
Towards Stratified Medicine in Plasma Cell Myeloma

Abstract
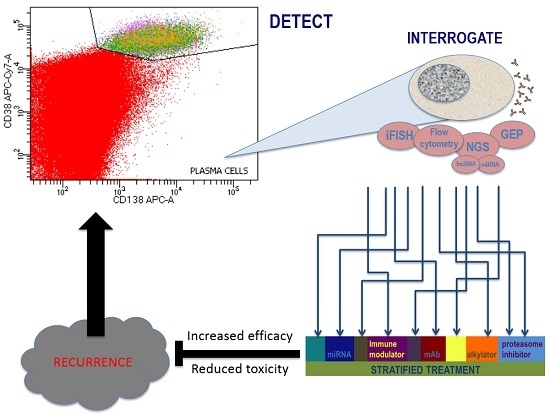
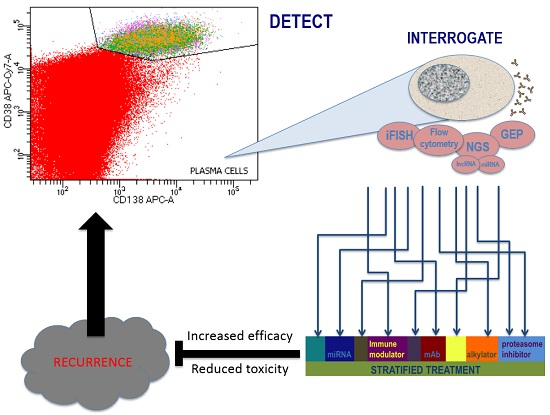
:
1. Introduction
2. Tumour Biology
3. Investigation of Suspected Plasma Cell Dyscrasias
4. Risk Stratification
4.1. Types of Risk Stratification Parameters
4.1.1. Patient Factors
4.1.2. Tumour Burden
4.1.3. Cytogenetics
4.1.4. Gene Expression Profiles
5. Treatment
5.1. Treatment Response
5.2. Link between Response Depth/Duration and Outcome
5.3. Overcoming Prognostic Disadvantage
6. Minimal Residual Disease (MRD)
6.1. MRD Detection by Flow Cytometry
6.2. MRD Detection Using Molecular Methods
6.3. Characterisation of MRD
6.3.1. Surface Markers with Known Prognostic Significance
6.3.2. Altered Gene Expression in MRD or at Relapse
7. Future Developments
8. Summary
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the world health organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Siegel, R.; Xu, J.; Ward, E. Cancer statistics, 2010. CA Cancer J. Clin. 2010, 60, 277–300. [Google Scholar] [CrossRef] [PubMed]
- Biran, N.; Ely, S.; Chari, A. Controversies in the assessment of minimal residual disease in multiple myeloma: Clinical significance of minimal residual disease negativity using highly sensitive techniques. Curr. Hematol. Malig. Rep. 2014, 9, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Rotolo, A.; Caputo, V.; Karadimitris, A. The prospects and promise of chimeric antigen receptor immunotherapy in multiple myeloma. Br. J. Haematol. 2016, 173, 350–364. [Google Scholar] [CrossRef] [PubMed]
- Kuehl, W.M.; Bergsagel, P.L. Multiple myeloma: Evolving genetic events and host interactions. Nat. Rev. Cancer 2002, 2, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International myeloma working group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, 538–548. [Google Scholar] [CrossRef]
- Wadhera, R.K.; Rajkumar, S.V. Prevalence of monoclonal gammopathy of undetermined significance: A systematic review. Mayo Clin. Proc. 2010, 85, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Terpos, E.; Niesvizky, R.; Palumbo, A. Clinical characteristics of patients with relapsed multiple myeloma. Cancer Treat. Rev. 2015, 41, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, J.R. The prognostic significance of cytogenetics and molecular profiling in multiple myeloma. Cancer Genet. 2011, 204, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Fonseca, R.; Ketterling, R.P.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Hayman, S.R.; Buadi, F.K.; Dingli, D.; Knudson, R.A.; et al. Trisomies in multiple myeloma: Impact on survival in patients with high-risk cytogenetics. Blood 2012, 119, 2100–2105. [Google Scholar] [CrossRef] [PubMed]
- Sonneveld, P.; Avet-Loiseau, H.; Lonial, S.; Usmani, S.; Siegel, D.; Anderson, K.C.; Chng, W.J.; Moreau, P.; Attal, M.; Kyle, R.A.; et al. Treatment of multiple myeloma with high-risk cytogenetics: A consensus of the international myeloma working group. Blood 2016, 127, 2955–2962. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Mikhael, J.R.; Buadi, F.K.; Dingli, D.; Dispenzieri, A.; Fonseca, R.; Gertz, M.A.; Greipp, P.R.; Hayman, S.R.; Kyle, R.A.; et al. Management of newly diagnosed symptomatic multiple myeloma: Updated mayo stratification of myeloma and risk-adapted therapy (mSMART) consensus guidelines. Mayo Clin. Proc. 2009, 84, 1095–1110. [Google Scholar] [CrossRef] [PubMed]
