
p-Hydroxyphenyl-pyranoanthocyanins: An Experimental and Theoretical Investigation of Their Acid—Base Properties and Molecular Interactions



Abstract
:
1. Introduction
2. Results and Discussion
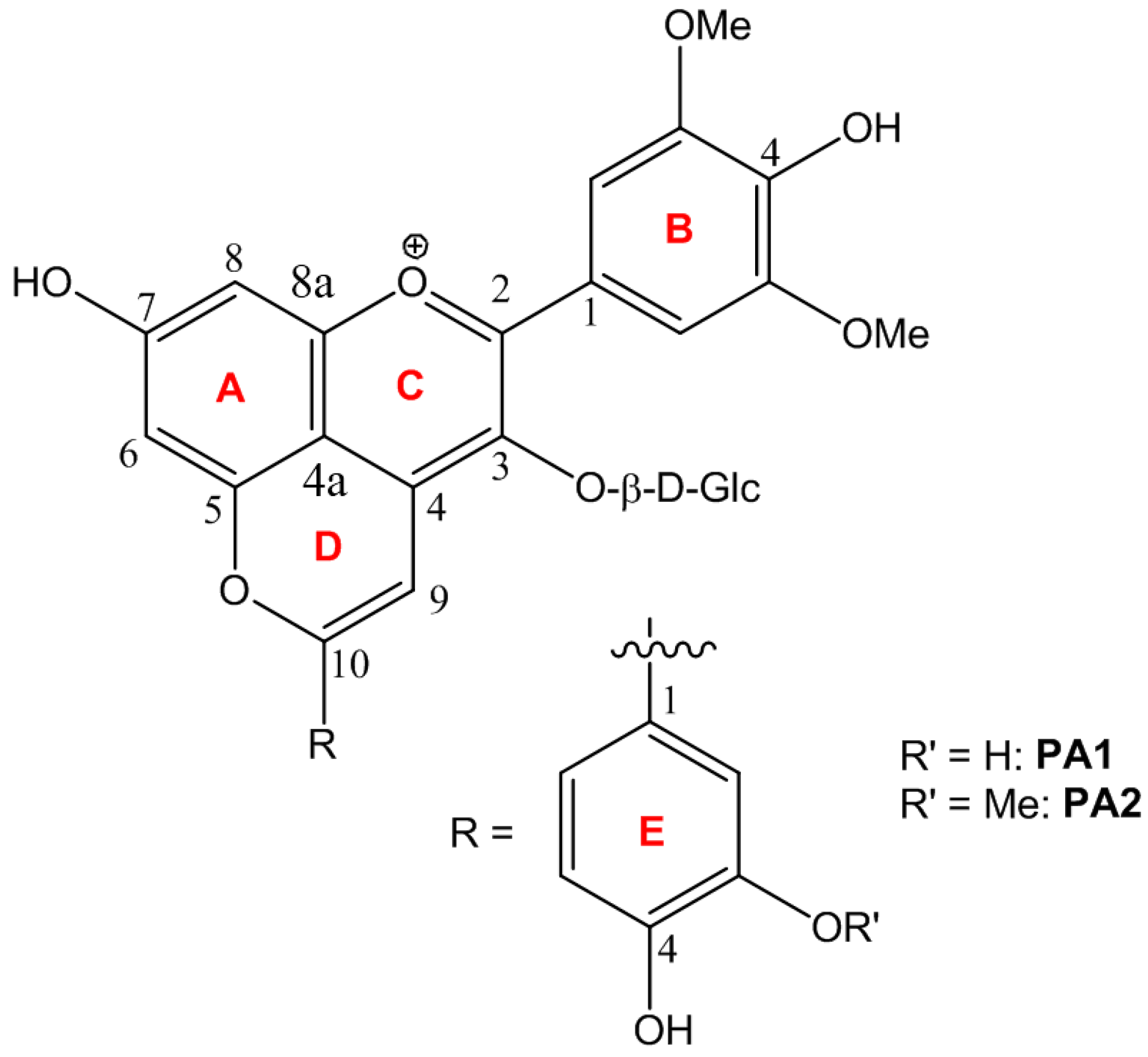
2.1. Characterization of PA1 and PA2
2.2. Structural Transformations
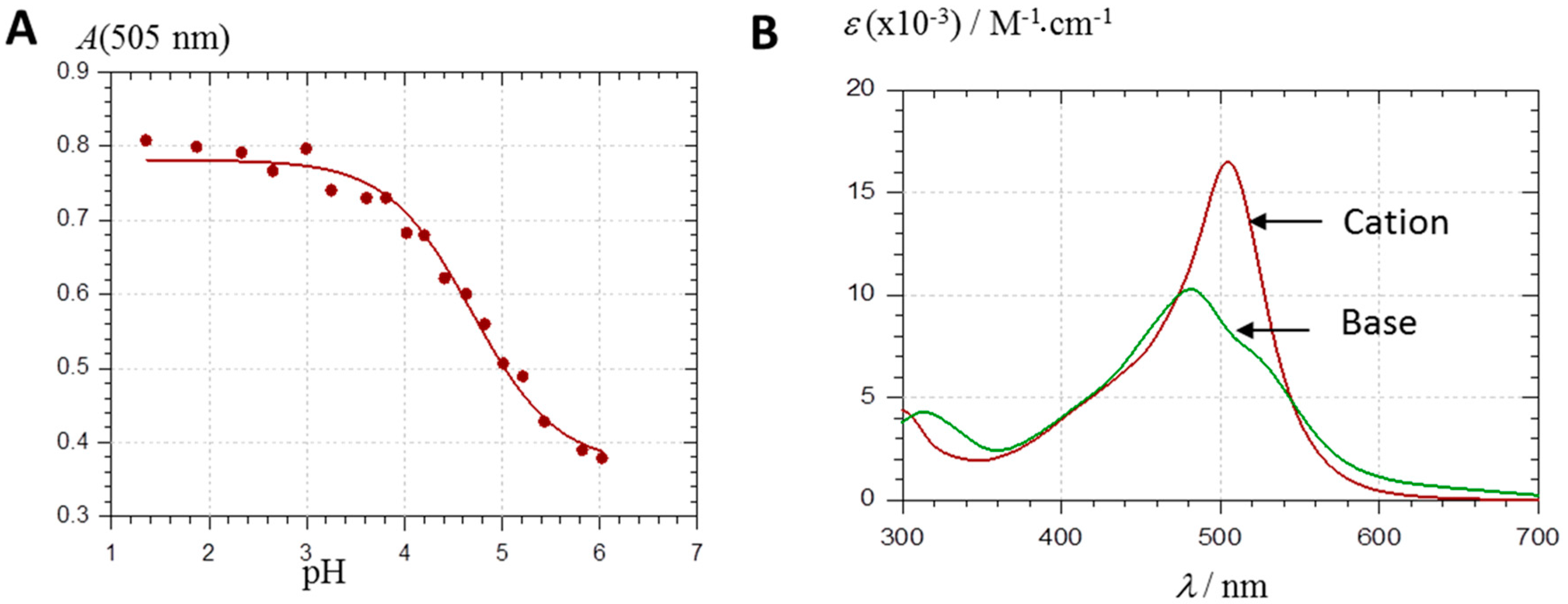
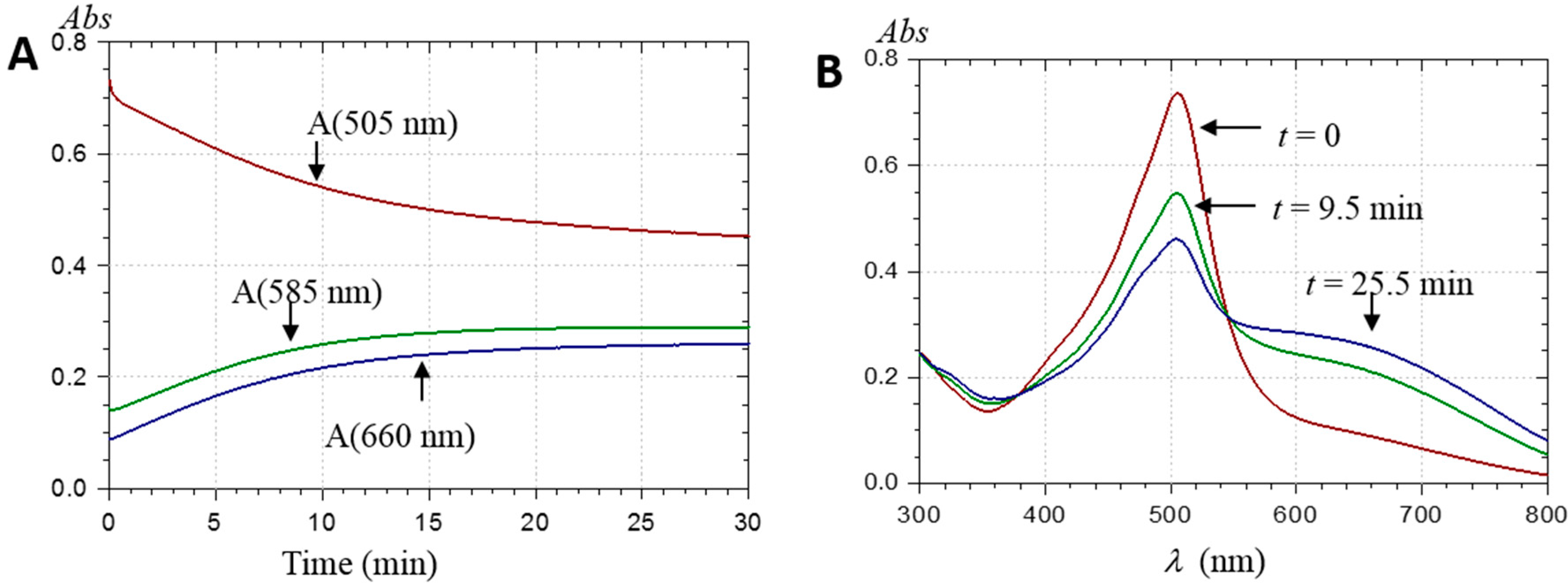
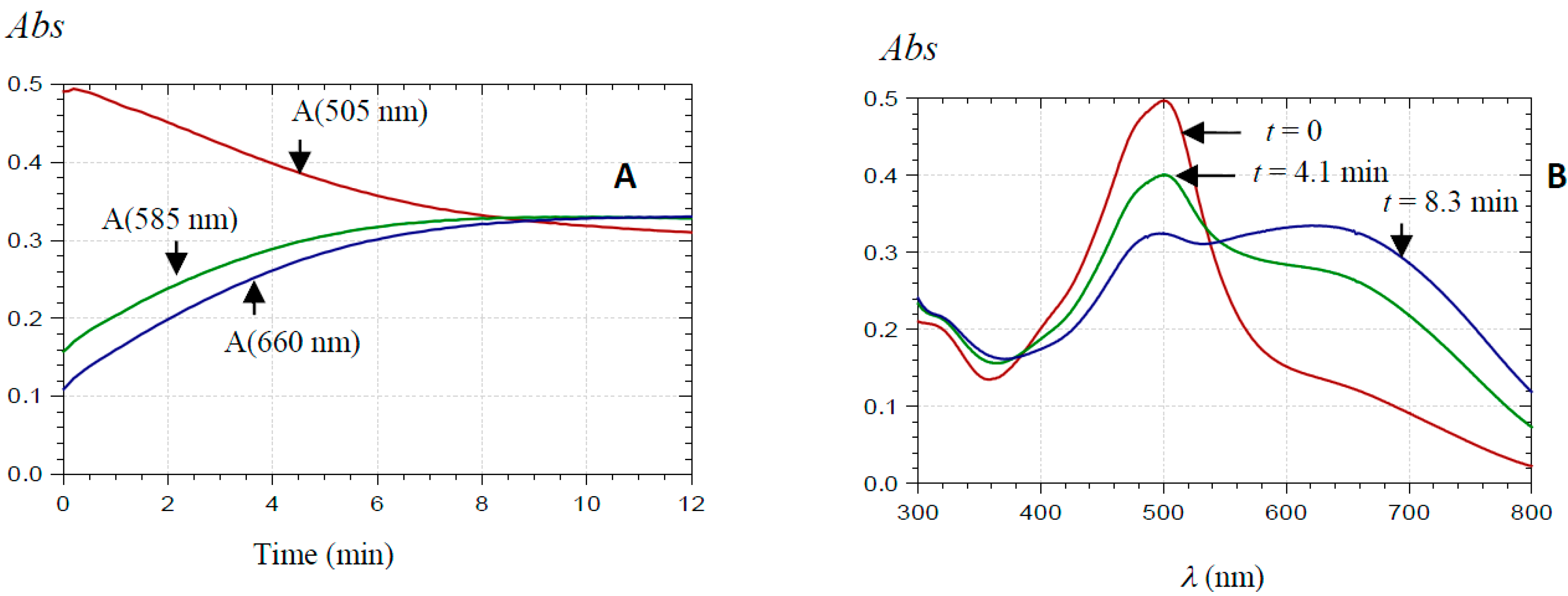
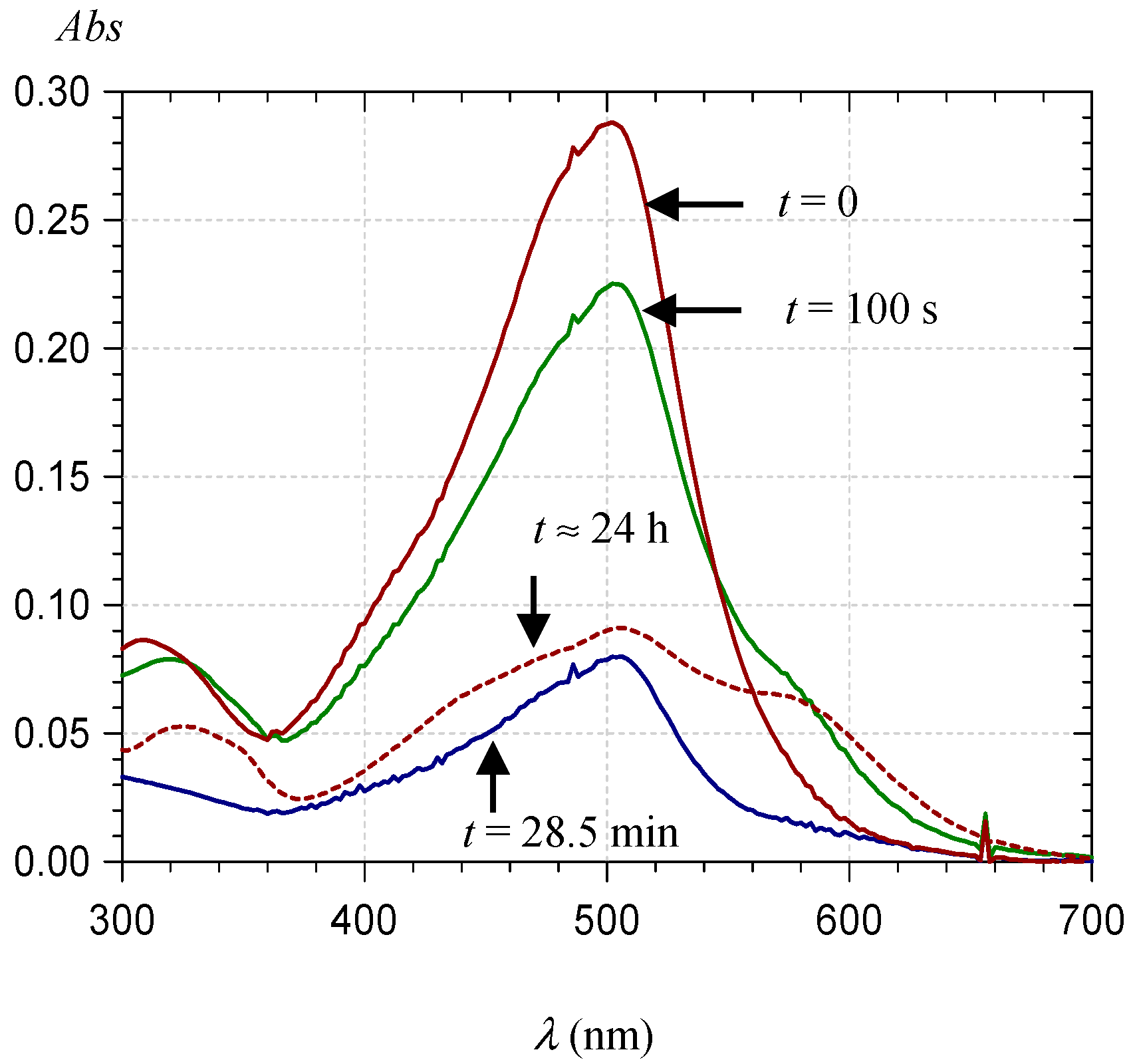
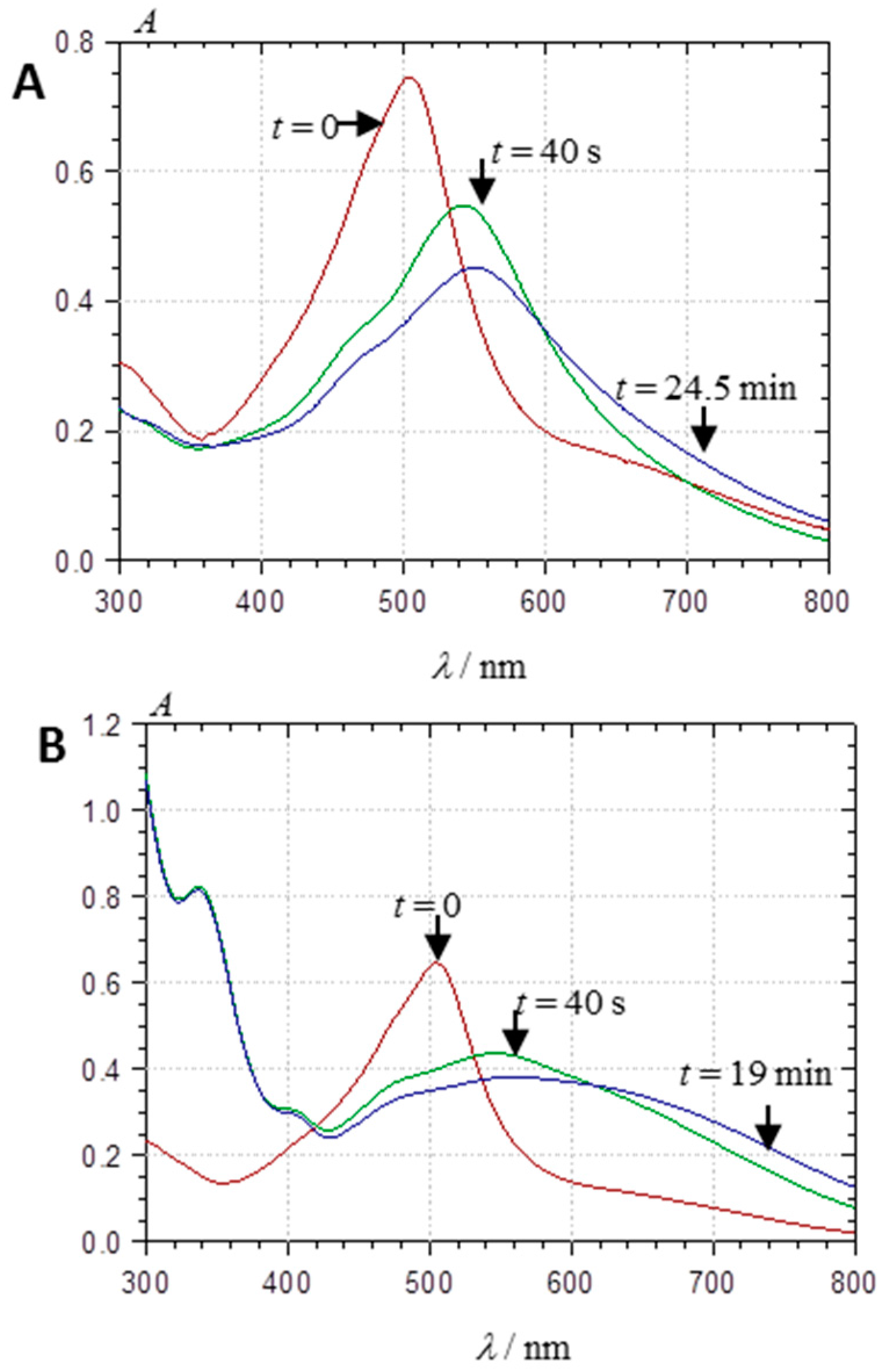
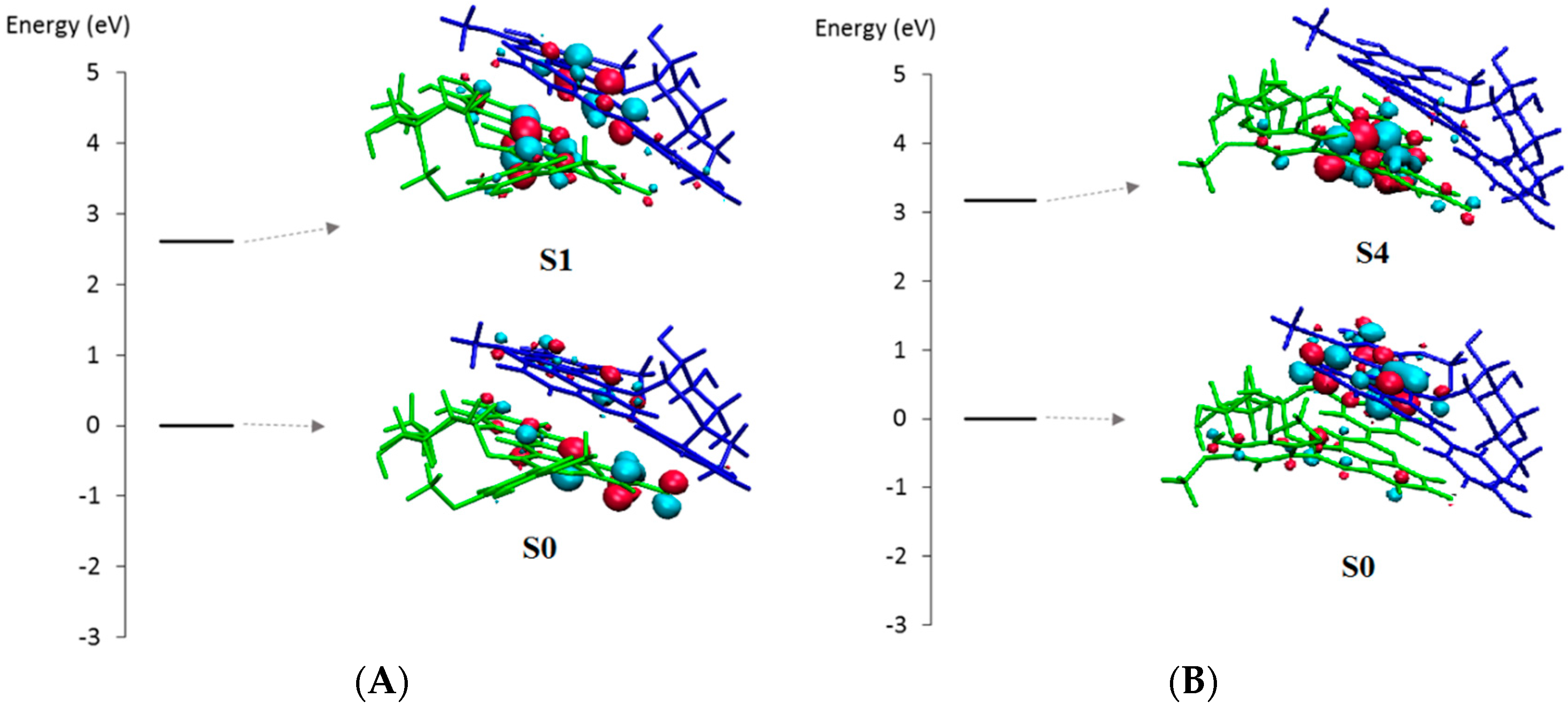
2.2.1. UV-Visible Spectroscopy
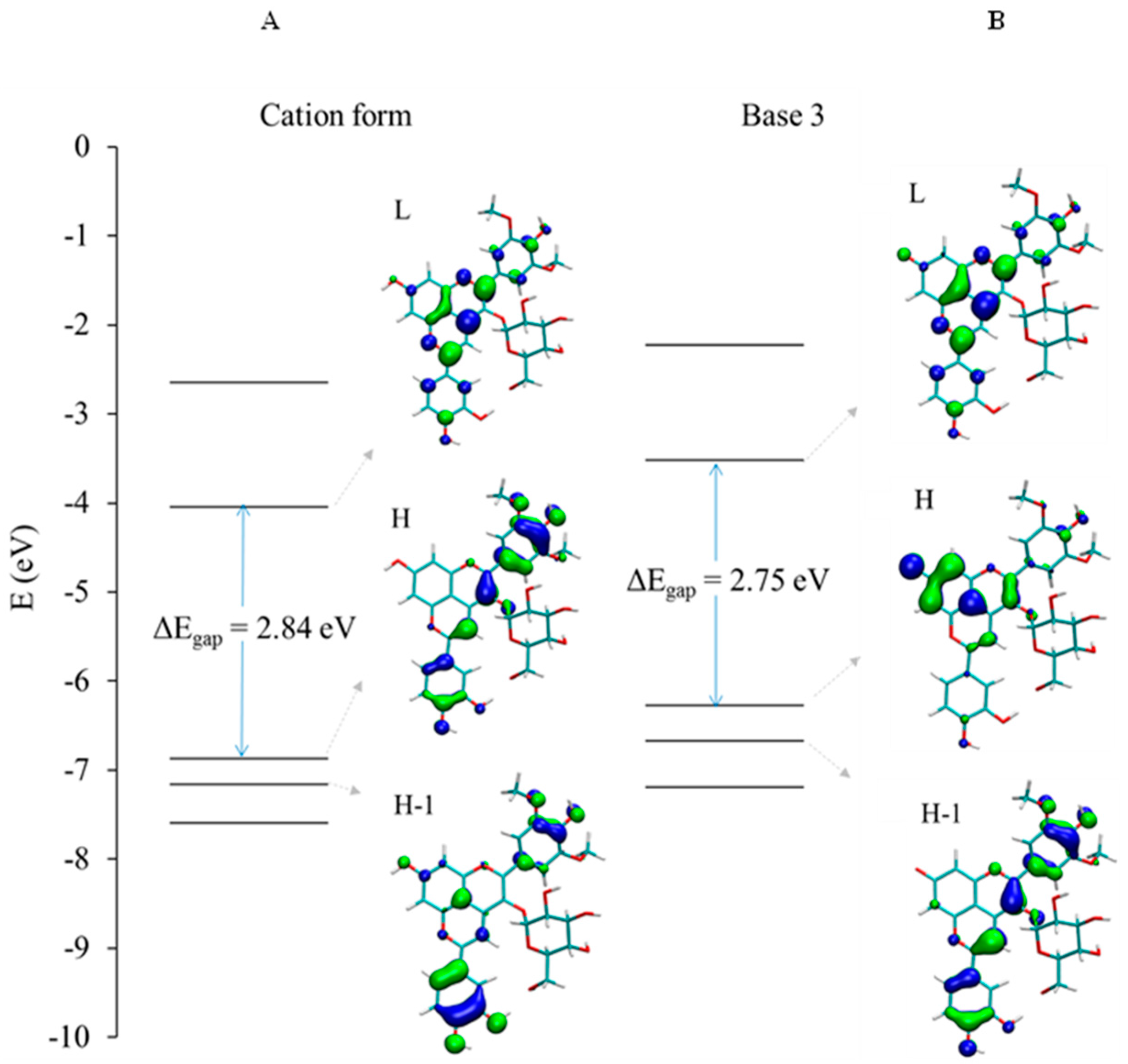
2.2.2. DFT-Based Rationalization
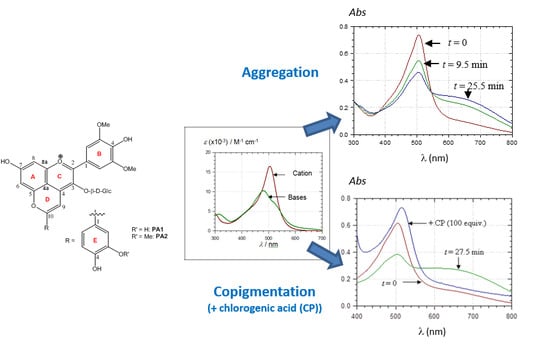
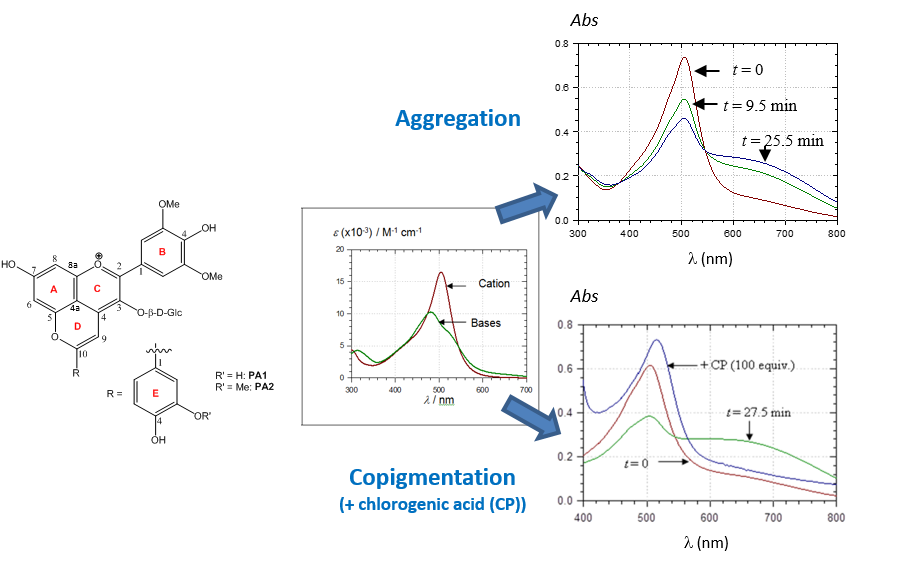
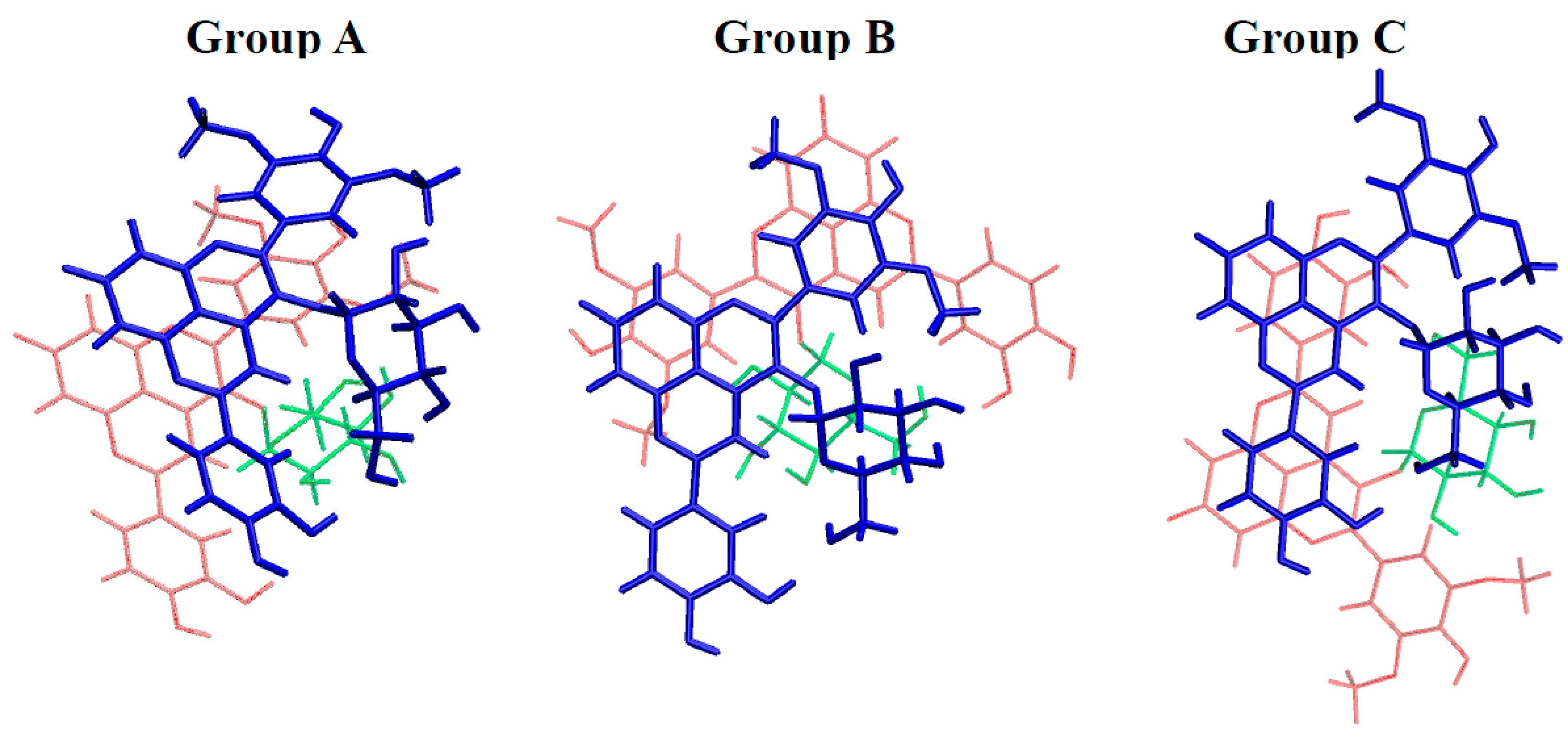
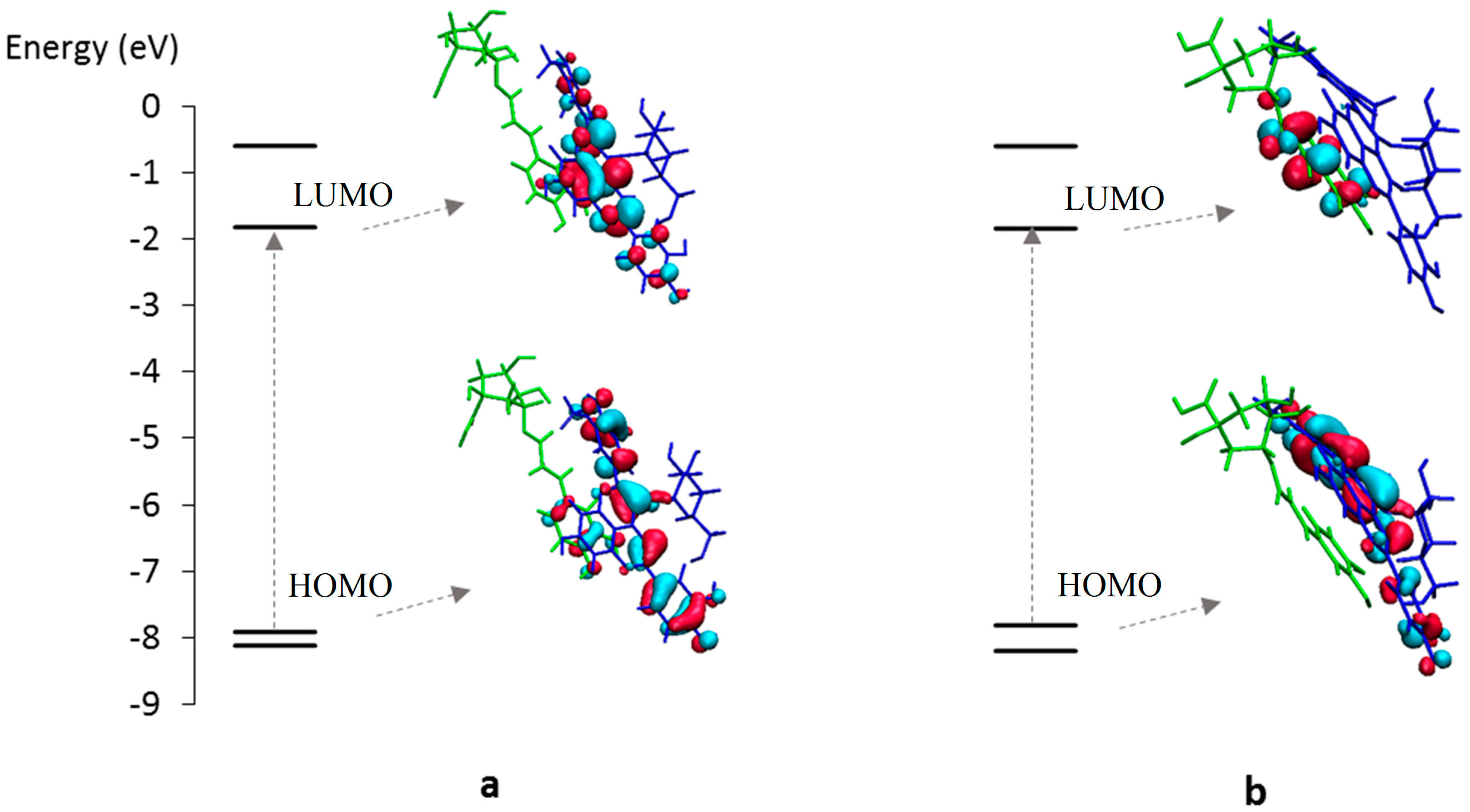
2.3. Noncovalent Assemblies of Pyranoanthocyanins
2.3.1. Self-Association
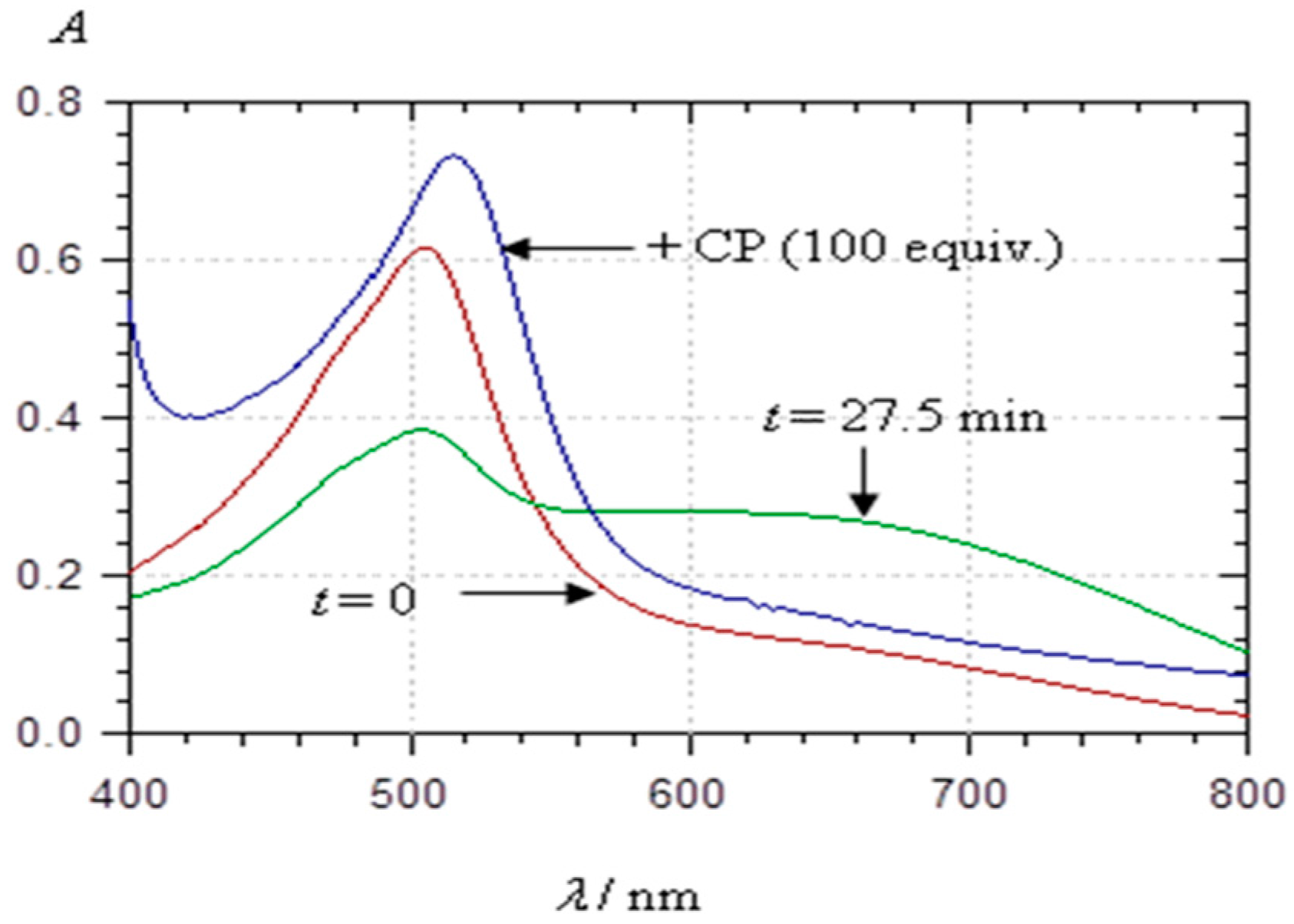
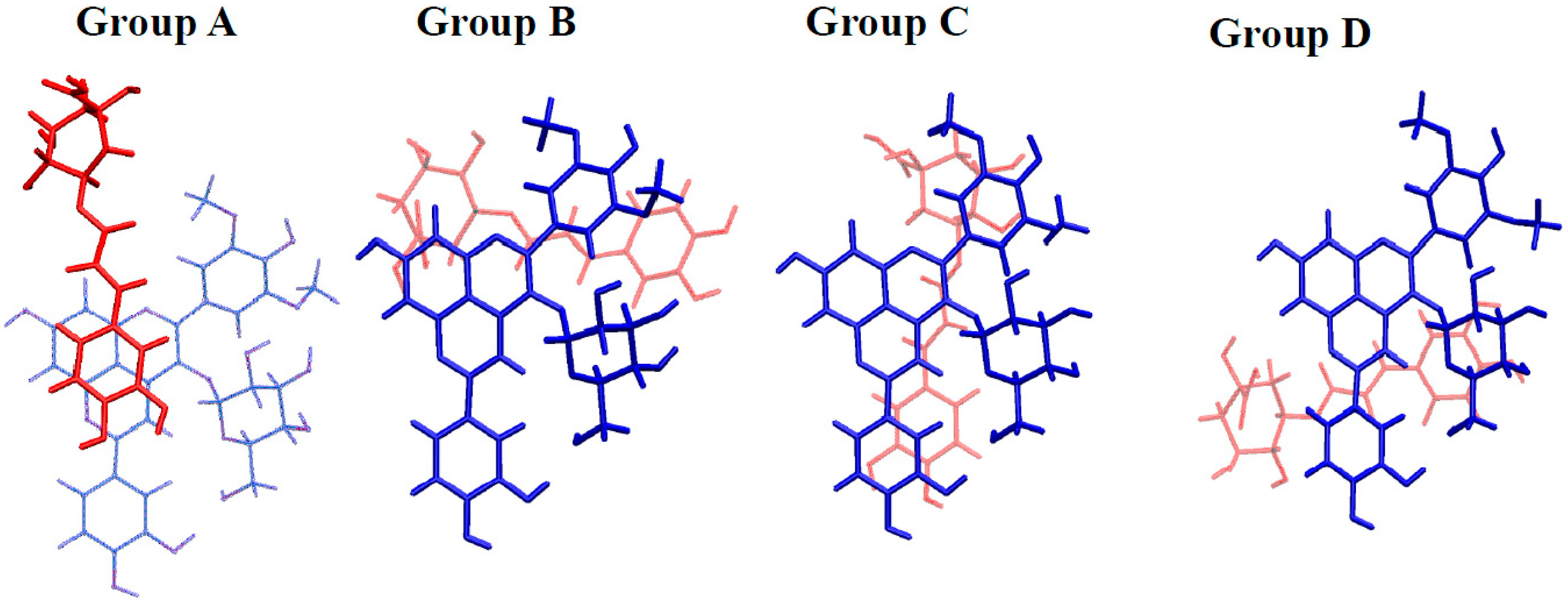
2.3.2. Copigmentation
2.3.3. Metal Complexation
2.4. Spectral Shifts Related to Noncovalent Association
3. Materials and Methods
3.1. Chemicals
3.2. Production of Vinylcatechol
3.3. Pyranoanthocyanin Synthesis
3.4. Purification of PA1 and PA2
3.5. NMR Analysis
3.6. UV-Visible Spectroscopy
3.7. Dynamic Light Scattering (DLS)
3.8. Methods of Calculation for UV-Visible Properties of Single Compounds
