
p53 Proteoforms and Intrinsic Disorder: An Illustration of the Protein Structure–Function Continuum Concept


Abstract
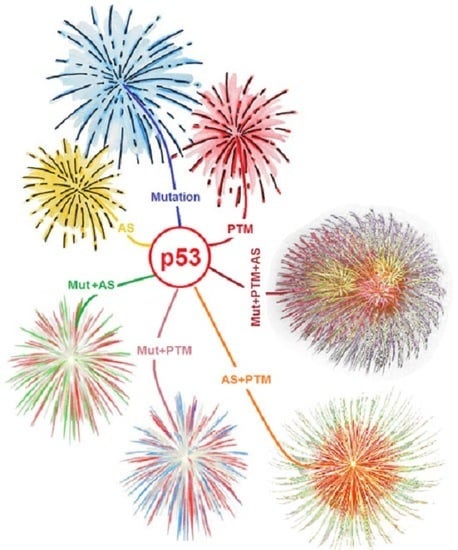
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
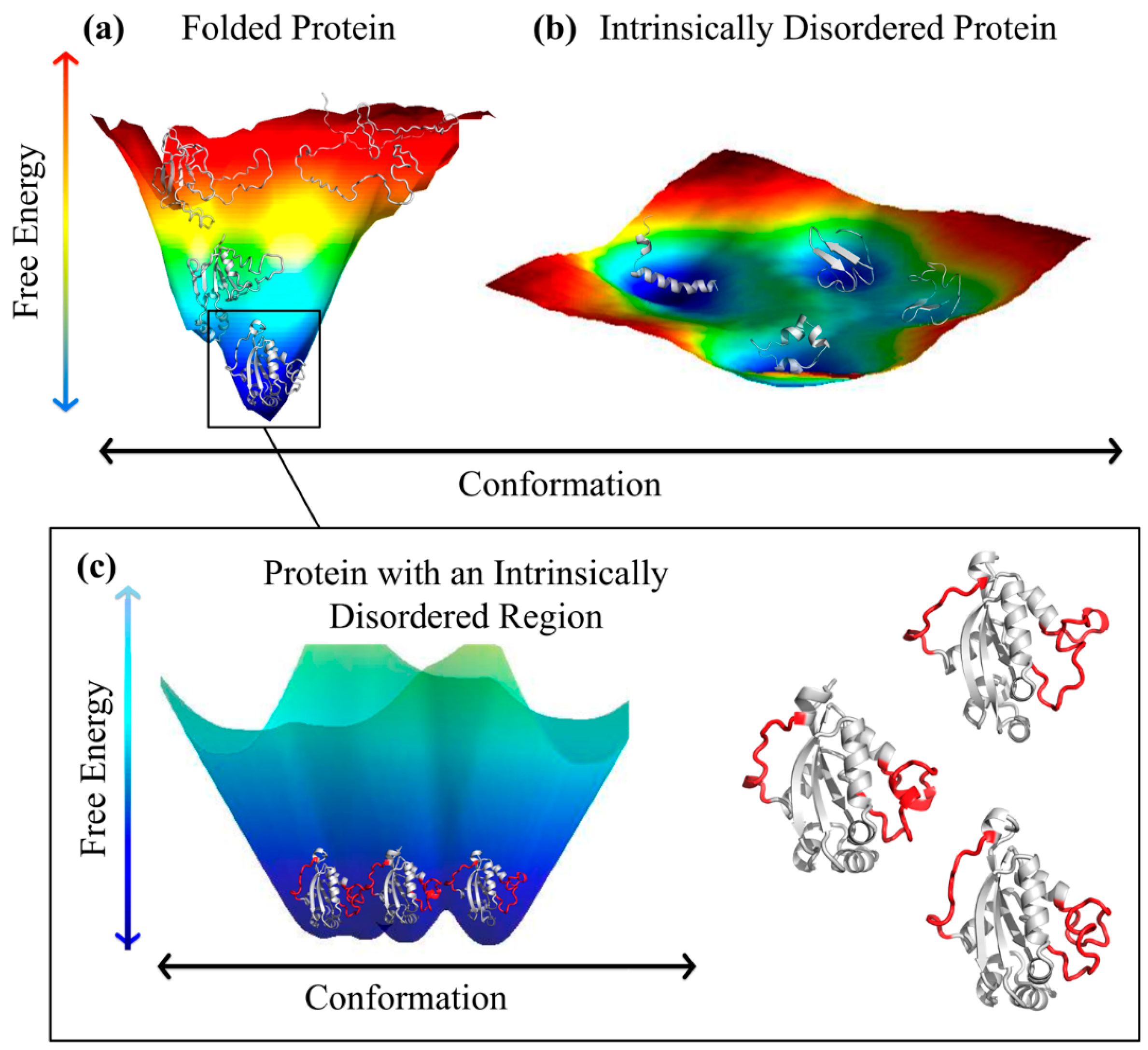
2. Intrinsically Disordered Proteins