- Mikhael, J.R.; Dingli, D.; Roy, V.; Reeder, C.B.; Buadi, F.K.; Hayman, S.R.; Dispenzieri, A.; Fonseca, R.; Sher, T.; Kyle, R.A.; et al. Management of newly diagnosed symptomatic multiple myeloma: Updated mayo stratification of myeloma and risk-adapted therapy (mSMART) consensus guidelines 2013. Mayo Clin. Proc. 2013, 88, 360–376. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.Y.; Huang, X.; Chim, C.S. DNA methylation of microRNA genes in multiple myeloma. Carcinogenesis 2012, 33, 1629–1638. [Google Scholar] [CrossRef] [PubMed]
- Calura, E.; Bisognin, A.; Manzoni, M.; Todoerti, K.; Taiana, E.; Sales, G.; Morgan, G.J.; Tonon, G.; Amodio, N.; Tassone, P.; et al. Disentangling the microRNA regulatory milieu in multiple myeloma: Integrative genomics analysis outlines mixed miRNA-TF circuits and pathway-derived networks modulated in t(4;14) patients. Oncotarget 2016, 7, 2367–2378. [Google Scholar] [PubMed]
- Ahmad, N.; Haider, S.; Jagannathan, S.; Anaissie, E.; Driscoll, J.J. MicroRNA theragnostics for the clinical management of multiple myeloma. Leukemia 2014, 28, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, M.T.; Gulla, A.; Cantafio, M.E.; Lionetti, M.; Leone, E.; Amodio, N.; Guzzi, P.H.; Foresta, U.; Conforti, F.; Cannataro, M.; et al. In vitro and in vivo anti-tumor activity of miR-221/222 inhibitors in multiple myeloma. Oncotarget 2013, 4, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Tagliaferri, P.; Rossi, M.; di Martino, M.T.; Amodio, N.; Leone, E.; Gulla, A.; Neri, A.; Tassone, P. Promises and challenges of microRNA-based treatment of multiple myeloma. Curr. Cancer Drug Targets 2012, 12, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, M.T.; Arbitrio, M.; Guzzi, P.H.; Cannataro, M.; Tagliaferri, P.; Tassone, P. Experimental treatment of multiple myeloma in the era of precision medicine. Expert Rev. Precis. Med. Drug Dev. 2016, 1, 37–51. [Google Scholar] [CrossRef]
- Ronchetti, D.; Lionetti, M.; Mosca, L.; Agnelli, L.; Andronache, A.; Fabris, S.; Deliliers, G.L.; Neri, A. An integrative genomic approach reveals coordinated expression of intronic miR-335, miR-342, and miR-561 with deregulated host genes in multiple myeloma. BMC Med. Genom. 2008, 1, 37. [Google Scholar] [CrossRef] [PubMed]
- Ronchetti, D.; Agnelli, L.; Taiana, E.; Galletti, S.; Manzoni, M.; Todoerti, K.; Musto, P.; Strozzi, F.; Neri, A. Distinct lncRNA transcriptional fingerprints characterize progressive stages of multiple myeloma. Oncotarget 2016, 7, 14814–14830. [Google Scholar] [PubMed]
- Li, B.; Chen, P.; Qu, J.; Shi, L.; Zhuang, W.; Fu, J.; Li, J.; Zhang, X.; Sun, Y.; Zhuang, W. Activation of LTBP3 gene by a long noncoding RNA (lncRNA) MALAT1 transcript in mesenchymal stem cells from multiple myeloma. J. Biol. Chem. 2014, 289, 29365–29375. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Zhao, H.; Wang, Z.; Cheng, L.; Yang, L.; Shi, H.; Yang, H.; Sun, J. Identification and validation of potential prognostic lncRNA biomarkers for predicting survival in patients with multiple myeloma. J. Exp. Clin. Cancer Res. 2015, 34, 102. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.; Ballabio, E.; Chen, X.H.; Kusec, R.; Taylor, S.; Hay, D.; Tramonti, D.; Saunders, N.J.; Littlewood, T.; Pezzella, F.; et al. MicroRNA expression in multiple myeloma is associated with genetic subtype, isotype and survival. Biol. Direct. 2011, 6, 23–40. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Fakhri, B.; Vij, R. Clonal evolution in multiple myeloma. Clin. Lymphoma Myeloma Leuk. 2016, 16, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Magrangeas, F.; Avet-Loiseau, H.; Gouraud, W.; Lodé, L.; Decaux, O.; Godmer, P.; Garderet, L.; Voillat, L.; Facon, T.; Stoppa, A.M.; et al. Minor clone provides a reservoir for relapse in multiple myeloma. Leukemia 2013, 27, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.S.; Lehners, N.; Xu, J.; Ho, A.D.; Schirmacher, P.; Goldschmidt, H.; Andrulis, M. Spatially divergent clonal evolution in multiple myeloma: Overcoming resistance to BRAF inhibition. Blood 2016, 127, 2155–2157. [Google Scholar] [CrossRef] [PubMed]
