
Progress and Prospects of Anti-HBV Gene Therapy Development

Abstract
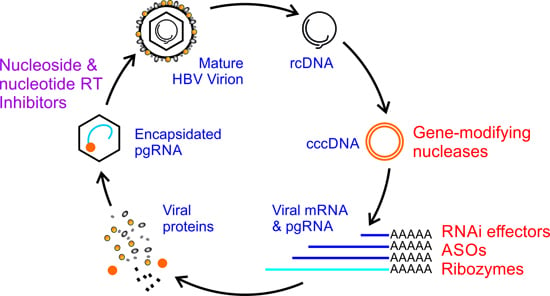
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction



2. Antisense Oligonucleotides
3. Ribozymes Targeted to Hepatitis B Virus (HBV)
4. Manipulation of RNA Interference (RNAi) to Counter HBV Infection
5. Genome and Epigenome Editing for HBV Gene Therapy
5.1. Zinc Finger Proteins and Their Derivatives
5.2. Transcription Activator-Like Effectors and Their Derivatives
5.3. CRISPR-Cas
5.4. Application of Genome Editing to HBV Therapy
6. Conclusions and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Franco, E.; Bagnato, B.; Marino, M.G.; Meleleo, C.; Serino, L.; Zaratti, L. Hepatitis B: Epidemiology and prevention in developing countries. World J. Hepatol. 2012, 4, 74. [Google Scholar] [CrossRef] [PubMed]
- Montuclard, C.; Hamza, S.; Rollot, F.; Evrard, P.; Faivre, J.; Hillon, P.; Martino, V.D.; Minello, A. Causes of death in people with chronic HBV infection: A population-based cohort study. J. Hepatol. 2015, 62, 1265–1271. [Google Scholar] [CrossRef] [PubMed]
- Arbuthnot, P.; Carmona, S.; Ely, A. Exploiting the RNA interference pathway to counter hepatitis B virus replication. Liver Int. 2005, 25, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Ivacik, D.; Ely, A.; Arbuthnot, P. Countering hepatitis B virus infection using RNAi: How far are we from the clinic? Rev. Med. Virol. 2011, 21, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Karayiannis, P. Hepatitis B virus: Old, new and future approaches to antiviral treatment. J. Antimicrob. Chemother. 2003, 51, 761–785. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Lin, L.; Xu, G.; Zhuang, Y.; Guo, Q.; Liu, Y.; Wang, H.; Zhou, X.; Wu, S.; Bao, S.; et al. Long-term lamivudine treatment achieves regression of advanced liver fibrosis/cirrhosis in patients with chronic hepatitis B. J. Gastroenterol. Hepatol. 2015, 30, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Gebbing, M.; Bergmann, T.; Schulz, E.; Ehrhardt, A. Gene therapeutic approaches to inhibit hepatitis B virus replication. World J. Hepatol. 2015, 7, 150–164. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Gripon, P.; Urban, S. Hepatitis B virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans. Hepatology 2007, 46, 1759–1768. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012, 1, e00049. [Google Scholar] [CrossRef] [PubMed]
- Katen, S.P.; Tan, Z.; Chirapu, S.R.; Finn, M.G.; Zlotnick, A. Assembly-directed antivirals differentially bind quasiequivalent pockets to modify hepatitis B virus capsid tertiary and quaternary structure. Structure 2013, 21, 1406–1416. [Google Scholar] [CrossRef] [PubMed]
- Feld, J.J.; Colledge, D.; Sozzi, V.; Edwards, R.; Littlejohn, M.; Locarnini, S.A. The phenylpropenamide derivative AT-130 blocks HBV replication at the level of viral RNA packaging. Antivir. Res. 2007, 76, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Delaney, W.E.T.; Edwards, R.; Colledge, D.; Shaw, T.; Furman, P.; Painter, G.; Locarnini, S. Phenylpropenamide derivatives AT-61 and AT-130 inhibit replication of wild-type and lamivudine-resistant strains of hepatitis B virus in vitro. Antimicrob. Agents Chemother. 2002, 46, 3057–3060. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.Y.; Xu, X.Q.; Guan, H.; Wang, L.L.; Wu, Q.; Zhao, G.M.; Li, S. A new series of HAPs as anti-HBV agents targeting at capsid assembly. Bioorg. Med. Chem. Lett. 2014, 24, 4247–4249. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Peng, B.; He, W.; Zhong, G.; Qi, Y.; Ren, B.; Gao, Z.; Jing, Z.; Song, M.; Xu, G.; et al. Molecular determinants of hepatitis B and D virus entry restriction in mouse sodium taurocholate cotransporting polypeptide. J. Virol. 2013, 87, 7977–7991. [Google Scholar] [CrossRef] [PubMed]
