
Mitochondrial Oxidative Stress, Mitochondrial DNA Damage and Their Role in Age-Related Vascular Dysfunction


Abstract
:
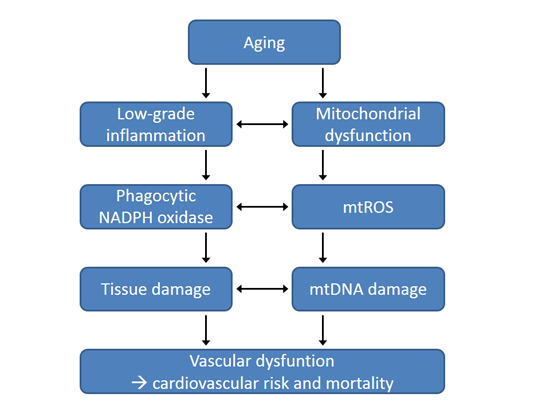{kind=link}
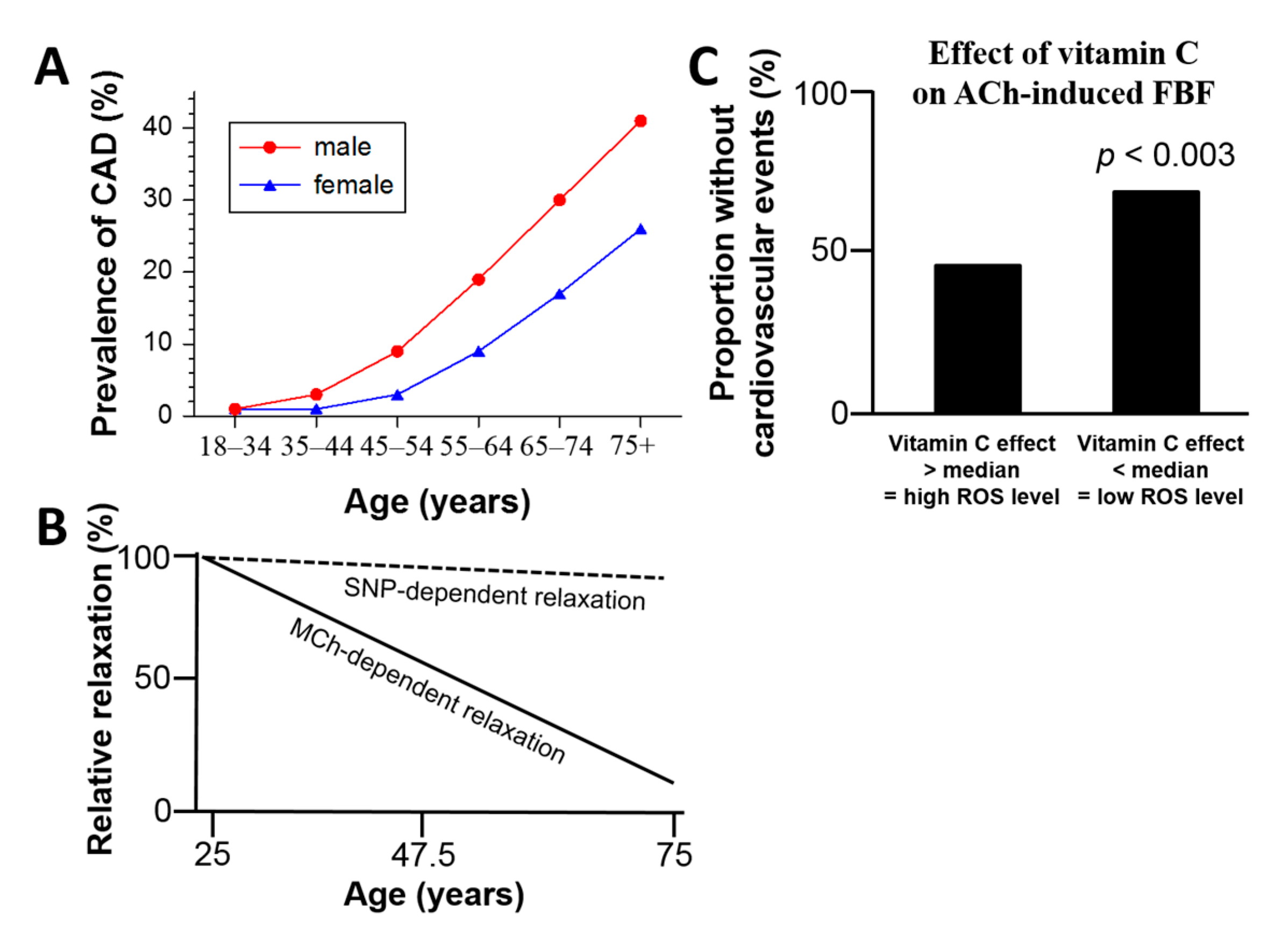{kind=link}
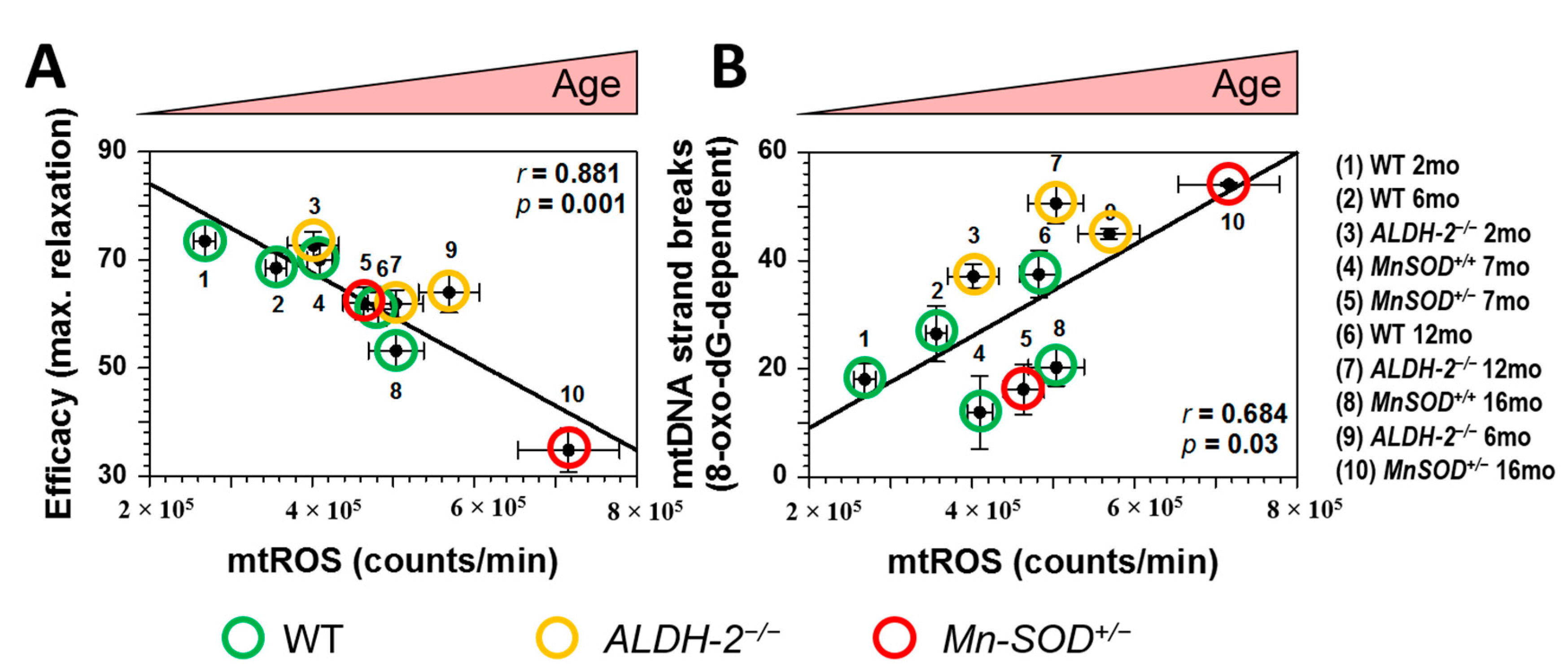{kind=link}
{kind=link}
{kind=link}
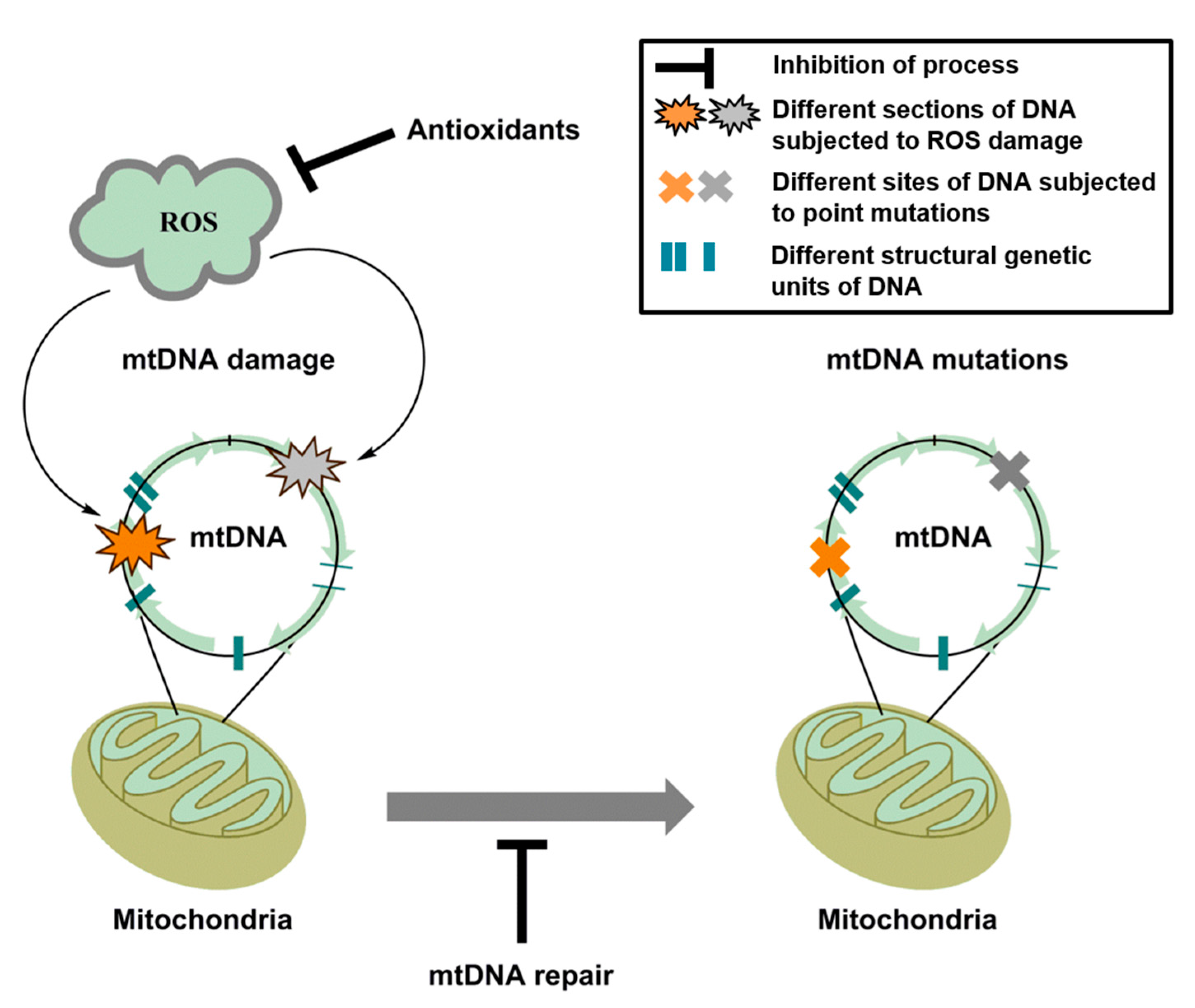{kind=link}
{kind=link}
{kind=link}
1. Introduction

2. The Cardiovascular System
3. Aging and Oxidative Stress
4. Vascular Function, Oxidative Stress and •NO Bioavailability in Aging
5. Aging, Mitochondrial Oxidative Stress, Mitochondrial DNA Damage and Endothelial Dysfunction





6. Aging and Mitochondrial DNA Repair
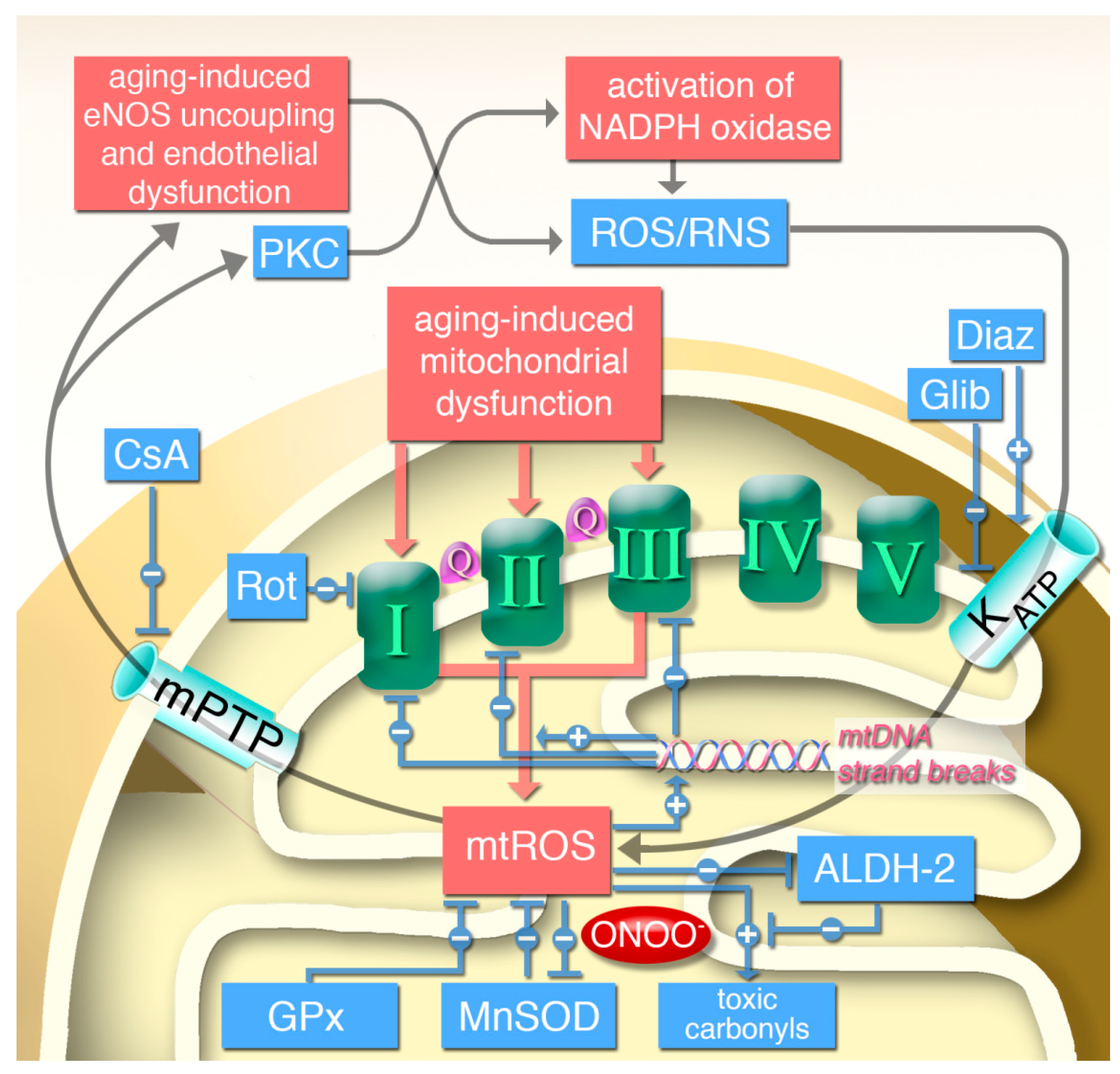
7. Crosstalk between Mitochondrial and NADPH Oxidase-Generated Reactive Oxygen and Nitrogen Species and Impact on Endothelial Function

8. Emerging Concepts of Aging
9. Clinical Impact
10. Perspective
Acknowledgments
Conflicts of Interest
References
- Kelly, D.T. Paul dudley white international lecture. Our future society. A global challenge. Circulation 1997, 95, 2459–2464. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G.; Levy, D. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part I: Aging arteries: A “set up” for vascular disease. Circulation 2003, 107, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Ras, R.T.; Streppel, M.T.; Draijer, R.; Zock, P.L. Flow-mediated dilation and cardiovascular risk prediction: A systematic review with meta-analysis. Int. J. Cardiol. 2013, 168, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.D.; Mingorance, C.; Rodriguez-Rodriguez, R.; Alvarez de Sotomayor, M. Endothelial dysfunction and aging: An update. Ageing Res. Rev. 2010, 9, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, B.; Silber, S.; Richartz, B.M.; Pieper, L.; Klotsche, J.; Wittchen, H.U. Inadequate medical treatment of patients with coronary artery disease by primary care physicians in germany. Clin. Res. Cardiol. 2006, 95, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.L. The role of nitric oxide in erectile dysfunction: Implications for medical therapy. J. Clin. Hypertens. 2006, 8, 53–62. [Google Scholar] [CrossRef]
- Csiszar, A.; Toth, J.; Peti-Peterdi, J.; Ungvari, Z. The aging kidney: Role of endothelial oxidative stress and inflammation. Acta Physiol. Hung. 2007, 94, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Price, J.M.; Hellermann, A.; Hellermann, G.; Sutton, E.T. Aging enhances vascular dysfunction induced by the alzheimer’s peptide β-amyloid. Neurol. Res. 2004, 26, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Coleman, H.R.; Chan, C.C.; Ferris, F.L., III; Chew, E.Y. Age-related macular degeneration. Lancet 2008, 372, 1835–1845. [Google Scholar] [CrossRef]
- El Assar, M.; Angulo, J.; Rodriguez-Manas, L. Oxidative stress and vascular inflammation in aging. Free Radic. Biol. Med. 2013, 65, 380–401. [Google Scholar] [CrossRef] [PubMed]
- Cesari, M.; Onder, G.; Russo, A.; Zamboni, V.; Barillaro, C.; Ferrucci, L.; Pahor, M.; Bernabei, R.; Landi, F. Comorbidity and physical function: Results from the aging and longevity study in the Sirente geographic area (iLSIRENTE study). Gerontology 2006, 52, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Yancik, R.; Ershler, W.; Satariano, W.; Hazzard, W.; Cohen, H.J.; Ferrucci, L. Report of the national institute on aging task force on comorbidity. J. Gerontol. Ser. A 2007, 62, 275–280. [Google Scholar] [CrossRef]
- Wieland, G.D. From bedside to bench: Research in comorbidity and aging. Sci. Aging Knowl. Environ. 2005, 2005, pe29. [Google Scholar] [CrossRef] [PubMed]
- Munzel, T.; Daiber, A.; Mulsch, A. Explaining the phenomenon of nitrate tolerance. Circ. Res. 2005, 97, 618–628. [Google Scholar] [CrossRef] [PubMed]
- Munzel, T.; Daiber, A.; Ullrich, V.; Mulsch, A. Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cGMP-dependent protein kinase. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Oelze, M.; Kroller-Schon, S.; Steven, S.; Lubos, E.; Doppler, C.; Hausding, M.; Tobias, S.; Brochhausen, C.; Li, H.; Torzewski, M.; et al. Glutathione peroxidase-1 deficiency potentiates dysregulatory modifications of endothelial nitric oxide synthase and vascular dysfunction in aging. Hypertension 2014, 63, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Van der Loo, B.; Labugger, R.; Skepper, J.N.; Bachschmid, M.; Kilo, J.; Powell, J.M.; Palacios-Callender, M.; Erusalimsky, J.D.; Quaschning, T.; Malinski, T.; et al. Enhanced peroxynitrite formation is associated with vascular aging. J. Exp. Med. 2000, 192, 1731–1744. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Daiber, A.; Oelze, M.; Daub, S.; Steven, S.; Schuff, A.; Kroller-Schon, S.; Hausding, M.; Wenzel, P.; Schulz, E.; Gori, T.; et al. Vascular redox signaling, redox switches in endothelial nitric oxide synthase and endothelial dysfunction. In Systems Biology of Free Radicals and Antioxidants; Laher, I., Ed.; Springer-Verlag: Berlin, Germany; Heidelberg, Germany, 2014; pp. 1177–1211. [Google Scholar]
- Donato, A.J.; Gano, L.B.; Eskurza, I.; Silver, A.E.; Gates, P.E.; Jablonski, K.; Seals, D.R. Vascular endothelial dysfunction with aging: Endothelin-1 and endothelial nitric oxide synthase. Am. J. Phys. Heart Circ. Physiol. 2009, 297, H425–H432. [Google Scholar] [CrossRef] [PubMed]
- Donato, A.J.; Magerko, K.A.; Lawson, B.R.; Durrant, J.R.; Lesniewski, L.A.; Seals, D.R. SIRT-1 and vascular endothelial dysfunction with ageing in mice and humans. J. Physiol. 2011, 589, 4545–4554. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Sasaki, S.; Nakagawa, K.; Kimura, M.; Noma, K.; Hara, K.; Jitsuiki, D.; Goto, C.; Oshima, T.; Chayama, K.; et al. Tetrahydrobiopterin improves aging-related impairment of endothelium-dependent vasodilation through increase in nitric oxide production. Atherosclerosis 2006, 186, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, P.; Schuhmacher, S.; Kienhofer, J.; Muller, J.; Hortmann, M.; Oelze, M.; Schulz, E.; Treiber, N.; Kawamoto, T.; Scharffetter-Kochanek, K.; et al. Manganese superoxide dismutase and aldehyde dehydrogenase deficiency increase mitochondrial oxidative stress and aggravate age-dependent vascular dysfunction. Cardiovasc. Res. 2008, 80, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, M.; Roddy, M.A.; Creager, S.J.; Creager, M.A. Aging progressively impairs endothelium-dependent vasodilation in forearm resistance vessels of humans. Hypertension 1996, 27, 849–853. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, T.; Schlinzig, T.; Krohn, K.; Meinertz, T.; Munzel, T. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation 2001, 104, 2673–2678. [Google Scholar] [CrossRef] [PubMed]