- Bergsagel, P.L.; Chesi, M.V. Molecular classification and risk stratification of myeloma. Hematol. Oncol. 2013, 31, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Keats, J.J.; Chesi, M.; Egan, J.B.; Garbitt, V.M.; Palmer, S.E.; Braggio, E.; van Wier, S.; Blackburn, P.R.; Baker, A.S.; Dispenzieri, A.; et al. Clonal competition with alternating dominance in multiple myeloma. Blood 2012, 120, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Bird, J.M.; Owen, R.G.; D’Sa, S.; Snowden, J.A.; Pratt, G.; Ashcroft, J.; Yong, K.; Cook, G.; Feyler, S.; Davies, F.; et al. Guidelines for diagnosis and management of multiple myeloma, 2013. Available online: www.ukmf.org.uk/guidelines-page/bshukmf-guidelines (accessed on 24 June 2016).
- Biran, N.; Jagannath, S.; Chari, A. Risk stratification in multiple myeloma, part 1: Characterization of high-risk disease. Clin. Adv. Hematol. Oncol. 2013, 11, 489–503. [Google Scholar] [PubMed]
- Biran, N.; Jagannath, S.; Chari, A. Risk stratification in multiple myeloma, part 2: The significance of genetic risk factors in the era of currently available therapies. Clin. Adv. Hematol. Oncol. 2013, 11, 578–583. [Google Scholar] [PubMed]
- Puig, N.; Sarasquete, M.E.; Balanzategui, A.; Martinez, J.; Paiva, B.; Garcia, H.; Fumero, S.; Jimenez, C.; Alcoceba, M.; Chillon, M.C.; et al. Critical evaluation of ASO RQ-PCR for minimal residual disease evaluation in multiple myeloma. A comparative analysis with flow cytometry. Leukemia 2014, 28, 391–397. [Google Scholar] [CrossRef] [PubMed]
- De Mel, S.; Lim, S.H.; Tung, M.L.; Chng, W.J. Implications of heterogeneity in multiple myeloma. BioMed Res. Int. 2014, 2014, 232546. [Google Scholar] [CrossRef] [PubMed]
- Chng, W.J.; Dispenzieri, A.; Chim, C.S.; Fonseca, R.; Goldschmidt, H.; Lentzsch, S.; Munshi, N.; Palumbo, A.; Miguel, J.S.; Sonneveld, P.; et al. IMWG consensus on risk stratification in multiple myeloma. Leukemia 2014, 28, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, R.; Monge, J.; Dimopoulos, M.A. Staging and prognostication of multiple myeloma. Expert Rev. Hematol. 2014, 7, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Greipp, P.R.; San Miguel, J.; Durie, B.G.; Crowley, J.J.; Barlogie, B.; Blade, J.; Boccadoro, M.; Child, J.A.; Avet-Loiseau, H.; Kyle, R.A.; et al. International staging system for multiple myeloma. J. Clin. Oncol. 2005, 23, 3412–3420. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, M.; Terpos, E.; Kleber, M.; Gay, F.; Wasch, R.; Morgan, G.; Cavo, M.; van de Donk, N.; Beilhack, A.; Bruno, B.; et al. European myeloma network recommendations on the evaluation and treatment of newly diagnosed patients with multiple myeloma. Haematologica 2014, 99, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised international staging system for multiple myeloma: A report from international myeloma working group. J. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef] [PubMed]
- Usmani, S.Z.; Rodriguez-Otero, P.; Bhutani, M.; Mateos, M.V.; Miguel, J.S. Defining and treating high-risk multiple myeloma. Leukemia 2015, 29, 2119–2125. [Google Scholar] [CrossRef] [PubMed]