- Zhong, G.; Yan, H.; Wang, H.; He, W.; Jing, Z.; Qi, Y.; Fu, L.; Gao, Z.; Huang, Y.; Xu, G.; et al. Sodium taurocholate cotransporting polypeptide mediates woolly monkey hepatitis B virus infection of tupaia hepatocytes. J. Virol. 2013, 87, 7176–7184. [Google Scholar] [CrossRef] [PubMed]
- Watashi, K.; Urban, S.; Li, W.; Wakita, T. NTCP and beyond: Opening the door to unveil hepatitis B virus entry. Int. J. Mol. Sci. 2014, 15, 2892–2905. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Yang, J.; Wang, S. Antisense oligonucleotides targeting abhydrolase domain containing 2 block human hepatitis B virus propagation. Oligonucleotides 2011, 21, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Ketting, R.F. MicroRNA biogenesis and function: An overview. Adv. Exp. Med. Biol. 2011, 700, 1–14. [Google Scholar] [PubMed]
- Hasselblatt, P.; Hockenjos, B.; Thoma, C.; Blum, H.E.; Offensperger, W.B. Translation of stable hepadnaviral mRNA cleavage fragments induced by the action of phosphorothioate-modified antisense oligodeoxynucleotides. Nucleic Acids Res. 2005, 33, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.U. Ribozyme: A clinical tool. Clin. Chim. Acta 2006, 367, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Doherty, E.A.; Doudna, J.A. Ribozyme structures and mechanisms. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 457–475. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhao, Z.; Guo, J.; Huang, P.; Zhu, X.; Zhou, X.; Yang, Z.; Zhao, L.; Xu, L.; Xu, J.; et al. Creation of a six-fingered artificial transcription factor that represses the hepatitis B virus HBx gene integrated into a human hepatocellular carcinoma cell line. J. Biomol. Screen. 2013, 18, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Jiang, G.; Lu, J.; Chen, S.; Cui, L.; Jiao, J.; Wang, Y. Inhibition of hepatitis B virus cccDNA by siRNA in transgenic mice. Cell Biochem. Biophys. 2014, 69, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Cradick, T.J.; Keck, K.; Bradshaw, S.; Jamieson, A.C.; McCaffrey, A.P. Zinc-finger nucleases as a novel therapeutic strategy for targeting hepatitis B virus DNAs. Mol. Ther. 2010, 18, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Gao, T.; Wang, X.; Hu, Y.; Hu, X.; Hu, Z.; Pang, J.; Li, Z.; Xue, J.; Feng, M.; et al. TALE nickase mediates high efficient targeted transgene integration at the human multi-copy ribosomal DNA locus. Biochem. Biophys. Res. Commun. 2014, 446, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Bloom, K.; Ely, A.; Mussolino, C.; Cathomen, T.; Arbuthnot, P. Inactivation of hepatitis B virus replication in cultured cells and in vivo with engineered transcription activator-like effector nucleases. Mol. Ther. 2013, 21, 1889–1897. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Lei, R.; Le Duff, Y.; Li, J.; Guo, F.; Wainberg, M.A.; Liang, C. The CRISPR-cas9 system inactivates latent HIV-1 proviral DNA. Retrovirology 2015, 12, 22. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, E.M.; Bassit, L.C.; Mueller, H.; Kornepati, A.V.; Bogerd, H.P.; Nie, T.; Chatterjee, P.; Javanbakht, H.; Schinazi, R.F.; Cullen, B.R. Suppression of hepatitis B virus DNA accumulation in chronically infected cells using a bacterial CRISPR-cas RNA-guided DNA endonuclease. Virology 2015, 476, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Zhen, S.; Hua, L.; Liu, Y.H.; Gao, L.C.; Fu, J.; Wan, D.Y.; Dong, L.H.; Song, H.F.; Gao, X. Harnessing the clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated Cas9 system to disrupt the hepatitis B virus. Gene Ther. 2015, 22, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Van Hauwermeiren, F.; Vandenbroucke, R.E.; Grine, L.; Puimege, L.; van Wonterghem, E.; Zhang, H.; Libert, C. Antisense oligonucleotides against TNFR1 prevent toxicity of TNF/IFNγ treatment in mouse tumor models. Int. J. Cancer 2014, 135, 742–750. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, G.; Gross, S.C.; Tewari, A.; Watabe, K. Antisense oligodeoxyribonucleotides inhibit the expression of the gene for hepatitis B virus surface antigen. J. Gen. Virol. 1990, 71 Pt 12, 3021–3025. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.Y.; Wu, C.H. Specific inhibition of hepatitis B viral gene expression in vitro by targeted antisense oligonucleotides. J. Biol. Chem. 1992, 267, 12436–12439. [Google Scholar] [PubMed]