3.9. Methods of Calculation for Self-Association and Copigmentation
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bayer, E.; Egeter, H.; Fink, A.; Nether, K.; Wegmann, K. Complex Formation and Flower Colors. Angew. Chem. Int. Ed. 1966, 5, 791–798. [Google Scholar] [CrossRef]
- Brouillard, R. The in vivo expression of anthocyanin colour in plants. Phytochemistry 1983, 22, 1311–1323. [Google Scholar] [CrossRef]
- Trouillas, P.; Sancho-García, J.C.; de Freitas, V.; Gierschner, J.; Otyepka, M.; Dangles, O. Stabilizing and Modulating Color by Copigmentation: Insights from Theory and Experiment. Chem. Rev. 2016, 116, 4937–4982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, T.; Kondo, T. Structure and Molecular Stacking of Anthocyanins. Flower Color Variation. Angew. Chem. Int. Ed. 1991, 30, 17–33. [Google Scholar] [CrossRef]
- Yoshida, K. Metal Complex Pigment Involved in the Blue Sepal Color Development of Hydrangea. J. Agric. Food Chem. 2015, 63, 7630–7635. [Google Scholar]
- Fulcrand, H.; Cameira dos Santos, P.J.; Sarni-Manchado, P.; Cheynier, V.; Favre-Bonvin, J.J. Structure of new anthocyanin-derived wine pigments. Chem. Soc. Perkin Trans. 1996, 1, 735–739. [Google Scholar] [CrossRef]