- Derlin, T.; Bannas, P. Imaging of multiple myeloma: Current concepts. World J. Orthop. 2014, 5, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Magnano, L.; Fernandez de Larrea, C.; Elena, M.; Cibeira, M.T.; Tovar, N.; Arostegui, J.I.; Pedrosa, F.; Rosinol, L.; Filella, X.; Yague, J.; et al. Prognostic impact of serum heavy/light chain pairs in patients with monoclonal gammopathy of undetermined significance and smoldering myeloma: Long-term results from a single institution. Clin. Lymphoma Myeloma Leuk. 2016, 16, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Harutyunyan, N.M.; Vardanyan, S.; Ghermezi, M.; Gottlieb, J.; Berenson, A.; Andreu-Vieyra, C.; Berenson, J.R. Levels of uninvolved immunoglobulins predict clinical status and progression-free survival for multiple myeloma patients. Br. J. Haematol. 2016, 174, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Korde, N.; Kristinsson, S.Y.; Landgren, O. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM): Novel biological insights and development of early treatment strategies. Blood 2011, 117, 5573–5581. [Google Scholar] [CrossRef] [PubMed]
- Go, R.S.; Gundrum, J.D.; Neuner, J.M. Determining the clinical significance of monoclonal gammopathy of undetermined significance: A seer-medicare population analysis. Clin. Lymphoma Myeloma Leuk. 2015, 15, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Durie, B.; Palumbo, A.; San-Miguel, J. Monoclonal antibodies in the treatment of multiple myeloma: Current status and future perspectives. Leukemia 2016, 30, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Graziani, M.S.; Merlini, G. Serum free light chain analysis in the diagnosis and management of multiple myeloma and related conditions. Expert Rev. Mol. Diagn. 2014, 14, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, E.; Li, M.; Kitto, A.; Li, J.; Wang, C.S.; Kirk, D.T.; Yellin, O.; Nichols, C.M.; Dreyer, M.P.; Ahles, C.P.; et al. Serum B-cell maturation antigen is elevated in multiple myeloma and correlates with disease status and survival. Br. J. Haematol. 2012, 158, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, R.; Bailey, R.J.; Ahmann, G.J.; Rajkumar, S.V.; Hoyer, J.D.; Lust, J.A.; Kyle, R.A.; Gertz, M.A.; Greipp, P.R.; Dewald, G.W. Genomic abnormalities in monoclonal gammopathy of undetermined significance. Blood 2002, 100, 1417–1424. [Google Scholar] [PubMed]
- Sergentanis, T.N.; Kastritis, E.; Terpos, E.; Dimopoulos, M.A.; Psaltopoulou, T. Cytogenetics and survival of multiple myeloma: Isolated and combined effects. Clin. Lymphoma Myeloma Leuk 2016, 16, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Shaughnessy, J.D.; Zhan, F.; Burington, B.E.; Huang, Y.; Colla, S.; Hanamura, I.; Stewart, J.P.; Kordsmeier, B.; Randolph, C.; Williams, D.R.; et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood 2007, 109, 2276–2284. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, R.; van Duin, M.; van Vliet, M.H.; Broijl, A.; van der Holt, B.; El Jarari, L.; van Beers, E.H.; Mulligan, G.; Avet-Loiseau, H.; Gregory, W.M.; et al. Prediction of high- and low-risk multiple myeloma based on gene expression and the international staging system. Blood 2015, 126, 1996–2004. [Google Scholar] [CrossRef] [PubMed]
- Paiva, B.; Corchete, L.A.; Vidriales, M.B.; Puig, N.; Maiso, P.; Rodriguez, I.; Alignani, D.; Burgos, L.; Sanchez, M.L.; Barcena, P.; et al. Phenotypic and genomic analysis of multiple myeloma minimal residual disease tumor cells: A new model to understand chemoresistance. Blood 2016, 127, 1896–1906. [Google Scholar] [CrossRef] [PubMed]
- Drain, S.; Flannely, L.; Drake, M.B.; Kettle, P.; Orr, N.; Bjourson, A.J.; Catherwood, M.A.; Alexander, H.D. Multidrug resistance gene expression and ABCB1 SNPs in plasma cell myeloma. Leuk. Res. 2011, 35, 1457–1463. [Google Scholar] [CrossRef] [PubMed]