- Offensperger, W.B.; Offensperger, S.; Walter, E.; Teubner, K.; Igloi, G.; Blum, H.E.; Gerok, W. In vivo inhibition of duck hepatitis B virus replication and gene expression by phosphorothioate modified antisense oligodeoxynucleotides. EMBO J. 1993, 12, 1257–1262. [Google Scholar] [PubMed]
- Yao, Z.; Zhou, Y.; Feng, X.; Chen, C.; Guo, J. In vivo inhibition of hepatitis B viral gene expression by antisense phosphorothioate oligodeoxynucleotides in athymic nude mice. J. Viral Hepat. 1996, 3, 19–22. [Google Scholar] [PubMed]
- Yao, Z.Q.; Zhou, Y.X.; Guo, J.; Feng, Z.H.; Feng, X.M.; Chen, C.X.; Jiao, J.Z.; Wang, S.Q. Inhibition of hepatitis B virus in vitro by antisense oligonucleotides. Acta Virol. 1996, 40, 35–39. [Google Scholar] [PubMed]
- Deng, Y.B.; Nong, L.G.; Huang, W.; Pang, G.G.; Wang, Y.F. Inhibition of hepatitis B virus (HBV) replication using antisense LNA targeting to both S and C genes in HBV. Zhonghua Gan Zang Bing Za Zhi 2009, 17, 900–904. [Google Scholar] [PubMed]
- Korba, B.E.; Gerin, J.L. Antisense oligonucleotides are effective inhibitors of hepatitis B virus replication in vitro. Antivir. Res. 1995, 28, 225–242. [Google Scholar] [CrossRef]
- Nakazono, K.; Ito, Y.; Wu, C.H.; Wu, G.Y. Inhibition of hepatitis B virus replication by targeted pretreatment of complexed antisense DNA in vitro. Hepatology 1996, 23, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- De Smet, M.D.; Meenken, C.J.; van den Horn, G.J. Fomivirsen—A phosphorothioate oligonucleotide for the treatment of CMV retinitis. Ocul. Immunol. Inflamm. 1999, 7, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.D.; Kopeckova, P.; Kopecek, J. Antisense oligonucleotides delivered to the lysosome escape and actively inhibit the hepatitis B virus. Bioconjug. Chem. 2002, 13, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Prakash, T.P.; Siwkowski, A.; Allerson, C.R.; Migawa, M.T.; Lee, S.; Gaus, H.J.; Black, C.; Seth, P.P.; Swayze, E.E.; Bhat, B. Antisense oligonucleotides containing conformationally constrained 2′,4′-(N-methoxy)aminomethylene and 2′,4′-aminooxymethylene and 2′-O,4′-C-aminomethylene bridged nucleoside analogues show improved potency in animal models. J. Med. Chem. 2010, 53, 1636–1650. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Nong, L.; Wei, Y. Antiviral effects of dual-target antisense LNA by cationic liposomes in transgenic mice. Sheng Wu Yi Xue Gong Cheng Xue Za Zhi 2013, 30, 828–832, 837. [Google Scholar] [PubMed]
- Sun, X.; Marque, L.O.; Cordner, Z.; Pruitt, J.L.; Bhat, M.; Li, P.P.; Kannan, G.; Ladenheim, E.E.; Moran, T.H.; Margolis, R.L.; et al. Phosphorodiamidate morpholino oligomers suppress mutant huntingtin expression and attenuate neurotoxicity. Hum. Mol. Genet. 2014, 23, 6302–6317. [Google Scholar] [CrossRef] [PubMed]
- Cirak, S.; Arechavala-Gomeza, V.; Guglieri, M.; Feng, L.; Torelli, S.; Anthony, K.; Abbs, S.; Garralda, M.E.; Bourke, J.; Wells, D.J.; et al. Exon skipping and dystrophin restoration in patients with duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: An open-label, phase 2, dose-escalation study. Lancet 2011, 378, 595–605. [Google Scholar] [CrossRef]
- Zhang, Y.; Qi, X.R.; Gao, Y.; Wei, L. Transfection and anti-HBV effect mediated by the hepatocytes-targeting cationic liposomes co-modified with β-sitosterol-β-d-glucoside and Brij 35. Yao Xue Xue Bao 2006, 41, 1111–1115. [Google Scholar] [PubMed]