- Savji, N.; Rockman, C.B.; Skolnick, A.H.; Guo, Y.; Adelman, M.A.; Riles, T.; Berger, J.S. Association between advanced age and vascular disease in different arterial territories: A population database of over 3.6 million subjects. J. Am. Coll. Cardiol. 2013, 61, 1736–1743. [Google Scholar] [CrossRef] [PubMed]
- Ong, K.L.; Cheung, B.M.; Man, Y.B.; Lau, C.P.; Lam, K.S. Prevalence, awareness, treatment, and control of hypertension among united states adults 1999–2004. Hypertension 2007, 49, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Daiber, A.; Kienhoefer, J.; Zee, R.; Ullrich, V.; van der Loo, B.; Bachschmid, M. The role of mitochondrial reactive oxygen species formation for age-induced vascular dysfunction. In Aging and Age-Related Disorders; Bondy, S.C., Maiese, K., Eds.; Humana Press: Clifton, NJ, USA, 2010; pp. 237–257. [Google Scholar]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Lenaz, G.; Bovina, C.; D’Aurelio, M.; Fato, R.; Formiggini, G.; Genova, M.L.; Giuliano, G.; Pich, M.M.; Paolucci, U.; Castelli, G.P.; et al. Role of mitochondria in oxidative stress and aging. Ann. N. Y. Acad. Sci. 2002, 959, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Boveris, A.; Cadenas, E. Mitochondrial energy metabolism and redox signaling in brain aging and neurodegeneration. Antioxid. Redox Signal. 2014, 20, 353–371. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.J.; Vapaatalo, H.; Mervaala, E. Angiotensin II and vascular inflammation. Med. Sci. Monit. 2005, 11, RA194–RA205. [Google Scholar] [PubMed]
- Lau, D.; Baldus, S. Myeloperoxidase and its contributory role in inflammatory vascular disease. Pharmacol. Ther. 2006, 111, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Willerson, J.T.; Golino, P.; Eidt, J.; Campbell, W.B.; Buja, L.M. Specific platelet mediators and unstable coronary artery lesions. Experimental evidence and potential clinical implications. Circulation 1989, 80, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Seals, D.R.; Jablonski, K.L.; Donato, A.J. Aging and vascular endothelial function in humans. Clin. Sci. 2011, 120, 357–375. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Dinenno, F.A.; Seals, D.R. Age-related increase in femoral intima-media thickness in healthy humans. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2172. [Google Scholar] [CrossRef] [PubMed]
- Crimi, E.; Ignarro, L.J.; Napoli, C. Microcirculation and oxidative stress. Free Radic. Res. 2007, 41, 1364–1375. [Google Scholar] [CrossRef] [PubMed]
- Mayhan, W.G.; Arrick, D.M.; Sharpe, G.M.; Sun, H. Age-related alterations in reactivity of cerebral arterioles: Role of oxidative stress. Microcirculation 2008, 15, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Militante, J.; Lombardini, J.B. Age-related retinal degeneration in animal models of aging: Possible involvement of taurine deficiency and oxidative stress. Neurochem. Res. 2004, 29, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Maier, O. Interrelation of oxidative stress and inflammation in neurodegenerative disease: Role of TNF. Oxidative Med. Cell. Longev. 2015, 2015, 610813. [Google Scholar] [CrossRef] [PubMed]
- Blasiak, J.; Petrovski, G.; Vereb, Z.; Facsko, A.; Kaarniranta, K. Oxidative stress, hypoxia, and autophagy in the neovascular processes of age-related macular degeneration. BioMed. Res. Int. 2014, 2014, 768026. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Waters, W.A. Some recent developments in the chemistry of free radicals. J. Chem. Soc. 1946, 409–415. [Google Scholar] [CrossRef]
- Rogell, B.; Dean, R.; Lemos, B.; Dowling, D.K. Mito-nuclear interactions as drivers of gene movement on and off the X-chromosome. BMC Genomics 2014, 15, 330. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.D.; Ridnour, L.A.; Isenberg, J.S.; Flores-Santana, W.; Switzer, C.H.; Donzelli, S.; Hussain, P.; Vecoli, C.; Paolocci, N.; Ambs, S.; et al. The chemical biology of nitric oxide: Implications in cellular signaling. Free Radic. Biol. Med. 2008, 45, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Van der Loo, B.; Bachschmid, M.; Skepper, J.N.; Labugger, R.; Schildknecht, S.; Hahn, R.; Mussig, E.; Gygi, D.; Luscher, T.F. Age-associated cellular relocation of Sod 1 as a self-defense is a futile mechanism to prevent vascular aging. Biochem. Biophys. Res. Commun. 2006, 344, 972–980. [Google Scholar] [CrossRef] [PubMed]
- De Haan, J.B.; Bladier, C.; Lotfi-Miri, M.; Taylor, J.; Hutchinson, P.; Crack, P.J.; Hertzog, P.; Kola, I. Fibroblasts derived from Gpx1 knockout mice display senescent-like features and are susceptible to H2O2-mediated cell death. Free Radic. Biol. Med. 2004, 36, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Altschmied, J.; Haendeler, J. Thioredoxin-1 and endothelial cell aging: Role in cardiovascular diseases. Antioxid. Redox Signal. 2009, 11, 1733–1740. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.M.; Jones, D.P. Redox control systems in the nucleus: Mechanisms and functions. Antioxid. Redox Signal. 2010, 13, 489–509. [Google Scholar] [CrossRef] [PubMed]
- Salmon, A.B.; Richardson, A.; Perez, V.I. Update on the oxidative stress theory of aging: Does oxidative stress play a role in aging or healthy aging? Free Radic. Biol. Med. 2010, 48, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.A.; Didion, S.P.; Andresen, J.J.; Faraci, F.M. Effect of aging, MnSOD deficiency, and genetic background on endothelial function: Evidence for MnSOD haploinsufficiency. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1941–1946. [Google Scholar] [CrossRef] [PubMed]
- Didion, S.P.; Kinzenbaw, D.A.; Schrader, L.I.; Faraci, F.M. Heterozygous CuZn superoxide dismutase deficiency produces a vascular phenotype with aging. Hypertension 2006, 48, 1072–1079. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, S.; Czapski, G.; Lind, J.; Merenyi, G. Tyrosine nitration by simultaneous generation of •NO and O•2 under physiological conditions. How the radicals do the job. J. Biol. Chem. 2000, 275, 3031–3036. [Google Scholar] [CrossRef] [PubMed]