- Brioli, A.; Melchor, L.; Cavo, M.; Morgan, G.J. The impact of intra-clonal heterogeneity on the treatment of multiple myeloma. Br. J. Haematol. 2014, 165, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Ocio, E.M.; Richardson, P.G.; Rajkumar, S.V.; Palumbo, A.; Mateos, M.V.; Orlowski, R.; Kumar, S.; Usmani, S.; Roodman, D.; Niesvizky, R.; et al. New drugs and novel mechanisms of action in multiple myeloma in 2013: A report from the international myeloma working group (IMWG). Leukemia 2014, 28, 525–542. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.; Shin, S.; Lee, A.; Yoon, D.H.; Suh, C.; Park, C.-J.; Huh, J.; Park, C.-S. RGS1 expression is associated with poor prognosis in multiple myeloma. J. Clin. Pathol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014, 5, 2997–3010. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Boyle, E.M.; Wardell, C.P.; Murison, A.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Johnson, D.C.; Kaiser, M.F.; Melchor, L.; et al. Mutational spectrum, copy number changes, and outcome: Results of a sequencing study of patients with newly diagnosed myeloma. J. Clin. Oncol. 2015, 33, 3911–3920. [Google Scholar] [CrossRef] [PubMed]
- Lipchick, B.C.; Fink, E.E.; Nikiforov, M.A. Oxidative stress and proteasome inhibitors in multiple myeloma. Pharmacol. Res. 2016, 105, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Naymagon, L.; Abdul-Hay, M. Novel agents in the treatment of multiple myeloma: A review about the future. J. Hematol. Oncol. 2016, 9, 52–72. [Google Scholar] [CrossRef] [PubMed]
- Nathwani, N.; Larsen, J.T.; Kapoor, P. Consolidation and maintenance therapies for newly diagnosed multiple myeloma in the era of novel agents. Curr. Hematol. Malig. Rep. 2016, 11, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Zangari, M.; Suva, L.J. The effects of proteasome inhibitors on bone remodeling in multiple myeloma. Bone 2016, 86, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Muchtar, E.; Gertz, M.A.; Magen, H. A practical review on carfilzomib in multiple myeloma. Eur. J. Haematol. 2016, 96, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Nikesitch, N.; Ling, S.C. Molecular mechanisms in multiple myeloma drug resistance. J. Clin. Pathol. 2016, 69, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Levin, N.; Spencer, A.; Harrison, S.J.; Chauhan, D.; Burrows, F.J.; Anderson, K.C.; Reich, S.D.; Richardson, P.G.; Trikha, M. Marizomib irreversibly inhibits proteasome to overcome compensatory hyperactivation in multiple myeloma and solid tumour patients. Br. J. Haematol. 2016, 174, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Mark, T.M.; Shah, J.J. Practical approaches to the management of dual refractory multiple myeloma. Curr. Hematol. Malig. Rep. 2016, 11, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Paiva, B.; Martinez-Lopez, J.; Vidriales, M.B.; Mateos, M.V.; Montalban, M.A.; Fernandez-Redondo, E.; Alonso, L.; Oriol, A.; Teruel, A.I.; de Paz, R.; et al. Comparison of immunofixation, serum free light chain, and immunophenotyping for response evaluation and prognostication in multiple myeloma. J. Clin. Oncol. 2011, 29, 1627–1633. [Google Scholar] [CrossRef] [PubMed]
- Wester, R.; Sonneveld, P. Innovations in treatment and response evaluation in multiple myeloma. Haematologica 2016, 101, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Rawstron, A.C.; Gregory, W.M.; de Tute, R.M.; Davies, F.E.; Bell, S.E.; Drayson, M.T.; Cook, G.; Jackson, G.H.; Morgan, G.J.; Child, J.A.; et al. Minimal residual disease in myeloma by flow cytometry: Independent prediction of survival benefit per log reduction. Blood 2015, 125, 1932–1935. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Paiva, B.; Anderson, K.C.; Durie, B.; Landgren, O.; Moreau, P.; Munshi, N.; Lonial, S.; Blade, J.; Mateos, M.V.; et al. International myeloma working group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016, 17, e328–346. [Google Scholar] [CrossRef]