- Robaczewska, M.; Guerret, S.; Remy, J.S.; Chemin, I.; Offensperger, W.B.; Chevallier, M.; Behr, J.P.; Podhajska, A.J.; Blum, H.E.; Trepo, C.; et al. Inhibition of hepadnaviral replication by polyethylenimine-based intravenous delivery of antisense phosphodiester oligodeoxynucleotides to the liver. Gene Ther. 2001, 8, 874–881. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.Y.; Walton, C.M.; Wu, C.H. Targeted polynucleotides for inhibition of hepatitis B and C viruses. Croat. Med. J. 2001, 42, 463–466. [Google Scholar] [PubMed]
- Wang, C.X.; Lu, Y.Q.; Qi, P.; Chen, L.H.; Han, J.X. Efficient inhibition of hepatitis B virus replication by hepatitis delta virus ribozymes delivered by targeting retrovirus. Virol. J. 2010, 7, 61. [Google Scholar] [CrossRef] [PubMed]
- Fedor, M.J. Structure and function of the hairpin ribozyme. J. Mol. Biol. 2000, 297, 269–291. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Vu, G.P.; Qian, H.; Chen, Y.C.; Wang, Y.; Reeves, M.; Zen, K.; Liu, F. Engineered RNase P ribozymes effectively inhibit human cytomegalovirus gene expression and replication. Viruses 2014, 6, 2376–2391. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, S.; Kashani-Sabet, M. Ribozymes in the age of molecular therapeutics. Curr. Mol. Med. 2004, 4, 489–506. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Chen, Y.C.; Gong, H.; Zeng, W.; Vu, G.P.; Trang, P.; Lu, S.; Wu, J.; Liu, F. Inhibition of hepatitis B virus gene expression and replication by ribonuclease P. Mol. Ther. 2013, 21, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.H.; Lin, J.S.; Liu, N.Z.; Kong, X.J.; Xie, N.; Wang, N.X.; Jin, Y.X.; Liang, K.H. Anti-HBV hairpin ribozyme-mediated cleavage of target RNA in vitro. World J. Gastroenterol. 2002, 8, 91–94. [Google Scholar] [PubMed]
- Welch, P.J.; Tritz, R.; Yei, S.; Barber, J.; Yu, M. Intracellular application of hairpin ribozyme genes against hepatitis B virus. Gene Ther. 1997, 4, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Passman, M.; Weinberg, M.; Kew, M.; Arbuthnot, P. In situ demonstration of inhibitory effects of hammerhead ribozymes that are targeted to the hepatitis Bx sequence in cultured cells. Biochem. Biophys. Res. Commun. 2000, 268, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Nash, K.L.; Alexander, G.J.; Lever, A.M. Inhibition of hepatitis B virus by lentiviral vector delivered antisense RNA and hammerhead ribozymes. J. Viral Hepat. 2005, 12, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, D.V.; Lee, P.A.; Johnson, D.A.; Overly, S.L.; McSwiggen, J.A.; Beigelman, L.; Mokler, V.R.; Maloney, L.; Vargeese, C.; Bowman, K.; et al. Characterization of nuclease-resistant ribozymes directed against hepatitis B virus RNA. J. Viral Hepat. 2002, 9, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Kong, Y.Y.; Wang, Y.; Qi, G.R. Inhibition of hepatitis B virus by hammerhead ribozyme targeted to the poly(a) signal sequence in cultured cells. Biol. Chem. 2001, 382, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Kong, Y.Y.; Wang, Y.; Qi, G.R. Intracellular inhibition of the replication of hepatitis B virus by hammerhead ribozymes. J. Gastroenterol. Hepatol. 2001, 16, 1125–1130. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.H.; Xin, P.; Morrey, J.D.; Clawson, G.A. A self-processing ribozyme cassette: Utility against human papillomavirus 11 E6/E7 mRNA and hepatitis B virus. Mol. Ther. 2004, 9, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kuang, E.; Dai, W.; Zhou, B.; Yang, F. Efficient inhibition of hepatitis B virus replication by hammerhead ribozymes delivered by hepatitis delta virus. Virus Res. 2005, 114, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.J.; Xiang, K.J.; Huang, Z.H.; Zhou, R.; Qi, X.Z. Construction of HBV-specific ribozyme and its recombinant with HDV and their cleavage activity in vitro. World J. Gastroenterol. 2000, 6, 377–380. [Google Scholar] [PubMed]