- Perez, V.I.; Bokov, A.; van Remmen, H.; Mele, J.; Ran, Q.; Ikeno, Y.; Richardson, A. Is the oxidative stress theory of aging dead? Biochim. Biophys. Acta 2009, 1790, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.L.; Lustgarten, M.S.; Jang, Y.; Richardson, A.; van Remmen, H. Trends in oxidative aging theories. Free Radic. Biol. Med. 2007, 43, 477–503. [Google Scholar] [CrossRef] [PubMed]
- Lebovitz, R.M.; Zhang, H.; Vogel, H.; Cartwright, J., Jr.; Dionne, L.; Lu, N.; Huang, S.; Matzuk, M.M. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9782–9787. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, T.T.; Carlson, E.J.; Melov, S.; Ursell, P.C.; Olson, J.L.; Noble, L.J.; Yoshimura, M.P.; Berger, C.; Chan, P.H.; et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 1995, 11, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.C.; Remmen, H.V. The mitochondrial theory of aging: Insight from transgenic and knockout mouse models. Exp. Gerontol. 2009, 44, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Chiao, Y.A.; Marcinek, D.J.; Szeto, H.H.; Rabinovitch, P.S. Mitochondrial oxidative stress in aging and healthspan. Longev. Healthspan 2014, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, R.T.; Walsh, M.E.; van Remmen, H. Mouse models of oxidative stress indicate a role for modulating healthy aging. J. Clin. Exp. Pathol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Berry, A.; Cirulli, F. The p66Shc gene paves the way for healthspan: Evolutionary and mechanistic perspectives. Neurosci. Biobehav. Rev. 2013, 37, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Wanagat, J.; Dai, D.F.; Rabinovitch, P. Mitochondrial oxidative stress and mammalian healthspan. Mech. Ageing Dev. 2010, 131, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and ugly. Am. J. Physiol. 1996, 271, C1424–C1437. [Google Scholar] [PubMed]
- Daiber, A.; Bachschmid, M. Enzyme inhibition by peroxynitrite-mediated tyrosine nitration and thiol oxidation. Curr. Enzym. Inhib. 2007, 3, 103–117. [Google Scholar] [CrossRef]
- Beckman, J.S. Protein tyrosine nitration and peroxynitrite. FASEB J. 2002, 16, 1144. [Google Scholar] [CrossRef] [PubMed]
- Quijano, C.; Alvarez, B.; Gatti, R.M.; Augusto, O.; Radi, R. Pathways of peroxynitrite oxidation of thiol groups. Biochem. J. 1997, 322, 167–173. [Google Scholar] [PubMed]
- MacMillan-Crow, L.A.; Crow, J.P.; Kerby, J.D.; Beckman, J.S.; Thompson, J.A. Nitration and inactivation of manganese superoxide dismutase in chronic rejection of human renal allografts. Proc. Natl. Acad. Sci. USA 1996, 93, 11853–11858. [Google Scholar] [CrossRef] [PubMed]
- Kuzkaya, N.; Weissmann, N.; Harrison, D.G.; Dikalov, S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: Implications for uncoupling endothelial nitric-oxide synthase. J. Biol. Chem. 2003, 278, 22546–22554. [Google Scholar] [CrossRef] [PubMed]
- Schulz, E.; Jansen, T.; Wenzel, P.; Daiber, A.; Munzel, T. Nitric oxide, tetrahydrobiopterin, oxidative stress, and endothelial dysfunction in hypertension. Antioxid. Redox Signal. 2008, 10, 1115–1126. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.I.; Eda, S.; Masada, M. Alterations of tetrahydrobiopterin biosynthesis and pteridine levels in mouse tissues during growth and aging. Brain Dev. 2000, 22, S45–S49. [Google Scholar] [CrossRef]
- Blackwell, K.A.; Sorenson, J.P.; Richardson, D.M.; Smith, L.A.; Suda, O.; Nath, K.; Katusic, Z.S. Mechanisms of aging-induced impairment of endothelium-dependent relaxation: Role of tetrahydrobiopterin. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2448–H2453. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.A.; Wang, T.Y.; Varadharaj, S.; Reyes, L.A.; Hemann, C.; Talukder, M.A.; Chen, Y.R.; Druhan, L.J.; Zweier, J.L. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 2010, 468, 1115–1118. [Google Scholar] [CrossRef] [PubMed]
- Schulz, E.; Wenzel, P.; Munzel, T.; Daiber, A. Mitochondrial redox signaling: Interaction of mitochondrial reactive oxygen species with other sources of oxidative stress. Antioxid. Redox Signal. 2014, 20, 308–324. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.H.; Shi, C.; Cohen, R.A. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J. Clin. Investig. 2002, 109, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [PubMed]
- Soucy, K.G.; Ryoo, S.; Benjo, A.; Lim, H.K.; Gupta, G.; Sohi, J.S.; Elser, J.; Aon, M.A.; Nyhan, D.; Shoukas, A.A.; et al. Impaired shear stress-induced nitric oxide production through decreased NOS phosphorylation contributes to age-related vascular stiffness. J. Appl. Physiol. 2006, 101, 1751–1759. [Google Scholar] [CrossRef] [PubMed]
- Cave, A.C.; Brewer, A.C.; Narayanapanicker, A.; Ray, R.; Grieve, D.J.; Walker, S.; Shah, A.M. Nadph oxidases in cardiovascular health and disease. Antioxid. Redox signal. 2006, 8, 691–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Paneni, F.; Osto, E.; Costantino, S.; Mateescu, B.; Briand, S.; Coppolino, G.; Perna, E.; Mocharla, P.; Akhmedov, A.; Kubant, R.; et al. Deletion of the activated protein-1 transcription factor JunD induces oxidative stress and accelerates age-related endothelial dysfunction. Circulation 2013, 127, 1229–1240. [Google Scholar] [CrossRef] [PubMed]
- Roubenoff, R.; Harris, T.B.; Abad, L.W.; Wilson, P.W.; Dallal, G.E.; Dinarello, C.A. Monocyte cytokine production in an elderly population: Effect of age and inflammation. J. Gerontol. Ser. A 1998, 53, M20–M26. [Google Scholar] [CrossRef]
- Moe, K.T.; Aulia, S.; Jiang, F.; Chua, Y.L.; Koh, T.H.; Wong, M.C.; Dusting, G.J. Differential upregulation of Nox homologues of NADPH oxidase by tumor necrosis factor-α in human aortic smooth muscle and embryonic kidney cells. J. Cell. Mol. Med. 2006, 10, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Karbach, S.; Wenzel, P.; Waisman, A.; Munzel, T.; Daiber, A. eNOS uncoupling in cardiovascular diseases—The role of oxidative stress and inflammation. Curr. Pharm. Des. 2014, 20, 3579–3594. [Google Scholar] [CrossRef] [PubMed]
- Nandi, J.; Saud, B.; Zinkievich, J.M.; Yang, Z.J.; Levine, R.A. TNF-α modulates INOS expression in an experimental rat model of indomethacin-induced jejunoileitis. Mol. Cell. Biochem. 2010, 336, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Csiszar, A.; Edwards, J.G.; Kaminski, P.M.; Wolin, M.S.; Kaley, G.; Koller, A. Increased superoxide production in coronary arteries in hyperhomocysteinemia: Role of tumor necrosis factor-α, NAD(P)H oxidase, and inducible nitric oxide synthase. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Busik, J.V.; Mohr, S.; Grant, M.B. Hyperglycemia-induced reactive oxygen species toxicity to endothelial cells is dependent on paracrine mediators. Diabetes 2008, 57, 1952–1965. [Google Scholar] [CrossRef] [PubMed]