- Rawstron, A.C.; Child, J.A.; de Tute, R.M.; Davies, F.E.; Gregory, W.M.; Bell, S.E.; Szubert, A.J.; Navarro-Coy, N.; Drayson, M.T.; Feyler, S.; et al. Minimal residual disease assessed by multiparameter flow cytometry in multiple myeloma: Impact on outcome in the medical research council myeloma IX study. J. Clin. Oncol. 2013, 31, 2540–2547. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Weisel, K.C.; Song, K.W.; Delforge, M.; Karlin, L.; Goldschmidt, H.; Moreau, P.; Banos, A.; Oriol, A.; Garderet, L.; et al. Cytogenetics and long-term survival of patients with refractory or relapsed and refractory multiple myeloma treated with pomalidomide and low-dose dexamethasone. Haematologica 2015, 100, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Avet-Loiseau, H.; Attal, M.; Campion, L.; Caillot, D.; Hulin, C.; Marit, G.; Stoppa, A.M.; Voillat, L.; Wetterwald, M.; Pegourie, B.; et al. Long-term analysis of the IFM 99 trials for myeloma: Cytogenetic abnormalities (t(4;14), del(17p), 1q gains) play a major role in defining long-term survival. J. Clin. Oncol. 2012, 30, 1949–1952. [Google Scholar] [CrossRef] [PubMed]
- Vincent Rajkumar, S. Multiple myeloma: 2014 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2014, 89, 999–1009. [Google Scholar] [PubMed]
- Wildes, T.M.; Rosko, A.; Tuchman, S.A. Multiple myeloma in the older adult: Better prospects, more challenges. J. Clin. Oncol. 2014, 32, 2531–2540. [Google Scholar] [CrossRef] [PubMed]
- Radich, J.P. How I monitor residual disease in chronic myeloid leukemia. Blood 2009, 114, 3376–3381. [Google Scholar] [CrossRef] [PubMed]
- Coustan-Smith, E.; Campana, D. Immunologic minimal residual disease detection in acute lymphoblastic leukemia: A comparative approach to molecular testing. Best Pract. Res. Clin. Haematol. 2010, 23, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Bader, P.; Kreyenberg, H.; von Stackelberg, A.; Eckert, C.; Salzmann-Manrique, E.; Meisel, R.; Poetschger, U.; Stachel, D.; Schrappe, M.; Alten, J.; et al. Monitoring of minimal residual disease after allogeneic stem-cell transplantation in relapsed childhood acute lymphoblastic leukemia allows for the identification of impending relapse: Results of the ALL-BFM-SCT 2003 trial. J. Clin. Oncol. 2015, 33, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- van de Velde, H.J.; Liu, X.; Chen, G.; Cakana, A.; Deraedt, W.; Bayssas, M. Complete response correlates with long-term survival and progression-free survival in high-dose therapy in multiple myeloma. Haematologica 2007, 92, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Paiva, B.; Vidriales, M.B.; Cerveró, J.; Mateo, G.; Pérez, J.J.; Montalbán, M.A.; Sureda, A.; Montejano, L.; Gutiérrez, N.C.; García de Coca, A.; et al. Multiparameter flow cytometric remission is the most relevant prognostic factor for multiple myeloma patients who undergo autologous stem cell transplantation. Blood 2008, 112, 4017–4023. [Google Scholar] [CrossRef] [PubMed]
- Gonsalves, W.I.; Morice, W.G.; Rajkumar, V.; Gupta, V.; Timm, M.M.; Dispenzieri, A.; Buadi, F.K.; Lacy, M.Q.; Singh, P.P.; Kapoor, P.; et al. Quantification of clonal circulating plasma cells in relapsed multiple myeloma. Br. J. Haematol. 2014, 167, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Witzig, T.E.; Dhodapkar, M.V.; Kyle, R.A.; Greipp, P.R. Quantitation of circulating peripheral blood plasma cells and their relationship to disease activity in patients with multiple myeloma. Cancer 1993, 72, 108–113. [Google Scholar] [CrossRef]