- Tan, T.M.; Zhou, L.; Houssais, S.; Seet, B.L.; Jaenicke, S.; Peter, F.; Lim, S.G. Intracellular inhibition of hepatitis B virus S gene expression by chimeric DNA-RNA phosphorothioate minimized ribozyme. Antisense Nucleic Acid Drug Dev. 2002, 12, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Sioud, M.; Opstad, A.; Hendry, P.; Lockett, T.J.; Jennings, P.A.; McCall, M.J. A minimised hammerhead ribozyme with activity against interleukin-2 in human cells. Biochem. Biophys. Res. Commun. 1997, 231, 397–402. [Google Scholar] [CrossRef] [PubMed]
- McCall, M.J.; Hendry, P.; Jennings, P.A. Minimal sequence requirements for ribozyme activity. Proc. Natl. Acad. Sci. USA 1992, 89, 5710–5714. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, M.S.; Ely, A.; Passman, M.; Mufamadi, S.M.; Arbuthnot, P. Effective anti-hepatitis B virus hammerhead ribozymes derived from multimeric precursors. Oligonucleotides 2007, 17, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, J.; Wu, C.H.; Ito, Y.; Wu, G.Y. Design and preparation of a multimeric self-cleaving hammerhead ribozyme. BioTechniques 1997, 22, 338–345. [Google Scholar] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.M. Dicing and slicing: The core machinery of the rna interference pathway. FEBS Lett. 2005, 579, 5822–5829. [Google Scholar] [CrossRef] [PubMed]
- Shabalina, S.A.; Koonin, E.V. Origins and evolution of eukaryotic RNA interference. Trends Ecol. Evol. 2008, 23, 578–587. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Angaji, S.A.; Hedayati, S.S.; Poor, R.H.; Madani, S.; Poor, S.S.; Panahi, S. Application of RNA interference in treating human diseases. J. Genet. 2010, 89, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Haussecker, D.; Kay, M.A. RNA interference. Drugging RNAi. Science 2015, 347, 1069–1070. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Qian, Y.; Yan, F.; Tu, J.; Yang, X.; Xing, Y.; Chen, Z. 5′-Triphosphate-siRNA activates RIG-I-dependent type I interferon production and enhances inhibition of hepatitis B virus replication in HepG2.2.15 cells. Eur. J. Pharmacol. 2013, 721, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, R.A.; Suter, S.R.; Ball-Jones, A.A.; Ibarra-Soza, J.M.; Zheng, Y.; Beal, P.A. Base modification strategies to modulate immune stimulation by an siRNA. ChemBioChem 2015, 16, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wang, X.; He, H.; Zhou, J.; Li, X.; Ma, H.; Li, Z.; Zeng, Y.; Shao, R.; Cen, S.; et al. Structure-based design of novel chemical modification of the 3′-overhang for optimization of short interfering RNA performance. Biochemistry 2015, 54, 1268–1277. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Li, X.; Yi, M.; Zhu, S.; Chen, W. Targeted delivery of siRNA against hepatitis B virus by preS1 peptide molecular ligand. Hepatol. Res. 2014, 44, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.H.; Bae, K.H.; Jo, S.D.; Kim, J.S.; Park, T.G. Cationic lipid-coated gold nanoparticles as efficient and non-cytotoxic intracellular siRNA delivery vehicles. Pharm. Res. 2012, 29, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Wooddell, C.I.; Rozema, D.B.; Hossbach, M.; John, M.; Hamilton, H.L.; Chu, Q.; Hegge, J.O.; Klein, J.J.; Wakefield, D.H.; Oropeza, C.E.; et al. Hepatocyte-targeted RNAi therapeutics for the treatment of chronic hepatitis B virus infection. Mol. Ther. 2013, 21, 973–985. [Google Scholar] [CrossRef] [PubMed]
- Sebestyen, M.G.; Wong, S.C.; Trubetskoy, V.; Lewis, D.L.; Wooddell, C.I. Targeted in vivo delivery of siRNA and an endosome-releasing agent to hepatocytes. Methods Mol. Biol. 2015, 1218, 163–186. [Google Scholar] [PubMed]
- Arrowhead Presents Data on ARC-520 and ARC-AAT at AASLD the Liver Meeting® 2014. Available online: http://ir.arrowheadresearch.com/releasedetail.cfm?releaseid=881791 (accessed on 28 July 2015).