and Intrinsically Disordered Protein Regions
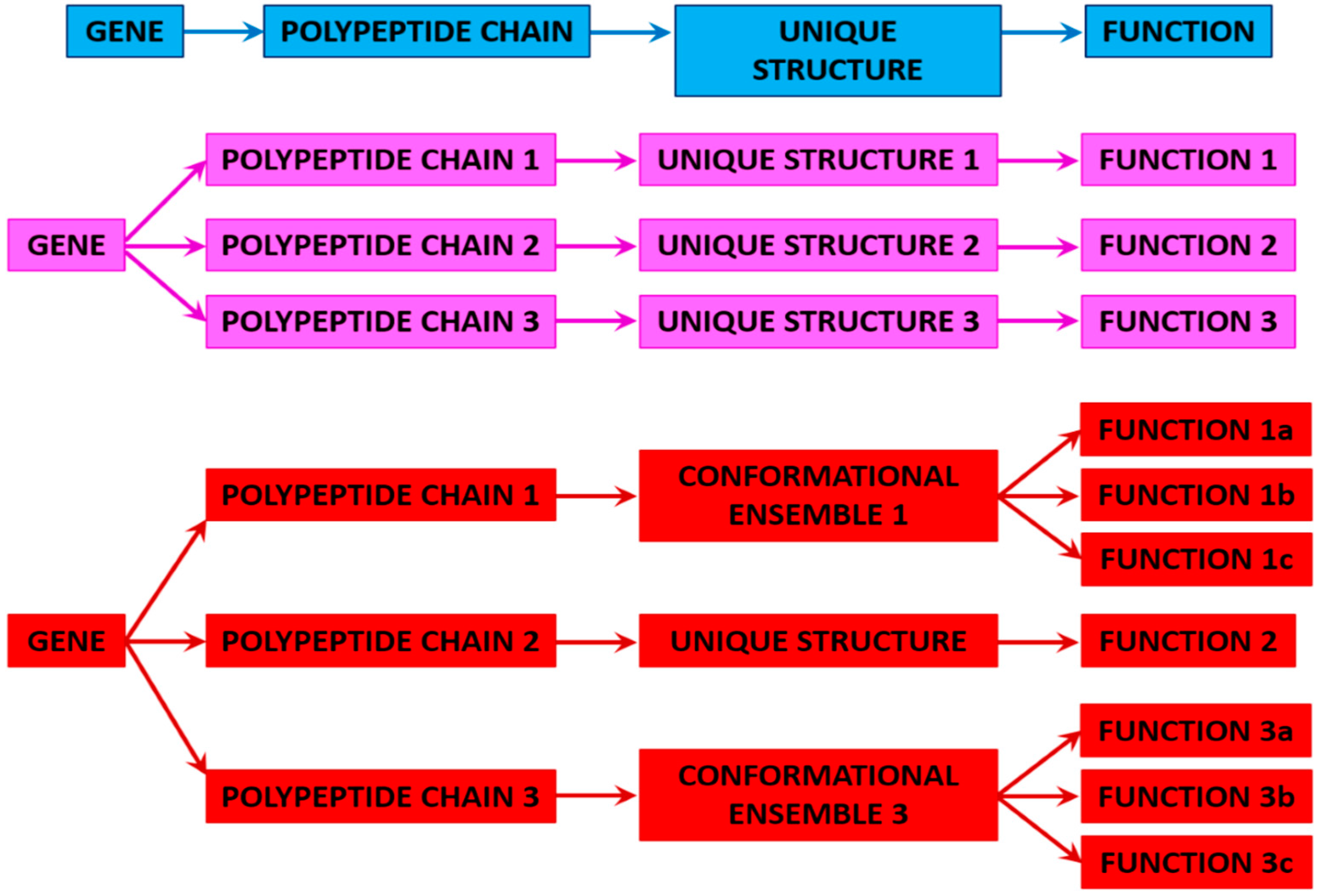
3. Proteoforms
3.1. Ordered and Intrinsically Disordered Proteoforms