- Moreau, P.; Robillard, N.; Jego, G.; Pellat, C.; Le Gouill, S.; Thoumi, S.; Avet-Loiseau, H.; Harousseau, J.L.; Bataille, R. Lack of CD27 in myeloma delineates different presentation and outcome. Br. J. Haematol. 2006, 132, 168–170. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Hieber, M.; Pérez-Andrés, M.; Paiva, B.; Flores-Montero, J.; Perez, J.J.; Gutierrez, N.C.; Vidriales, M.-B.; Matarraz, S.; San Miguel, J.F.; Orfao, A. CD117 expression in gammopathies is associated with an altered maturation of the myeloid and lymphoid hematopoietic cell compartments and favorable disease features. Haematologica 2011, 96, 328–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruneri, G.; Ponzoni, M.; Ferreri, A.J.M.; Freschi, M.; Tresoldi, M.; Baldini, L.; Mattioli, M.; Agnelli, L.; Govi, S.; Mancuso, P.; et al. The prevalence and clinical implications of C-kit expression in plasma cell myeloma. Histopathology 2006, 48, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Zismanov, V.; Lishner, M.; Tartakover-Matalon, S.; Radnay, J.; Shapiro, H.; Drucker, L. Tetraspanin-induced death of myeloma cell lines is autophagic and involves increased UPR signalling. Br. J. Cancer 2009, 101, 1402–1409. [Google Scholar] [CrossRef] [PubMed]
- Paiva, B.; Gutierrez, N.C.; Chen, X.; Vidriales, M.B.; Montalban, M.A.; Rosinol, L.; Oriol, A.; Martinez-Lopez, J.; Mateos, M.V.; Lopez-Corral, L.; et al. Clinical significance of CD81 expression by clonal plasma cells in high-risk smoldering and symptomatic multiple myeloma patients. Leukemia 2012, 26, 1862–1869. [Google Scholar] [CrossRef] [PubMed]
- Mateo, G.; Montalban, M.A.; Vidriales, M.B.; Lahuerta, J.J.; Mateos, M.V.; Gutierrez, N.; Rosinol, L.; Montejano, L.; Blade, J.; Martinez, R.; et al. Prognostic value of immunophenotyping in multiple myeloma: A study by the pethema/gem cooperative study groups on patients uniformly treated with high-dose therapy. J. Clin. Oncol. 2008, 26, 2737–2744. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.J.; Lee, H.; Jung, G.; Gil, M.; Park, H.; Park, Y.S.; Yoon, D.H.; Suh, C.; Park, C.J.; Huh, J.; et al. Expression of CD99 in multiple myeloma: A clinicopathologic and immunohistochemical study of 170 cases. Korean J. Pathol. 2014, 48, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Pellat-Deceunynck, C.; Barillé, S.; Puthier, D.; Rapp, M.J.; Harousseau, J.L.; Bataille, R.; Amiot, M. Adhesion molecules on human myeloma cells: Significant changes in expression related to malignancy, tumor spreading, and immortalization. Cancer Res. 1995, 55, 3647–3653. [Google Scholar] [PubMed]
- Zheng, W.; Liu, D.; Fan, X.; Powers, L.; Goswami, M.; Hu, Y.; Lin, P.; Medeiros, L.J.; Wang, S.A. Potential therapeutic biomarkers in plasma cell myeloma: A flow cytometry study. Cytometry B Clin. Cytom. 2013, 84, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Paino, T.; Paiva, B.; Sayagues, J.M.; Mota, I.; Carvalheiro, T.; Corchete, L.A.; Aires-Mejia, I.; Perez, J.J.; Sanchez, M.L.; Barcena, P.; et al. Phenotypic identification of subclones in multiple myeloma with different chemoresistant, cytogenetic and clonogenic potential. Leukemia 2015, 29, 1186–1194. [Google Scholar] [CrossRef] [PubMed]
- Atanackovic, D.; Radhakrishnan, S.V.; Bhardwaj, N.; Luetkens, T. Chimeric antigen receptor (CAR) therapy for multiple myeloma. Br. J. Haematol. 2016, 172, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Dowling, P.; Hayes, C.; Ting, K.R.; Hameed, A.; Meiller, J.; Mitsiades, C.; Anderson, K.C.; Clynes, M.; Clarke, C.; Richardson, P.; et al. Identification of proteins found to be significantly altered when comparing the serum proteome from multiple myeloma patients with varying degrees of bone disease. BMC Genom. 2014, 15, 904–916. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.T.; Tian, E.B.; Chen, Y.L.; Deng, H.T.; Wang, Q.T. Proteomic analysis for finding serum pathogenic factors and potential biomarkers in multiple myeloma. Chin. Med. J. 2015, 128, 1108–1113. [Google Scholar] [PubMed]