- Alnylam Announces New RNAi Therapeutic Program for the Treatment of Hepatitis B Virus (HBV) Infection and Reports an Up to 2.3 Log10 Reduction of HBV Surface Antigen (HBsAg) in Chronically Infected Chimpanzees. Available online: http://investors.alnylam.com/releasedetail.cfm?ReleaseID=847055 (accessed on 28 July 2015).
- Tekmira Presents Results of Preclinical Studies with Hepatitis B Therapeutic. Available online: http://www.sec.gov/Archives/edgar/data/1447028/000117184314004780/newsrelease.htm (accessed on 28 July 2015).
- Tekmira Initiates Phase I Clinical Trial of TKM-HBV. Available online: http://investor.tekmirapharm.com/releasedetail.cfm?ReleaseID=892233 (accessed on 28 July 2015).
- Evaluation of TT-034: Safe and Durable Hepatic Expression of Anti-HCV ShRNA in a Non-Human Primate Model. Available online: http://www.benitec.com/documents/1311_tt_presn_hcv_aasld.pdf (accessed on 28 July 2015).
- Grimm, D.; Streetz, K.L.; Jopling, C.L.; Storm, T.A.; Pandey, K.; Davis, C.R.; Marion, P.; Salazar, F.; Kay, M.A. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 2006, 441, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.P.; Wu, T.H.; Chen, C.C.; Wu, P.Y.; Shih, Y.M.; Tsuneyama, K.; Tao, M.H. Studies of efficacy and liver toxicity related to adeno-associated virus-mediated RNA interference. Hum. Gene Ther. 2013, 24, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.; Wang, L.; Lee, J.S.; Schurmann, N.; Gu, S.; Borner, K.; Storm, T.A.; Kay, M.A. Argonaute proteins are key determinants of RNAi efficacy, toxicity, and persistence in the adult mouse liver. J. Clin. Investig. 2010, 120, 3106–3119. [Google Scholar] [CrossRef] [PubMed]
- McBride, J.L.; Boudreau, R.L.; Harper, S.Q.; Staber, P.D.; Monteys, A.M.; Martins, I.; Gilmore, B.L.; Burstein, H.; Peluso, R.W.; Polisky, B.; et al. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: Implications for the therapeutic development of RNAi. Proc. Natl. Acad. Sci. USA 2008, 105, 5868–5873. [Google Scholar] [CrossRef] [PubMed]
- Ely, A.; Naidoo, T.; Arbuthnot, P. Efficient silencing of gene expression with modular trimeric Pol II expression cassettes comprising microRNA shuttles. Nucleic Acids Res. 2009, 37, e91. [Google Scholar] [CrossRef] [PubMed]
- Ely, A.; Naidoo, T.; Mufamadi, S.; Crowther, C.; Arbuthnot, P. Expressed anti-HBV primary microRNA shuttles inhibit viral replication efficiently in vitro and in vivo. Mol. Ther. 2008, 16, 1105–1112. [Google Scholar] [CrossRef] [PubMed]
- Ivacik, D.; Ely, A.; Ferry, N.; Arbuthnot, P. Sustained inhibition of hepatitis B virus replication in vivo using RNAi-activating lentiviruses. Gene Ther. 2015, 22, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Mowa, M.B.; Crowther, C.; Ely, A.; Arbuthnot, P. Inhibition of hepatitis B virus replication by helper dependent adenoviral vectors expressing artificial anti-HBV pri-miRs from a liver-specific promoter. Biomed. Res. Int. 2014, 2014, 718743. [Google Scholar] [CrossRef] [PubMed]
- Giering, J.C.; Grimm, D.; Storm, T.A.; Kay, M.A. Expression of shRNA from a tissue-specific Pol II promoter is an effective and safe RNAi therapeutic. Mol. Ther. 2008, 16, 1630–1636. [Google Scholar] [CrossRef] [PubMed]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., 3rd. ZFN, TALEN, and CRISPR-cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, J.S. A guide to genome engineering with programmable nucleases. Nat. Rev. Genet. 2014, 15, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010, 11, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.M.; Musunuru, K. Expanding the genetic editing tool kit: ZFNs, TALENs, and CRISPR-Cas9. J. Clin. Investig. 2014, 124, 4154–4161. [Google Scholar] [CrossRef] [PubMed]