3.2. Different Types of Proteoforms: A General Overview
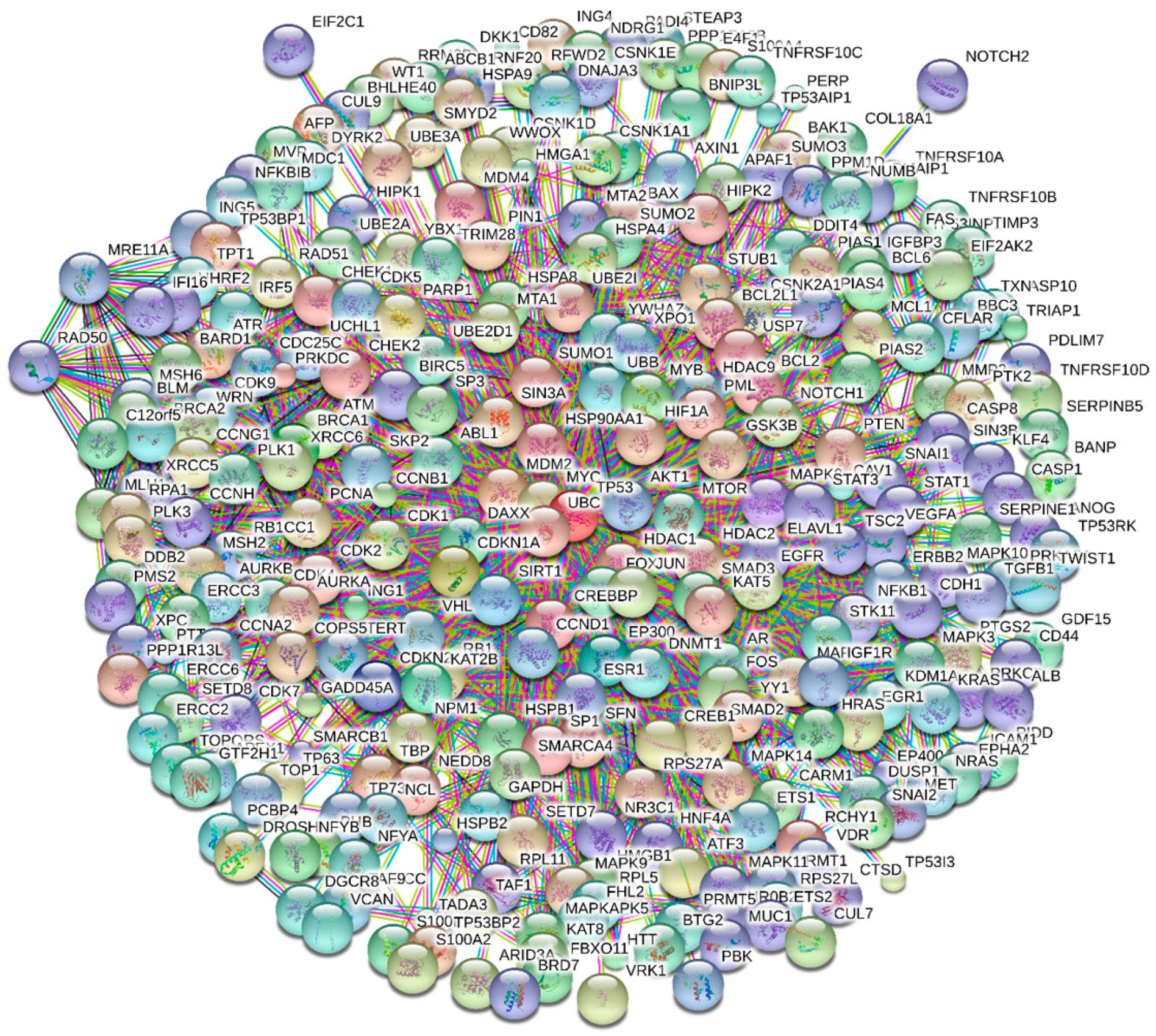
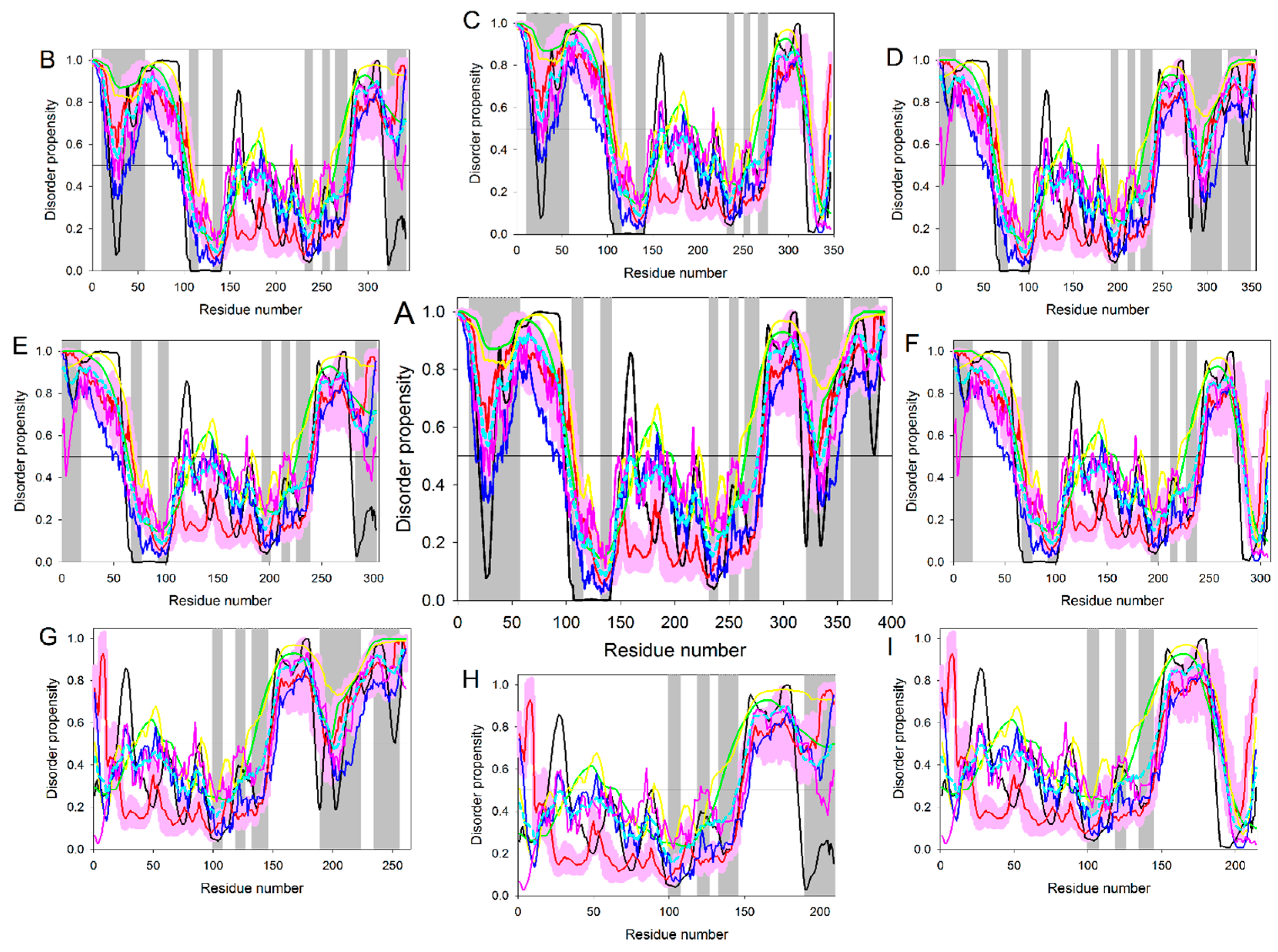