- Schwarz, M.; Wabnitz, T.C.; Winterhalter, P. Pathway Leading to the Formation of Anthocyanin—Vinylphenol Adducts and Related Pigments in Red Wines. J. Agric. Food Chem. 2003, 51, 3682–3687. [Google Scholar] [CrossRef] [PubMed]
- Vallverdú-Queralt, A.; Meudec, E.; Ferreira-Lima, N.; Sommerer, N.; Dangles, O.; Cheynier, V.; Guernevé, C.L. A comprehensive investigation of guaiacyl-pyranoanthocyanin synthesis by one-/two-dimensional NMR and UPLC-DAD-ESI-MSn. Food Chem. 2016, 199, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Quijada-Morín, N.; Dangles, O.; Rivas-Gonzalo, J.C.; Escribano-Bailón, M.T. Physico-Chemical and Chromatic Characterization of Malvidin 3-Glucoside-vinylcatechol and Malvidin 3-Glucoside-vinylguaiacol Wine Pigments. J. Agric. Food Chem. 2010, 58, 9744–9752. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.; Fernandes, V.; Miranda, C.; Santos-Buelga, C.; Silva, A.; Freitas, V.; Mateus, N. Color properties of four cyanidin-pyruvic acid adducts. J. Agric. Food Chem. 2006, 54, 6894–6903. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.; Petrov, V.; Teixeira, N.; Mateus, N.; Pina, F.; de Freitas, V. Establishment of the Chemical Equilibria of Different Types of Pyranoanthocyanins in Aqueous Solutions: Evidence for the Formation of Aggregation in Pyranomalvidin-3-O-coumaroylglucoside-(+)-catechin. J. Phys. Chem. B 2010, 114, 13232–13240. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Oliveira, J.; Silva, A.M.S.; Mateus, N.; de Freitas, V. Oxovitisins: A new class of neutral pyranone-anthocyanin derivatives in red wines. J. Agric. Food Chem. 2010, 58, 8814–8819. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Alonso, S.; Blanco-Vega, D.; Gómez, V.; Hermosín-Gutiérrez, I. Synthesis, Isolation, Structure Elucidation, and Color Properties of 10-Acetyl-pyranoanthocyanins. J. Agric. Food Chem. 2012, 60, 12210–12223. [Google Scholar] [CrossRef] [PubMed]
- Bakker, J.; Timberlake, C.F. Isolation, Identification, and Characterization of New Color-Stable Anthocyanins Occurring in Some Red Wines. J. Agric. Food Chem. 1997, 45, 35–43. [Google Scholar] [CrossRef]
- Asenstorfer, R.E.; Jones, G.P. Charge equilibria and pK values of 5-carboxypyranomalvidin-3-glucoside (vitisin A) by electrophoresis and absorption spectroscopy. Tetrahedron 2007, 63, 4788–4792. [Google Scholar] [CrossRef]
- Hakansson, A.E.; Pardon, K.; Hayasaka, Y.; de Sa, M.; Herderich, M. Structures and colour properties of new red wine pigments. Tetrahedron Lett. 2003, 44, 4887–4891. [Google Scholar] [CrossRef]
- Pina, F. Chemical Applications of Anthocyanins and Related Compounds. A Source of Bioinspiration. J. Agric. Food Chem. 2014, 62, 6885–6897. [Google Scholar] [CrossRef] [PubMed]