- Chu, V.T.; Weber, T.; Wefers, B.; Wurst, W.; Sander, S.; Rajewsky, K.; Kuhn, R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015, 33, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Dougan, S.K.; Truttmann, M.C.; Bilate, A.M.; Ingram, J.R.; Ploegh, H.L. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015, 33, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.B.; Platt, R.J.; Zhang, F. Therapeutic genome editing: Prospects and challenges. Nat. Med. 2015, 21, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Klug, A. The discovery of zinc fingers and their applications in gene regulation and genome manipulation. Annu. Rev. Biochem. 2010, 79, 213–231. [Google Scholar] [CrossRef] [PubMed]
- Alwin, S.; Gere, M.B.; Guhl, E.; Effertz, K.; Barbas, C.F., 3rd; Segal, D.J.; Weitzman, M.D.; Cathomen, T. Custom zinc-finger nucleases for use in human cells. Mol. Ther. 2005, 12, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Xu, K.; Hu, L.; Wang, L.; Zhang, T.; Ren, C.; Zhang, Z. A suicidal zinc finger nuclease expression coupled with a surrogate reporter for efficient genome engineering. Biotechnol. Lett. 2015, 37, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Townsend, J.A.; Wright, D.A.; Winfrey, R.J.; Fu, F.; Maeder, M.L.; Joung, J.K.; Voytas, D.F. High-frequency modification of plant genes using engineered zinc-finger nucleases. Nature 2009, 459, 442–445. [Google Scholar] [CrossRef] [PubMed]
- Osakabe, Y.; Osakabe, K. Genome editing with engineered nucleases in plants. Plant Cell Physiol. 2015, 56, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Guan, G.; Zhang, X.; Naruse, K.; Nagahama, Y.; Hong, Y. Gene replacement by zinc finger nucleases in medaka embryos. Mar. Biotechnol. 2014, 16, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Menke, D.B. Engineering subtle targeted mutations into the mouse genome. Genesis 2013, 51, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, K.A.; Fischer, K.P.; Joyce, M.A.; Tyrrell, D.L. Zinc finger proteins designed to specifically target duck hepatitis B virus covalently closed circular DNA inhibit viral transcription in tissue culture. J. Virol. 2008, 82, 8013–8021. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Kim, E.; Kim, J.S. Targeted chromosomal deletions in human cells using zinc finger nucleases. Genome Res. 2010, 20, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Kweon, J.; Kim, E.; Kim, S.; Kim, J.S. Targeted chromosomal duplications and inversions in the human genome using zinc finger nucleases. Genome Res. 2012, 22, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Friedman, G.; Doyon, Y.; Wang, N.S.; Li, C.J.; Miller, J.C.; Hua, K.L.; Yan, J.J.; Babiarz, J.E.; Gregory, P.D.; et al. Targeted gene addition to a predetermined site in the human genome using a ZFN-based nicking enzyme. Genome Res. 2012, 22, 1316–1326. [Google Scholar] [CrossRef] [PubMed]
- Kandavelou, K.; Mani, M.; Durai, S.; Chandrasegaran, S. “Magic” scissors for genome surgery. Nat. Biotechnol. 2005, 23, 686–687. [Google Scholar] [CrossRef] [PubMed]
- Urnov, F.D.; Miller, J.C.; Lee, Y.L.; Beausejour, C.M.; Rock, J.M.; Augustus, S.; Jamieson, A.C.; Porteus, M.H.; Gregory, P.D.; Holmes, M.C. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 2005, 435, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Zhao, H. Transcription activator-like effector nucleases (TALENs): A highly efficient and versatile tool for genome editing. Biotechnol. Bioeng. 2013, 110, 1811–1821. [Google Scholar] [CrossRef] [PubMed]
- Joung, J.K.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2013, 14, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Lamb, B.M.; Mercer, A.C.; Barbas, C.F., 3rd. Directed evolution of the TALE N-terminal domain for recognition of all 5′ bases. Nucleic Acids Res. 2013, 41, 9779–9785. [Google Scholar] [CrossRef] [PubMed]
- Ain, Q.U.; Chung, J.Y.; Kim, Y.H. Current and future delivery systems for engineered nucleases: ZFN, TALEN and RGEN. J. Control. Release 2015, 205, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Groner, A.C.; Meylan, S.; Ciuffi, A.; Zangger, N.; Ambrosini, G.; Denervaud, N.; Bucher, P.; Trono, D. KRAB-zinc finger proteins and KAP1 can mediate long-range transcriptional repression through heterochromatin spreading. PLoS Genet. 2010, 6, e1000869. [Google Scholar] [CrossRef] [PubMed]