- Csiszar, A.; Labinskyy, N.; Smith, K.; Rivera, A.; Orosz, Z.; Ungvari, Z. Vasculoprotective effects of anti-tumor necrosis factor-α treatment in aging. Am. J. Pathol. 2007, 170, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L.; Corsi, A.; Lauretani, F.; Bandinelli, S.; Bartali, B.; Taub, D.D.; Guralnik, J.M.; Longo, D.L. The origins of age-related proinflammatory state. Blood 2005, 105, 2294–2299. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, P.; Knorr, M.; Kossmann, S.; Stratmann, J.; Hausding, M.; Schuhmacher, S.; Karbach, S.H.; Schwenk, M.; Yogev, N.; Schulz, E.; et al. Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation 2011, 124, 1370–1381. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Hoch, N.E.; Brown, K.A.; McCann, L.A.; Rahman, A.; Dikalov, S.; Goronzy, J.; Weyand, C.; Harrison, D.G. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J. Exp. Med. 2007, 204, 2449–2460. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.; Stefanovic, N.; Pete, J.; Calkin, A.C.; Giunti, S.; Thallas-Bonke, V.; Jandeleit-Dahm, K.A.; Allen, T.J.; Kola, I.; Cooper, M.E.; et al. Lack of the antioxidant enzyme glutathione peroxidase-1 accelerates atherosclerosis in diabetic apolipoprotein e-deficient mice. Circulation 2007, 115, 2178–2187. [Google Scholar] [CrossRef] [PubMed]
- Forgione, M.A.; Cap, A.; Liao, R.; Moldovan, N.I.; Eberhardt, R.T.; Lim, C.C.; Jones, J.; Goldschmidt-Clermont, P.J.; Loscalzo, J. Heterozygous cellular glutathione peroxidase deficiency in the mouse: Abnormalities in vascular and cardiac function and structure. Circulation 2002, 106, 1154–1158. [Google Scholar] [CrossRef] [PubMed]
- Chrissobolis, S.; Didion, S.P.; Kinzenbaw, D.A.; Schrader, L.I.; Dayal, S.; Lentz, S.R.; Faraci, F.M. Glutathione peroxidase-1 plays a major role in protecting against angiotensin II-induced vascular dysfunction. Hypertension 2008, 51, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Blankenberg, S.; Rupprecht, H.J.; Bickel, C.; Torzewski, M.; Hafner, G.; Tiret, L.; Smieja, M.; Cambien, F.; Meyer, J.; Lackner, K.J. Glutathione peroxidase 1 activity and cardiovascular events in patients with coronary artery disease. N. Engl. J. Med. 2003, 349, 1605–1613. [Google Scholar] [CrossRef] [PubMed]
- Kroller-Schon, S.; Steven, S.; Kossmann, S.; Scholz, A.; Daub, S.; Oelze, M.; Xia, N.; Hausding, M.; Mikhed, Y.; Zinssius, E.; et al. Molecular mechanisms of the crosstalk between mitochondria and NADPH oxidase through reactive oxygen species-studies in white blood cells and in animal models. Antioxid. Redox Signal. 2014, 20, 247–266. [Google Scholar] [CrossRef] [PubMed]
- Hausding, M.; Jurk, K.; Daub, S.; Kroller-Schon, S.; Stein, J.; Schwenk, M.; Oelze, M.; Mikhed, Y.; Kerahrodi, J.G.; Kossmann, S.; et al. CD40L contributes to angiotensin II-induced pro-thrombotic state, vascular inflammation, oxidative stress and endothelial dysfunction. Basic Res. Cardiol. 2013, 108, 386. [Google Scholar] [CrossRef] [PubMed]
- Fleming, I.; Fisslthaler, B.; Dimmeler, S.; Kemp, B.E.; Busse, R. Phosphorylation of Thr495 regulates Ca2+/calmodulin-dependent endothelial nitric oxide synthase activity. Circ. Res. 2001, 88, E68–E75. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.I.; Fulton, D.; Babbitt, R.; Fleming, I.; Busse, R.; Pritchard, K.A., Jr.; Sessa, W.C. Phosphorylation of threonine 497 in endothelial nitric-oxide synthase coordinates the coupling of l-arginine metabolism to efficient nitric oxide production. J. Biol. Chem. 2003, 278, 44719–44726. [Google Scholar] [CrossRef] [PubMed]
- Loot, A.E.; Schreiber, J.G.; Fisslthaler, B.; Fleming, I. Angiotensin ii impairs endothelial function via tyrosine phosphorylation of the endothelial nitric oxide synthase. J. Exp. Med. 2009, 206, 2889–2896. [Google Scholar] [CrossRef] [PubMed]
- Brune, B.; Schmidt, K.U.; Ullrich, V. Activation of soluble guanylate cyclase by carbon monoxide and inhibition by superoxide anion. Eur. J. Biochem. 1990, 192, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Lauer, N.; Mulsch, A.; Kojda, G. The effect of peroxynitrite on the catalytic activity of soluble guanylyl cyclase. Free Radic. Biol. Med. 2001, 31, 1360–1367. [Google Scholar] [CrossRef]
- Artz, J.D.; Schmidt, B.; McCracken, J.L.; Marletta, M.A. Effects of nitroglycerin on soluble guanylate cyclase: Implications for nitrate tolerance. J. Biol. Chem. 2002, 277, 18253–18256. [Google Scholar] [CrossRef] [PubMed]
- Crassous, P.A.; Couloubaly, S.; Huang, C.; Zhou, Z.; Baskaran, P.; Kim, D.D.; Papapetropoulos, A.; Fioramonti, X.; Duran, W.N.; Beuve, A. Soluble guanylyl cyclase is a target of angiotensin II-induced nitrosative stress in a hypertensive rat model. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H597–H604. [Google Scholar] [CrossRef] [PubMed]
- Mayer, B.; Kleschyov, A.L.; Stessel, H.; Russwurm, M.; Munzel, T.; Koesling, D.; Schmidt, K. Inactivation of soluble guanylate cyclase by stoichiometric S-nitrosation. Mol. Pharmacol. 2009, 75, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Sayed, N.; Kim, D.D.; Fioramonti, X.; Iwahashi, T.; Duran, W.N.; Beuve, A. Nitroglycerin-induced S-nitrosylation and desensitization of soluble guanylyl cyclase contribute to nitrate tolerance. Circ. Res. 2008, 103, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Stasch, J.P.; Schmidt, P.M.; Nedvetsky, P.I.; Nedvetskaya, T.Y.; H S, A.K.; Meurer, S.; Deile, M.; Taye, A.; Knorr, A.; Lapp, H.; et al. Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J. Clin. Investig. 2006, 116, 2552–2561. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Daum, G.; Fischer, J.W.; Hawkins, S.; Bochaton-Piallat, M.L.; Gabbiani, G.; Clowes, A.W. Loss of expression of the β subunit of soluble guanylyl cyclase prevents nitric oxide-mediated inhibition of DNA synthesis in smooth muscle cells of old rats. Circ. Res. 2000, 86, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Ruetten, H.; Zabel, U.; Linz, W.; Schmidt, H.H. Downregulation of soluble guanylyl cyclase in young and aging spontaneously hypertensive rats. Circ. Res. 1999, 85, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Kloss, S.; Bouloumie, A.; Mulsch, A. Aging and chronic hypertension decrease expression of rat aortic soluble guanylyl cyclase. Hypertension 2000, 35, 43–47. [Google Scholar] [PubMed]