- Pina, F.; Melo, M.J.; Laia, C.A.T.; Parola, A.J.; Lima, J.C. Chemistry and Applications of Flavylium Compounds: A Handful of Colours. Chem. Soc. Rev. 2012, 41, 869–908. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.; Mateus, N.; de Freitas, V. Previous and recent advances in pyranoanthocyanins equilibria in aqueous solution. Dyes Pigm. 2014, 100, 190–200. [Google Scholar] [CrossRef]
- Trouillas, P.; di Meo, F.; Gierschner, J.; Linares, M.; Sancho-García, J.C.; Otyepka, M. Optical properties of wine pigments: Theoretical guidelines with new methodological perspectives. Tetrahedron 2015, 71, 3079–3088. [Google Scholar] [CrossRef]
- Hoshino, T. Self-Association of Flavylium Cations of Anthocyanidin 3,5-Diglucosides Studied by Circular Dichroism and 1H NMR. Phytochemistry 1992, 31, 647–653. [Google Scholar] [CrossRef]
- Hoshino, T. An Approximate Estimate of Self-Association Constants and the Self-Stacking Conformation of Malvin Quinonoidal Bases Studied by 1H NMR. Phytochemistry 1991, 30, 2049–2055. [Google Scholar] [CrossRef]
- Leydet, Y.; Gavara, R.; Petrov, V.; Diniz, A.M.; Parola, A.J.; Lima, J.C.; Pina, F. The Effect of Self-Aggregation on the Determination of the Kinetic and Thermodynamic Constants of the Network of Chemical Reactions in 3-Glucoside Anthocyanins. Phytochemistry 2012, 83, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Steyer, D.; Erny, C.; Claudel, P.; Riveill, G.; Karst, F.; Legras, J.