- Sripathy, S.P.; Stevens, J.; Schultz, D.C. The KAP1 corepressor functions to coordinate the assembly of de novo HP1-demarcated microenvironments of heterochromatin required for KRAB zinc finger protein-mediated transcriptional repression. Mol. Cell. Biol. 2006, 26, 8623–8638. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed]
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014, 32, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Bondy-Denomy, J.; Davidson, A.R. To acquire or resist: The complex biological effects of CRISPR-Cas systems. Trends Microbiol. 2014, 22, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Fineran, P.C.; Dy, R.L. Gene regulation by engineered CRISPR-Cas systems. Curr. Opin. Microbiol. 2014, 18, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Kew, M.C. Hepatitis B virus X protein in the pathogenesis of hepatitis B virus-induced hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2011, 26 (Suppl. 1), 144–152. [Google Scholar] [CrossRef] [PubMed]
- Weber, N.D.; Stone, D.; Sedlak, R.H.; de Silva Feelixge, H.S.; Roychoudhury, P.; Schiffer, J.T.; Aubert, M.; Jerome, K.R. AAV-mediated delivery of zinc finger nucleases targeting hepatitis B virus inhibits active replication. PLoS ONE 2014, 9, e97579. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, W.; Lin, J.; Wang, F.; Wu, M.; Chen, C.; Zheng, Y.; Peng, X.; Li, J.; Yuan, Z. An efficient antiviral strategy for targeting hepatitis B virus genome using transcription activator-like effector nucleases. Mol. Ther. 2014, 22, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.R.; Yang, H.C.; Kuo, Y.T.; Liu, C.J.; Yang, T.Y.; Sung, K.C.; Lin, Y.Y.; Wang, H.Y.; Wang, C.C.; Shen, Y.C.; et al. The CRISPR-cas9 system facilitates clearance of the intrahepatic HBV templates in vivo. Mol. Ther. Nucleic Acids 2014, 3, e186. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Sohn, J.A. Targeting hepatitis B virus with CRISPR-cas9. Mol. Ther. Nucleic Acids 2014, 3, e216. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Qu, L.; Wang, H.; Wei, L.; Dong, Y.; Xiong, S. Targeting hepatitis B virus cccDNA by CRISPR-cas9 nuclease efficiently inhibits viral replication. Antivir. Res. 2015, 118, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Li, G.; Hu, H.; Yang, C.; Zhang, X.; Leng, Q.; Xie, Y.; Yu, D.; Zhang, X.; Gao, Y.; et al. Recombinant covalently closed circular hepatitis B virus DNA induces prolonged viral persistence in immunocompetent mice. J. Virol. 2014, 88, 8045–8056. [Google Scholar] [CrossRef] [PubMed]
- Kinetics of Knockdown from RNAi Therapeutic ARC-520 on HBV RNA, DNA and Antigens in Mice and Chimpanzees. Available online: http://www.arrowheadresearch.com/sites/default/files/MolBiol_HBV_poster_102313.pdf (accessed on 28 July 2015).
- Yuan, T.H.; Li, M.Y.; Li, W.Y.; Li, H.; Jiang, Z.H. Combinatorial effects of ST6Gal I siRNA and antisense oligonucleotide-mediated gene silence on metastasis ability of cervical carcinoma cells. Sichuan Da Xue Xue Bao Yi Xue Ban 2007, 38, 217–221. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maepa, M.B.; Roelofse, I.; Ely, A.; Arbuthnot, P. Progress and Prospects of Anti-HBV Gene Therapy Development. Int. J. Mol. Sci. 2015, 16, 17589-17610. https://doi.org/10.3390/ijms160817589
Maepa MB, Roelofse I, Ely A, Arbuthnot P. Progress and Prospects of Anti-HBV Gene Therapy Development. International Journal of Molecular Sciences. 2015; 16(8):17589-17610. https://doi.org/10.3390/ijms160817589
Chicago/Turabian StyleMaepa, Mohube B., Ilke Roelofse, Abdullah Ely, and Patrick Arbuthnot. 2015. "Progress and Prospects of Anti-HBV Gene Therapy Development" International Journal of Molecular Sciences 16, no. 8: 17589-17610. https://doi.org/10.3390/ijms160817589
APA StyleMaepa, M. B., Roelofse, I., Ely, A., & Arbuthnot, P. (2015). Progress and Prospects of Anti-HBV Gene Therapy Development. International Journal of Molecular Sciences, 16(8), 17589-17610. https://doi.org/10.3390/ijms160817589