- Lubos, E.; Mahoney, C.E.; Leopold, J.A.; Zhang, Y.Y.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase-1 modulates lipopolysaccharide-induced adhesion molecule expression in endothelial cells by altering CD14 expression. FASEB J. 2010, 24, 2525–2532. [Google Scholar] [CrossRef] [PubMed]
- Kessler, P.; Bauersachs, J.; Busse, R.; Schini-Kerth, V.B. Inhibition of inducible nitric oxide synthase restores endothelium-dependent relaxations in proinflammatory mediator-induced blood vessels. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1746–1755. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, K.S.; Zamamiri-Davis, F.; Stewart, J.B.; Thompson, J.T.; Sordillo, L.M.; Reddy, C.C. Selenium deficiency increases the expression of inducible nitric oxide synthase in RAW 264.7 macrophages: Role of nuclear factor-κB in up-regulation. Biochem. J. 2002, 366, 203–209. [Google Scholar] [PubMed]
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 2011, 208, 519–533. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.R.; Visioli, F.; Frei, B.; Hagen, T.M. Age-related changes in endothelial nitric oxide synthase phosphorylation and nitric oxide dependent vasodilation: Evidence for a novel mechanism involving sphingomyelinase and ceramide-activated phosphatase 2A. Aging Cell 2006, 5, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, M.J.; Brixey, R.; Batchelor, H.; Hale, A.B.; Channon, K.M. Integrated redox sensor and effector functions for tetrahydrobiopterin- and glutathionylation-dependent endothelial nitric-oxide synthase uncoupling. J. Biol. Chem. 2013, 288, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Schriner, S.E.; Linford, N.J.; Martin, G.M.; Treuting, P.; Ogburn, C.E.; Emond, M.; Coskun, P.E.; Ladiges, W.; Wolf, N.; van Remmen, H.; et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005, 308, 1909–1911. [Google Scholar] [CrossRef] [PubMed]
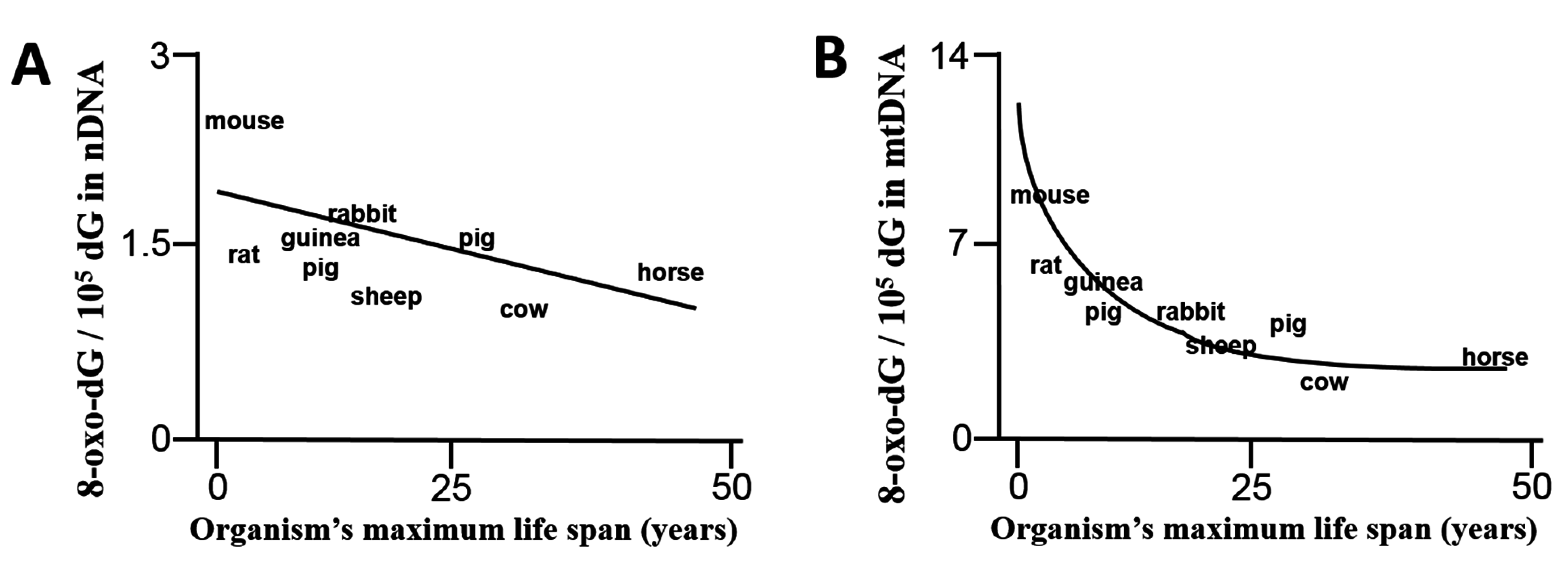
- Barja, G.; Herrero, A. Oxidative damage to mitochondrial DNA is inversely related to maximum life span in the heart and brain of mammals. FASEB J. 2000, 14, 312–318. [Google Scholar] [PubMed]
- Sastre, J.; Pallardo, F.V.; Vina, J. The role of mitochondrial oxidative stress in aging. Free Radic. Biol. Med. 2003, 35, 1–8. [Google Scholar] [CrossRef]
- Madamanchi, N.R.; Runge, M.S. Mitochondrial dysfunction in atherosclerosis. Circ. Res. 2007, 100, 460–473. [Google Scholar] [CrossRef] [PubMed]
- De Souza-Pinto, N.C.; Eide, L.; Hogue, B.A.; Thybo, T.; Stevnsner, T.; Seeberg, E.; Klungland, A.; Bohr, V.A. Repair of 8-oxodeoxyguanosine lesions in mitochondrial DNA depends on the oxoguanine DNA glycosylase (OGG1) gene and 8-oxoguanine accumulates in the mitochondrial dna of OGG1-defective mice. Cancer Res. 2001, 61, 5378–5381. [Google Scholar] [PubMed]
- De Souza-Pinto, N.C.; Hogue, B.A.; Bohr, V.A. DNA repair and aging in mouse liver: 8-oxodG glycosylase activity increase in mitochondrial but not in nuclear extracts. Free Radic. Biol. Med. 2001, 30, 916–923. [Google Scholar] [CrossRef]
- Souza-Pinto, N.C.; Croteau, D.L.; Hudson, E.K.; Hansford, R.G.; Bohr, V.A. Age-associated increase in 8-oxo-deoxyguanosine glycosylase/ap lyase activity in rat mitochondria. Nucleic Acids Res. 1999, 27, 1935–1942. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly, Y.M.; Gidlof, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Lewis, W.; Day, B.J.; Kohler, J.J.; Hosseini, S.H.; Chan, S.S.; Green, E.C.; Haase, C.P.; Keebaugh, E.S.; Long, R.; Ludaway, T.; et al. Decreased mtDNA, oxidative stress, cardiomyopathy, and death from transgenic cardiac targeted human mutant polymerase γ. Lab. Investig. 2007, 87, 326–335. [Google Scholar] [PubMed]
- Finsterer, J. Overview on visceral manifestations of mitochondrial disorders. Neth. J. Med. 2006, 64, 61–71. [Google Scholar] [PubMed]
- Anan, R.; Nakagawa, M.; Miyata, M.; Higuchi, I.; Nakao, S.; Suehara, M.; Osame, M.; Tanaka, H. Cardiac involvement in mitochondrial diseases. A study on 17 patients with documented mitochondrial DNA defects. Circulation 1995, 91, 955–961. [Google Scholar] [CrossRef] [PubMed]
- Pinsky, D.J.; Oz, M.C.; Koga, S.; Taha, Z.; Broekman, M.J.; Marcus, A.J.; Liao, H.; Naka, Y.; Brett, J.; Cannon, P.J.; et al. Cardiac preservation is enhanced in a heterotopic rat transplant model by supplementing the nitric oxide pathway. J. Clin. Investig. 1994, 93, 2291–2297. [Google Scholar] [CrossRef] [PubMed]
- Zeviani, M.; di Donato, S. Mitochondrial disorders. Brain 2004, 127, 2153–2172. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, S.W.; Patterson, C.; Knight-Lozano, C.A.; Burow, D.L.; Conklin, C.A.; Hu, Z.; Reuf, J.; Horaist, C.; Lebovitz, R.; Hunter, G.C.; et al. Mitochondrial integrity and function in atherogenesis. Circulation 2002, 106, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Sevini, F.; Giuliani, C.; Vianello, D.; Giampieri, E.; Santoro, A.; Biondi, F.; Garagnani, P.; Passarino, G.; Luiselli, D.; Capri, M.; et al. mtDNA mutations in human aging and longevity: Controversies and new perspectives opened by high-throughput technologies. Exp.Gerontol. 2014, 56, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Itsara, L.S.; Kennedy, S.R.; Fox, E.J.; Yu, S.; Hewitt, J.J.; Sanchez-Contreras, M.; Cardozo-Pelaez, F.; Pallanck, L.J. Oxidative stress is not a major contributor to somatic mitochondrial DNA mutations. PLoS Genet. 2014, 10, e1003974. [Google Scholar] [CrossRef] [PubMed]
- Larsson, N.G. Somatic mitochondrial DNA mutations in mammalian aging. Annu. Rev. Biochem. 2010, 79, 683–706. [Google Scholar] [CrossRef] [PubMed]
- De Bont, R.; van Larebeke, N. Endogenous DNA damage in humans: A review of quantitative data. Mutagenesis 2004, 19, 169–185. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.R.; Salk, J.J.; Schmitt, M.W.; Loeb, L.A. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 2013, 9, e1003794. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.Y.; Kim, D.K.; Mook-Jung, I. The role of mitochondrial DNA mutation on neurodegenerative diseases. Exp. Mol. Med. 2015, 47, e150. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H. Amyloid β, mitochondrial structural and functional dynamics in alzheimer’s disease. Exp. Neurol. 2009, 218, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Canugovi, C.; Shamanna, R.A.; Croteau, D.L.; Bohr, V.A. Base excision DNA repair levels in mitochondrial lysates of alzheimer’s disease. Neurobiol. Aging 2014, 35, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, K.J.; Ratnaike, T.E.; de Gruyter, H.L.; Jaros, E.; Turnbull, D.M. Mitochondrial DNA deletions cause the biochemical defect observed in alzheimer’s disease. Neurobiol. Aging 2012, 33, 2210–2214. [Google Scholar] [CrossRef] [PubMed]