-L. Genetic analysis of geraniol metabolism during fermentation. Food. Microbiol. 2013, 33, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, F.; Sancho-García, J.C.; Dangles, O.; Trouillas, P. Highlights on anthocyanin pigmentation and copigmentation: A matter of flavonoid π-stacking complexation to be described by DFT-D. J. Chem. Theory Comput. 2012, 8, 2034–2043. [Google Scholar] [CrossRef] [PubMed]
- Anouar, E.H.; Gierschner, J.; Duroux, J.L.; Trouillas, P. UV/Visible spectra of natural polyphenols: A time-dependent density functional theory study. Food Chem. 2012, 131, 79–89. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 (Revision A.02); Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pyranoanthocyanin Form | PA1 | PA2 |
|---|---|---|
| Base 1 | 4.28 | 3.60 |
| Base 2 | 4.31 | 3.14 |
| Base 3 | 0 | 0 |
| Group | Geometry | A···A | AH+···A | AH+···AH+ |
|---|---|---|---|---|
| A | 1 | −28.0 | −22.3 | −10.1 |
| 2 | −23.2 | −25.3 | −25.4 | |
| 3 | −22.8 | −22.2 | −11.4 | |
| 4 | −19.2 | −19.9 | −9.9 | |
| 5 | −18.2 | −22.4 | −14.9 | |
| 6 | −14.9 | −15.9 | −9.0 | |
| 7 | −12.7 | −13.4 | −7.9 | |
| B | 8 | −15.9 | −21.6 | −8.6 |
| 9 | −15.6 | −15.3 | −9.5 | |
| C | 10 | −17.9 | −8.5 | −6.0 |
| 11 | −15.2 | −14.0 | −10.0 |
| AH+···Chlorogenic Acid | A···Chlorogenic Acid | ||||
|---|---|---|---|---|---|
