.C.; Hu, G.X.; Yu, Q.S. Binding interaction of gatifloxacin with bovineserum albumin. Anal. Sci. 2004, 20, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.J.; Zhang, Y.T. Spectroscopic investigation on the interaction of salidroside with bovine serum albumin. J. Mol. Struct. 2008, 889, 20–27. [Google Scholar] [CrossRef]
- Joseph, R.L.; Gregorio, W. Quenching of fluorescence by oxygen. Probe for structural fluctuations in macromolecules. Biochemistry 1973, 12, 4161–4170. [Google Scholar]
- Cheng, Z.J. Comparative studies on the interactions of honokiol and magnolol with human serum albumin. J. Pharm. Biomed. 2012, 66, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Shahabadi, N.; Khorshidi, A.; Moghadam, N.H. Study on the interaction of the epilepsy drug, zonisamide with human serum albumin (HSA) by spectroscopic and molecular docking techniques. Spectrochim. Acta A 2013, 114, 627–632. [Google Scholar] [CrossRef] [PubMed]
- He, W.Y.; Li, Y.; Si, H.Z.; Dong, Y.M.; Sheng, F.L.; Yao, X.J.; Hu, Z.D. Molecular modeling and spectroscopic studies on the binding of guaiacol to human serum albumin. J. Photochem. Photobiol. A 2006, 182, 158–167. [Google Scholar] [CrossRef]
- He, W.Y.; Hu, Z.D.; Yao, X.J.; Chen, G.Y. Comparison of the interaction of alpinetin and cardamonin with human gammaglobulin. Acta Chim. Sin. 2010, 68, 679–688. [Google Scholar]
- Wu, P.G.; Brand, L. Resonance energy transfer: Methods and applications. Anal. Biochem. 1994, 218, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.D.; Subramanian, S. Thermodynamics of protein association reactions: Forces contributing to stability. Biochemistry 1981, 20, 3096–3102. [Google Scholar] [CrossRef] [PubMed]
- Toneatto, J.; Argüello, G.A. New advances in the study on the interaction of [Cr(phen)2(dppz)]3+ complex with biological models; association to transporting proteins. J. Inorg. Biochem. 2011, 105, 645–651. [Google Scholar]
- Wang, W.P.; Min, W.N.; Chen, J.R.; Wu, X.H.; Hu, Z.D. Binding study of diprophylline with lysozyme by spectroscopic methods. J. Lumin. 2011, 131, 820–824. [Google Scholar] [CrossRef]
- Stryer, L. Fluorescence energy transfer as a spectroscopic ruler. Annu. Rev. Biochem. 1978, 47, 819–846. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Lan, J.F.; Guan, M.; Sheng, F.L.; Zhang, H.X. Spectroscopic investigation of interaction between mangiferin and bovine serum albumin. Spectrochim. Acta A 2009, 73, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.W.; Que, Q.M.; Pan, J.H.; Guo, J.B. Study of the interaction between icariin and human serum albumin by fluorescence spectroscopy. J. Mol. Struct. 2008, 881, 132–138. [Google Scholar] [CrossRef]
- Kuznetsova, I.M.; Turoverov, K.K.; Uversky, V.N. Use of the phase diagram method to analyze the protein unfolding-refolding reactions: Fishing out the “Invisible” intermediates. J. Poteome Res. 2004, 3, 485–494. [Google Scholar] [CrossRef]
- Guo, M.; Zhang, L.Y.; Lv, W.J.; Cao, H.R. Analysis of the spectroscopic characteristics on the binding interaction between tosufloxacin and bovine lactoferrin. J. Lumin. 2011, 131, 768–775. [Google Scholar] [CrossRef]
- Weiner, S.J.; Kollman, P.A.; Case, D.A.; Singh, C.; Ghio, G.; Alagona, S.; Profeta, P.; Weiner, P. A new force field for molecular mechanical simulation of nucleic acids and proteins. J. Am. Chem. Soc. 1984, 106, 765–784. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a lamarckian genetic algorithm and empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Lu, S.Y. Molecular Simulation Studies on Structure and Function of Glycogen Synthase Kinase3β. Ph.D. Thesis, Zhejiang University, Hangzhou, China, 2012. [Google Scholar]
- Berhanu, W.M.; Masunov, A.E. Unique example of amyloid aggregates stabilized by main chain H-bond instead of the steric zipper: Molecular dynamics study of the amyloidogenic segment of amylin wild-type and mutants. J. Mol. Model. 2012, 18, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.L.; Zhao, X.; Yu, H.; Wang, Y.B.; Sun, T.D.; Huang, X.R. Molecular dynamics of an extremely thermophilic ribose binding protein. Acta Chim. Sin. 2012, 70, 606–610. [Google Scholar] [CrossRef]
- Zhan, D.; Yu, L.; Jin, H.; Guan, S.; Han, W. Molecular modeling and MM-PBSA free energy analysis of endo-1,4-β-xylanase from Ruminococcus albus 8. Int. J. Mol. Sci. 2014, 15, 17284–17303. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhan, M.; Guo, M.; Jiang, Y.; Wang, X. Characterization of the Interaction between Gallic Acid and Lysozyme by Molecular Dynamics Simulation and Optical Spectroscopy. Int. J. Mol. Sci. 2015, 16, 14786-14807. https://doi.org/10.3390/ijms160714786
Zhan M, Guo M, Jiang Y, Wang X. Characterization of the Interaction between Gallic Acid and Lysozyme by Molecular Dynamics Simulation and Optical Spectroscopy. International Journal of Molecular Sciences. 2015; 16(7):14786-14807. https://doi.org/10.3390/ijms160714786
Chicago/Turabian StyleZhan, Minzhong, Ming Guo, Yanke Jiang, and Xiaomeng Wang. 2015. "Characterization of the Interaction between Gallic Acid and Lysozyme by Molecular Dynamics Simulation and Optical Spectroscopy" International Journal of Molecular Sciences 16, no. 7: 14786-14807. https://doi.org/10.3390/ijms160714786
APA StyleZhan, M., Guo, M., Jiang, Y., & Wang, X. (2015). Characterization of the Interaction between Gallic Acid and Lysozyme by Molecular Dynamics Simulation and Optical Spectroscopy. International Journal of Molecular Sciences, 16(7), 14786-14807. https://doi.org/10.3390/ijms160714786