- Muftuoglu, M.; Mori, M.P.; de Souza-Pinto, N.C. Formation and repair of oxidative damage in the mitochondrial DNA. Mitochondrion 2014, 17, 164–181. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Demple, B. DNA repair in mammalian mitochondria: Much more than we thought? Environ. Mol. Mutagen. 2010, 51, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Bogenhagen, D.F. Repair of mtDNA in vertebrates. Am. J. Hum. Genet. 1999, 64, 1276–1281. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, K.; Ohtsubo, T.; Oda, H.; Fujiwara, T.; Kang, D.; Sugimachi, K.; Nakabeppu, Y. Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol. Biol. Cell 1999, 10, 1637–1652. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, H.; Otterlei, M.; Haug, T.; Solum, K.; Nagelhus, T.A.; Skorpen, F.; Krokan, H.E. Nuclear and mitochondrial uracil-DNA glycosylases are generated by alternative splicing and transcription from different positions in the UNG gene. Nucleic Acids Res. 1997, 25, 750–755. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Kohmoto, T.; Tabata, R.; Seki, Y. Differential intracellular localization of the human and mouse endonuclease III homologs and analysis of the sorting signals. DNA Repair 2002, 1, 847–854. [Google Scholar] [CrossRef]
- Hu, J.; de Souza-Pinto, N.C.; Haraguchi, K.; Hogue, B.A.; Jaruga, P.; Greenberg, M.M.; Dizdaroglu, M.; Bohr, V.A. Repair of formamidopyrimidines in DNA involves different glycosylases: Role of the OGG1, NTH1, and NEIL1 enzymes. J. Biol. Chem. 2005, 280, 40544–40551. [Google Scholar] [CrossRef] [PubMed]
- Ohtsubo, T.; Nishioka, K.; Imaiso, Y.; Iwai, S.; Shimokawa, H.; Oda, H.; Fujiwara, T.; Nakabeppu, Y. Identification of human muty homolog (hMYH) as a repair enzyme for 2-hydroxyadenine in DNA and detection of multiple forms of hMYH located in nuclei and mitochondria. Nucleic Acids Res. 2000, 28, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Kim, H.L.; Kim, Y.J.; Weon, J.I.; Sung, M.K.; Chung, H.W.; Seo, Y.R. Human AP endonuclease 1: A potential marker for the prediction of environmental carcinogenesis risk. Oxidative Med. Cell. Longev. 2014, 2014, 730301. [Google Scholar] [CrossRef] [PubMed]
- Mason, P.A.; Matheson, E.C.; Hall, A.G.; Lightowlers, R.N. Mismatch repair activity in mammalian mitochondria. Nucleic Acids Res. 2003, 31, 1052–1058. [Google Scholar] [CrossRef] [PubMed]
- De Souza-Pinto, N.C.; Mason, P.A.; Hashiguchi, K.; Weissman, L.; Tian, J.; Guay, D.; Lebel, M.; Stevnsner, T.V.; Rasmussen, L.J.; Bohr, V.A. Novel DNA mismatch-repair activity involving YB-1 in human mitochondria. DNA Repair 2009, 8, 704–719. [Google Scholar] [CrossRef] [PubMed]
- Pohjoismaki, J.L.; Goffart, S.; Tyynismaa, H.; Willcox, S.; Ide, T.; Kang, D.; Suomalainen, A.; Karhunen, P.J.; Griffith, J.D.; Holt, I.J.; et al. Human heart mitochondrial DNA is organized in complex catenated networks containing abundant four-way junctions and replication forks. J. Biol. Chem. 2009, 284, 21446–21457. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.J. Mechanism of homologous recombination and implications for aging-related deletions in mitochondrial DNA. Microbiol. Mol. Biol. Rev. 2013, 77, 476–496. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.J.; Guan, M.X.; Clark-Walker, G.D. MGM101, a nuclear gene involved in maintenance of the mitochondrial genome in saccharomyces cerevisiae. Nucleic Acids Res. 1993, 21, 3473–3477. [Google Scholar] [CrossRef] [PubMed]
- Gredilla, R.; Garm, C.; Stevnsner, T. Nuclear and mitochondrial DNA repair in selected eukaryotic aging model systems. Oxidative Med. Cell. Longev. 2012, 2012, 282438. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Cao, G.; Hastings, T.; Feng, Y.; Pei, W.; O’Horo, C.; Chen, J. Age-dependent decline of DNA repair activity for oxidative lesions in rat brain mitochondria. J. Neurochem. 2002, 81, 1273–1284. [Google Scholar] [CrossRef] [PubMed]
- Daiber, A.; Oelze, M.; Wenzel, P.; Wickramanayake, J.M.; Schuhmacher, S.; Jansen, T.; Lackner, K.J.; Torzewski, M.; Munzel, T. Nitrate tolerance as a model of vascular dysfunction: Roles for mitochondrial aldehyde dehydrogenase and mitochondrial oxidative stress. Pharmacol. Rep. 2009, 61, 33–48. [Google Scholar] [CrossRef]
- Daiber, A.; Oelze, M.; Coldewey, M.; Bachschmid, M.; Wenzel, P.; Sydow, K.; Wendt, M.; Kleschyov, A.L.; Stalleicken, D.; Ullrich, V.; et al. Oxidative stress and mitochondrial aldehyde dehydrogenase activity: A comparison of pentaerythritol tetranitrate with other organic nitrates. Mol. Pharmacol. 2004, 66, 1372–1382. [Google Scholar] [CrossRef] [PubMed]
- Sydow, K.; Daiber, A.; Oelze, M.; Chen, Z.; August, M.; Wendt, M.; Ullrich, V.; Mulsch, A.; Schulz, E.; Keaney, J.F., Jr.; et al. Central role of mitochondrial aldehyde dehydrogenase and reactive oxygen species in nitroglycerin tolerance and cross-tolerance. J. Clin. Investig. 2004, 113, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Esplugues, J.V.; Rocha, M.; Nunez, C.; Bosca, I.; Ibiza, S.; Herance, J.R.; Ortega, A.; Serrador, J.M.; D’Ocon, P.; Victor, V.M. Complex I dysfunction and tolerance to nitroglycerin: An approach based on mitochondrial-targeted antioxidants. Circ. Res. 2006, 99, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, P.; Mollnau, H.; Oelze, M.; Schulz, E.; Wickramanayake, J.M.; Muller, J.; Schuhmacher, S.; Hortmann, M.; Baldus, S.; Gori, T.; et al. First evidence for a crosstalk between mitochondrial and nadph oxidase-derived reactive oxygen species in nitroglycerin-triggered vascular dysfunction. Antioxid. Redox Signal. 2008, 10, 1435–1447. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.I.; Nazarewicz, R.R.; Bikineyeva, A.; Hilenski, L.; Lassegue, B.; Griendling, K.K.; Harrison, D.G.; Dikalova, A.E. Nox2-induced production of mitochondrial superoxide in angiotensin II-mediated endothelial oxidative stress and hypertension. Antioxid. Redox Signal. 2014, 20, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 2008, 102, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Nazarewicz, R.R.; Dikalova, A.E.; Bikineyeva, A.; Dikalov, S.I. Nox2 as a potential target of mitochondrial superoxide and its role in endothelial oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1131–H1140. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P. Triggering mitochondrial radical release: A new function for NADPH oxidases. Hypertension 2005, 45, 847–848. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Zhang, G.X.; Nishiyama, A.; Shokoji, T.; Yao, L.; Fan, Y.Y.; Rahman, M.; Abe, Y. Mitochondria-derived reactive oxygen species and vascular MAP kinases: Comparison of angiotensin II and diazoxide. Hypertension 2005, 45, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.; Harrison, D.G.; Dikalov, S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 2010, 107, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Fukui, T.; Ishizaka, N.; Rajagopalan, S.; Laursen, J.B.; Capers, Q.T.; Taylor, W.R.; Harrison, D.G.; de Leon, H.; Wilcox, J.N.; Griendling, K.K. p22phox mRNA expression and NADPH oxidase activity are increased in aortas from hypertensive rats. Circ. Res. 1997, 80, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Cheresh, P.; Kim, S.J.; Tulasiram, S.; Kamp, D.W. Oxidative stress and pulmonary fibrosis. Biochim. Biophys. Acta 2013, 1832, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Nazarewicz, R.R.; Dikalov, S.I. Mitochondrial ROS in the prohypertensive immune response. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R98–R100. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Hajieva, P.; Moosmann, B. Adaptive antioxidant methionine accumulation in respiratory chain complexes explains the use of a deviant genetic code in mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 16496–16501. [Google Scholar] [CrossRef] [PubMed]
- Moskovitz, J.; Bar-Noy, S.; Williams, W.M.; Requena, J.; Berlett, B.S.; Stadtman, E.R. Methionine sulfoxide reductase (MsrA) is a regulator of antioxidant defense and lifespan in mammals. Proc. Natl. Acad. Sci. USA 2001, 98, 12920–12925. [Google Scholar] [CrossRef] [PubMed]
- Stadtman, E.R.; Moskovitz, J.; Berlett, B.S.; Levine, R.L. Cyclic oxidation and reduction of protein methionine residues is an important antioxidant mechanism. Mol. Cell. Biochem. 2002, 234–235, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Taungjaruwinai, W.M.; Bhawan, J.; Keady, M.; Thiele, J.J. Differential expression of the antioxidant repair enzyme methionine sulfoxide reductase (MSRA and MSRB) in human skin. Am. J. Dermatopathol. 2009, 31, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Mochin, M.T.; Underwood, K.F.; Cooper, B.; McLenithan, J.C.; Pierce, A.D.; Nalvarte, C.; Arbiser, J.; Karlsson, A.I.; Moise, A.R.; Moskovitz, J.; et al. Hyperglycemia and redox status regulate RUNX2 DNA-binding and an angiogenic phenotype in endothelial cells. Microvasc. Res. 2015, 97, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Chen, W.; Yin, J.; Chen, S.; Zhang, J.; Gong, J. Methionine sulfoxide reductase A rs10903323 G/A polymorphism is associated with increased risk of coronary artery disease in a chinese population. Clin. Biochem. 2013, 46, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Haenold, R.; Wassef, R.; Brot, N.; Neugebauer, S.; Leipold, E.; Heinemann, S.H.; Hoshi, T. Protection of vascular smooth muscle cells by over-expressed methionine sulphoxide reductase A: Role of intracellular localization and substrate availability. Free Radic. Res. 2008, 42, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Moosmann, B.; Behl, C. Mitochondrially encoded cysteine predicts animal lifespan. Aging Cell 2008, 7, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Camici, G.G.; Cosentino, F.; Tanner, F.C.; Luscher, T.F. The role of p66Shc deletion in age-associated arterial dysfunction and disease states. J. Appl. Physiol. 2008, 105, 1628–1631. [Google Scholar] [CrossRef] [PubMed]
- Francia, P.; delli Gatti, C.; Bachschmid, M.; Martin-Padura, I.; Savoia, C.; Migliaccio, E.; Pelicci, P.G.; Schiavoni, M.; Luscher, T.F.; Volpe, M.; et al. Deletion of p66Shc gene protects against age-related endothelial dysfunction. Circulation 2004, 110, 2889–2895. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.; Fischer, F.; Wolters, D.; Steegborn, C. Activation of the lifespan regulator p66Shc through reversible disulfide bond formation. Proc. Natl. Acad. Sci. USA 2008, 105, 5705–5709. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Rimessi, A.; Marchi, S.; Orsini, F.; Migliaccio, E.; Giorgio, M.; Contursi, C.; Minucci, S.; Mantovani, F.; Wieckowski, M.R.; et al. Protein kinase C β and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science 2007, 315, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Rizzuto, R. P66Shc, oxidative stress and aging: Importing a lifespan determinant into mitochondria. Cell. Cycle 2008, 7, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Rota, M.; LeCapitaine, N.; Hosoda, T.; Boni, A.; De Angelis, A.; Padin-Iruegas, M.E.; Esposito, G.; Vitale, S.; Urbanek, K.; Casarsa, C.; et al. Diabetes promotes cardiac stem cell aging and heart failure, which are prevented by deletion of the p66Shc gene. Circ. Res. 2006, 99, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Yamamori, T.; White, A.R.; Mattagajasingh, I.; Khanday, F.A.; Haile, A.; Qi, B.; Jeon, B.H.; Bugayenko, A.; Kasuno, K.; Berkowitz, D.E.; et al. p66Shc regulates endothelial no production and endothelium-dependent vasorelaxation: Implications for age-associated vascular dysfunction. J. Mol. Cell. Cardiol. 2005, 39, 992–995. [Google Scholar] [CrossRef] [PubMed]
- Trinei, M.; Giorgio, M.; Cicalese, A.; Barozzi, S.; Ventura, A.; Migliaccio, E.; Milia, E.; Padura, I.M.; Raker, V.A.; Maccarana, M.; et al. A p53-p66Shc signalling pathway controls intracellular redox status, levels of oxidation-damaged DNA and oxidative stress-induced apoptosis. Oncogene 2002, 21, 3872–3878. [Google Scholar] [CrossRef] [PubMed]
- Di Lisa, F.; Kaludercic, N.; Carpi, A.; Menabo, R.; Giorgio, M. Mitochondrial pathways for ROS formation and myocardial injury: The relevance of p66Shc and monoamine oxidase. Basic Res. Cardiol. 2009, 104, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Spescha, R.D.; Klohs, J.; Semerano, A.; Giacalone, G.; Derungs, R.S.; Reiner, M.F.; Rodriguez Gutierrez, D.; Mendez-Carmona, N.; Glanzmann, M.; Savarese, G.; et al. Post-ischaemic silencing of p66Shc reduces ischaemia/reperfusion brain injury and its expression correlates to clinical outcome in stroke. Eur. Heart J. 2015, 36, 1590–1600. [Google Scholar] [CrossRef] [PubMed]