| Group | Geometry | Binding Energy | Group | Geometry | Binding Energy |
| A | 1 | −13.2 | A | 1 | −22.0 |
| 2 | −13.2 | 2 | −17.7 | ||
| 3 | −12.5 | 3 | −14.5 | ||
| B | 4 | −18.0 | B | 4 | −24.5 |
| 5 | −12.9 | C | 5 | −18.4 | |
| C | 6 | −12.9 | 6 | −14.6 | |
| D | 7 | −12.9 | D | 7 | −13.1 |
| Geometry | Δλ | MO | CT |
|---|---|---|---|
| 1 | 8.03 | HOMO → LUMO (0.84) | No |
| 2 | 8.60 | HOMO → LUMO (0.83) | Weak |
| 3 | 12.03 | HOMO → LUMO (0.83) | Yes |
| 4 | 4.45 | HOMO → LUMO (0.44) | Yes |
| 5 | 0.04 | HOMO → LUMO (0.63) | Yes |
| 6 | 16.79 | HOMO → LUMO (0.87) | No |
| 7 | 7.86 | HOMO → LUMO (0.85) | Yes |
| States | 1 * | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| S0→ S1 | 451.2 (0.155) | 462.1 (0.028) | 452.9 (0.072) | 454.1 (0.128) | 468.3 (0.019) |
| S0→ S2 | 435.2 (0.093) | 433.6 (0.46)9 | 449.1 (0.146) | 450.9 (0.047) | 449.3 (0.301) |
| S0→ S3 | 419.7 (0.495) | 410.7 (0.019) | 411.3 (0.358) | 420.5 (0.649) | 410.1 (0.045) |
| S0→ S4 | 405.1 (1.052) | 396.3 (0.213) | 399.1 (0.636) | 399.3 (0.747) | 393.5 (0.861) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vallverdú-Queralt, A.; Biler, M.; Meudec, E.; Guernevé, C.L.; Vernhet, A.; Mazauric, J.-P.; Legras, J.-L.; Loonis, M.; Trouillas, P.; Cheynier, V.; et al. p-Hydroxyphenyl-pyranoanthocyanins: An Experimental and Theoretical Investigation of Their Acid—Base Properties and Molecular Interactions. Int. J. Mol. Sci. 2016, 17, 1842. https://doi.org/10.3390/ijms17111842