- Dytfeld, D.; Rosebeck, S.; Kandarpa, M.; Mayampurath, A.; Mellacheruvu, D.; Alonge, M.M.; Ngoka, L.; Jasielec, J.; Richardson, P.G.; Volchenboum, S.; et al. Proteomic profiling of naïve multiple myeloma patient plasma cells identifies pathways associated with favourable response to bortezomib-based treatment regimens. Br. J. Haematol. 2015, 170, 66–79. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Plasma Cell Myeloma |
|---|
| One or both of |
| ≥10% clonal plasma cells in the BM |
| Extramedullary plasmacytoma |
| One or more of the following CRAB criteria and biomarkers of malignancy |
| Hypercalcaemia (C) as defined by serum calcium >0.25 mM above the normal range or >2.75 mM |
| Renal insufficiency (R) as defined by creatinine clearance <40 mL/min or serum creatinine >177 µM |
| Anaemia (A) as defined by haemoglobin >20 g/L below the normal range or <100 g/L |
| ≥1 bone lesion (B) as detected by radiography, computed tomography (CT) or Positron Emission Tomography-Computed Tomography (PET-CT) |
| Bone marrow consisting of ≥60% clonal plasma cells as calculated by ĸ/λ light chain restriction flow cytometry, immunohistochemistry (IHC) or immunofluorescence (IF) |
| Involved:uninvolved serum free light chain ration ≥100 |
| >1 focal lesion by magnetic resonance imaging (MRI) |
| Smouldering Multiple Myeloma |
| One or more of Monoclonal IgG or IgA ≥30 g/L in serum |
| Urinary monoclonal protein ≥500 mg/24 h in urine |
| Bone marrow consisting of 10%–60% clonal plasma cells as calculated by κ/λ light chain restriction flow cytometry, immunohistochemistry (IHC) or immunofluorescence (IF) |
| Not defined as plasma cell myeloma or amyloidosis |
| Stage | Criteria | 5 Year Survival |
|---|---|---|
| I | ISS-1 No high-risk chromosomal aberrations by iFISH Normal lactate dehydrogenase | 80% |
| II | Neither Stage I nor III | 60% |
| III | ISS-3 One or more high-risk chromosomal aberration by iFISH or high lactate dehydrogenase | 40% |
| High-risk chromosomal aberrations are defined as del(17p), t(4;14), t(14;16) | ||
| Risk | Criteria | Median OS |
|---|---|---|
| High | High-risk chromosomal aberrations del(17p), t(14;16), t(14;20) or high-risk GEP | 3 years |
| Intermediate | Chromosomal aberrations t(4;14), del 13, Hypodiploidy or PCLI >3% | 4–5 years |
| Standard | Not the above | 8–10 years |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Egan, P.; Drain, S.; Conway, C.; Bjourson, A.J.; Alexander, H.D. Towards Stratified Medicine in Plasma Cell Myeloma. Int. J. Mol. Sci. 2016, 17, 1760. https://doi.org/10.3390/ijms17101760
Egan P, Drain S, Conway C, Bjourson AJ, Alexander HD. Towards Stratified Medicine in Plasma Cell Myeloma. International Journal of Molecular Sciences. 2016; 17(10):1760. https://doi.org/10.3390/ijms17101760
Chicago/Turabian StyleEgan, Philip, Stephen Drain, Caroline Conway, Anthony J. Bjourson, and H. Denis Alexander. 2016. "Towards Stratified Medicine in Plasma Cell Myeloma" International Journal of Molecular Sciences 17, no. 10: 1760. https://doi.org/10.3390/ijms17101760
APA StyleEgan, P., Drain, S., Conway, C., Bjourson, A. J., & Alexander, H. D. (2016). Towards Stratified Medicine in Plasma Cell Myeloma. International Journal of Molecular Sciences, 17(10), 1760. https://doi.org/10.3390/ijms17101760