- Moskalev, A.A.; Aliper, A.M.; Smit-McBride, Z.; Buzdin, A.; Zhavoronkov, A. Genetics and epigenetics of aging and longevity. Cell. Cycle 2014, 13, 1063–1077. [Google Scholar] [CrossRef] [PubMed]
- Barbot, W.; Dupressoir, A.; Lazar, V.; Heidmann, T. Epigenetic regulation of an IAP retrotransposon in the aging mouse: Progressive demethylation and de-silencing of the element by its repetitive induction. Nucleic Acids Res. 2002, 30, 2365–2373. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Nunez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003, 113, 703–716. [Google Scholar] [CrossRef]
- Tsurumi, A.; Li, W.X. Global heterochromatin loss: A unifying theory of aging? Epigenetics 2012, 7, 680–688. [Google Scholar] [CrossRef] [PubMed]
- McCauley, B.S.; Dang, W. Histone methylation and aging: Lessons learned from model systems. Biochim. Biophys. Acta 2014, 1839, 1454–1462. [Google Scholar] [CrossRef] [PubMed]
- Bilsland, A.E.; Revie, J.; Keith, W. Microrna and senescence: The senectome, integration and distributed control. Crit. Rev. Oncog. 2013, 18, 373–390. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Iekushi, K.; Lechner, S.; Seeger, T.; Fischer, A.; Heydt, S.; Kaluza, D.; Treguer, K.; Carmona, G.; Bonauer, A.; et al. MicroRNA-34a regulates cardiac ageing and function. Nature 2013, 495, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Guarner, V.; Rubio-Ruiz, M.E. Low-grade systemic inflammation connects aging, metabolic syndrome and cardiovascular disease. Interdiscip. Top. Gerontol. 2015, 40, 99–106. [Google Scholar] [PubMed]
- Howcroft, T.K.; Campisi, J.; Louis, G.B.; Smith, M.T.; Wise, B.; Wyss-Coray, T.; Augustine, A.D.; McElhaney, J.E.; Kohanski, R.; Sierra, F. The role of inflammation in age-related disease. Aging 2013, 5, 84–93. [Google Scholar] [PubMed]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.J.; et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 2013, 4, 2192. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.W. Established risk factors and coronary artery disease: The framingham study. Am. J. Hypertens. 1994, 7, 7S–12S. [Google Scholar] [PubMed]
- Munzel, T.; Sinning, C.; Post, F.; Warnholtz, A.; Schulz, E. Pathophysiology, diagnosis and prognostic implications of endothelial dysfunction. Ann. Med. 2008, 40, 180–196. [Google Scholar] [CrossRef] [PubMed]
- Munzel, T.; Gori, T.; Bruno, R.M.; Taddei, S. Is oxidative stress a therapeutic target in cardiovascular disease? Eur. Heart J. 2010, 31, 2741–2748. [Google Scholar] [CrossRef] [PubMed]
- Schachinger, V.; Britten, M.B.; Zeiher, A.M. Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation 2000, 101, 1899–1906. [Google Scholar] [CrossRef] [PubMed]
- Gori, T.; Munzel, T. Oxidative stress and endothelial dysfunction: Therapeutic implications. Ann. Med. 2011, 43, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.F.; Chen, D.D.; Daiber, A.; Faraci, F.M.; Li, H.; Rembold, C.M.; Laher, I. Free radical biology of the cardiovascular system. Clin. Sci. 2012, 123, 73–91. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Matsumoto, M.; Den, Y.B.; Iwai, K.; Munehira, J.; Hattori, H.; Hoshino, T.; Yamada, K.; Kawanishi, K.; Tsuchiya, H. Impaired endothelial function in hypertensive elderly patients evaluated by high resolution ultrasonography. Can. J. Cardiol. 1999, 15, 563–568. [Google Scholar] [PubMed]
- Wray, D.W.; Nishiyama, S.K.; Harris, R.A.; Zhao, J.; McDaniel, J.; Fjeldstad, A.S.; Witman, M.A.; Ives, S.J.; Barrett-O’Keefe, Z.; Richardson, R.S. Acute reversal of endothelial dysfunction in the elderly after antioxidant consumption. Hypertension 2012, 59, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Jousilahti, P.; Vartiainen, E.; Tuomilehto, J.; Puska, P. Sex, age, cardiovascular risk factors, and coronary heart disease: A prospective follow-up study of 14 786 middle-aged men and women in Finland. Circulation 1999, 99, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Holzer, M.; Trieb, M.; Konya, V.; Wadsack, C.; Heinemann, A.; Marsche, G. Aging affects high-density lipoprotein composition and function. Biochim. Biophys. Acta 2013, 1831, 1442–1448. [Google Scholar] [CrossRef] [PubMed]
- Besler, C.; Heinrich, K.; Riwanto, M.; Luscher, T.F.; Landmesser, U. High-density lipoprotein-mediated anti-atherosclerotic and endothelial-protective effects: A potential novel therapeutic target in cardiovascular disease. Curr. Pharm. Des. 2010, 16, 1480–1493. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Siuda, D.; Xia, N.; Reifenberg, G.; Daiber, A.; Munzel, T.; Forstermann, U.; Li, H. Maternal treatment of spontaneously hypertensive rats with pentaerythritol tetranitrate reduces blood pressure in female offspring. Hypertension 2015, 65, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Gioscia-Ryan, R.A.; LaRocca, T.J.; Sindler, A.L.; Zigler, M.C.; Murphy, M.P.; Seals, D.R. Mitochondria-targeted antioxidant (MitoQ) ameliorates age-related arterial endothelial dysfunction in mice. J. Physiol. 2014, 592, 2549–2561. [Google Scholar] [CrossRef] [PubMed]
- Klipstein-Grobusch, K.; Geleijnse, J.M.; den Breeijen, J.H.; Boeing, H.; Hofman, A.; Grobbee, D.E.; Witteman, J.C. Dietary antioxidants and risk of myocardial infarction in the elderly: The rotterdam study. Am. J. Clin. Nutr. 1999, 69, 261–266. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikhed, Y.; Daiber, A.; Steven, S. Mitochondrial Oxidative Stress, Mitochondrial DNA Damage and Their Role in Age-Related Vascular Dysfunction. Int. J. Mol. Sci. 2015, 16, 15918-15953. https://doi.org/10.3390/ijms160715918
Mikhed Y, Daiber A, Steven S. Mitochondrial Oxidative Stress, Mitochondrial DNA Damage and Their Role in Age-Related Vascular Dysfunction. International Journal of Molecular Sciences. 2015; 16(7):15918-15953. https://doi.org/10.3390/ijms160715918
Chicago/Turabian StyleMikhed, Yuliya, Andreas Daiber, and Sebastian Steven. 2015. "Mitochondrial Oxidative Stress, Mitochondrial DNA Damage and Their Role in Age-Related Vascular Dysfunction" International Journal of Molecular Sciences 16, no. 7: 15918-15953. https://doi.org/10.3390/ijms160715918
APA StyleMikhed, Y., Daiber, A., & Steven, S. (2015). Mitochondrial Oxidative Stress, Mitochondrial DNA Damage and Their Role in Age-Related Vascular Dysfunction. International Journal of Molecular Sciences, 16(7), 15918-15953. https://doi.org/10.3390/ijms160715918