Vallverdú-Queralt A, Biler M, Meudec E, Guernevé CL, Vernhet A, Mazauric J-P, Legras J-L, Loonis M, Trouillas P, Cheynier V, et al. p-Hydroxyphenyl-pyranoanthocyanins: An Experimental and Theoretical Investigation of Their Acid—Base Properties and Molecular Interactions. International Journal of Molecular Sciences. 2016; 17(11):1842. https://doi.org/10.3390/ijms17111842
Chicago/Turabian StyleVallverdú-Queralt, Anna, Michal Biler, Emmanuelle Meudec, Christine Le Guernevé, Aude Vernhet, Jean-Paul Mazauric, Jean-Luc Legras, Michèle Loonis, Patrick Trouillas, Véronique Cheynier, and et al. 2016. "p-Hydroxyphenyl-pyranoanthocyanins: An Experimental and Theoretical Investigation of Their Acid—Base Properties and Molecular Interactions" International Journal of Molecular Sciences 17, no. 11: 1842. https://doi.org/10.3390/ijms17111842
APA StyleVallverdú-Queralt, A., Biler, M., Meudec, E., Guernevé, C. L., Vernhet, A., Mazauric, J. -P., Legras, J. -L., Loonis, M., Trouillas, P., Cheynier, V., & Dangles, O. (2016). p-Hydroxyphenyl-pyranoanthocyanins: An Experimental and Theoretical Investigation of Their Acid—Base Properties and Molecular Interactions. International Journal of Molecular Sciences, 17(11), 1842. https://doi.org/10.3390/ijms17111842