4. Multifunctionality of p53 and Intrinsic Disorder
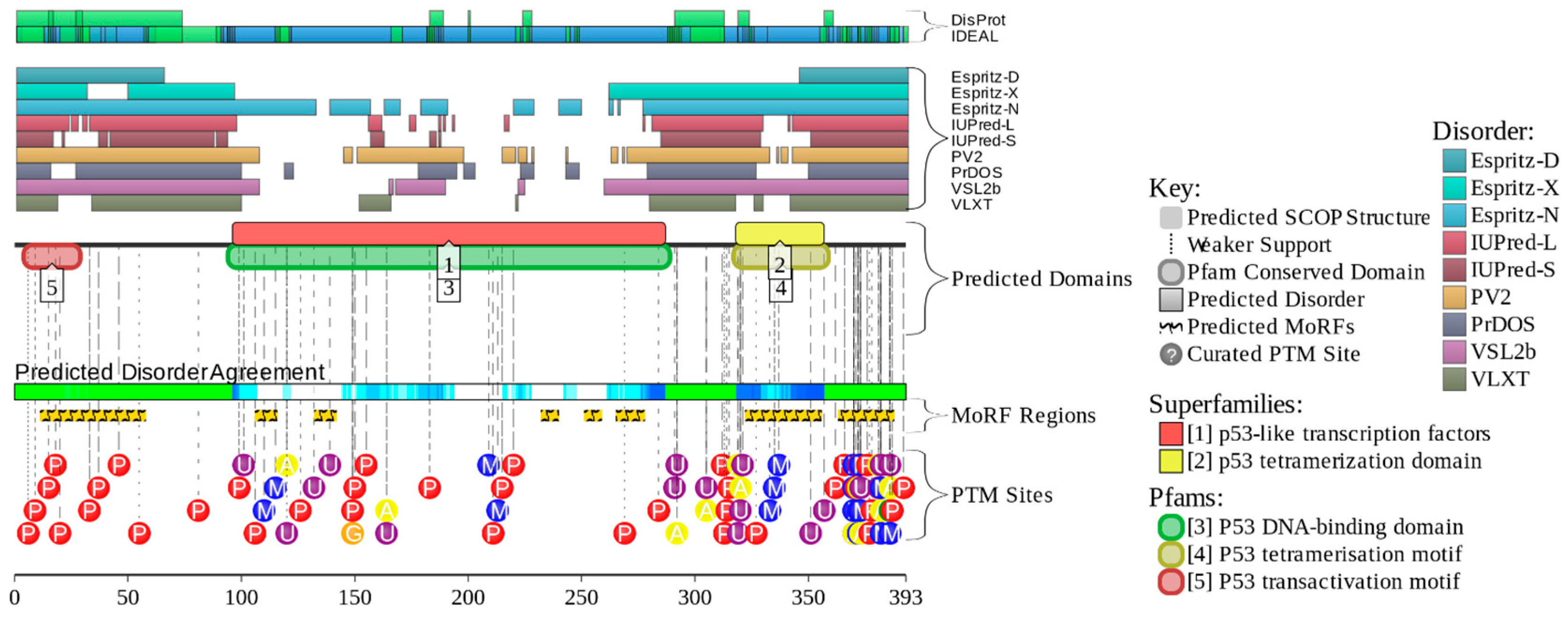
5. Conformational/Basic Proteoforms of p53
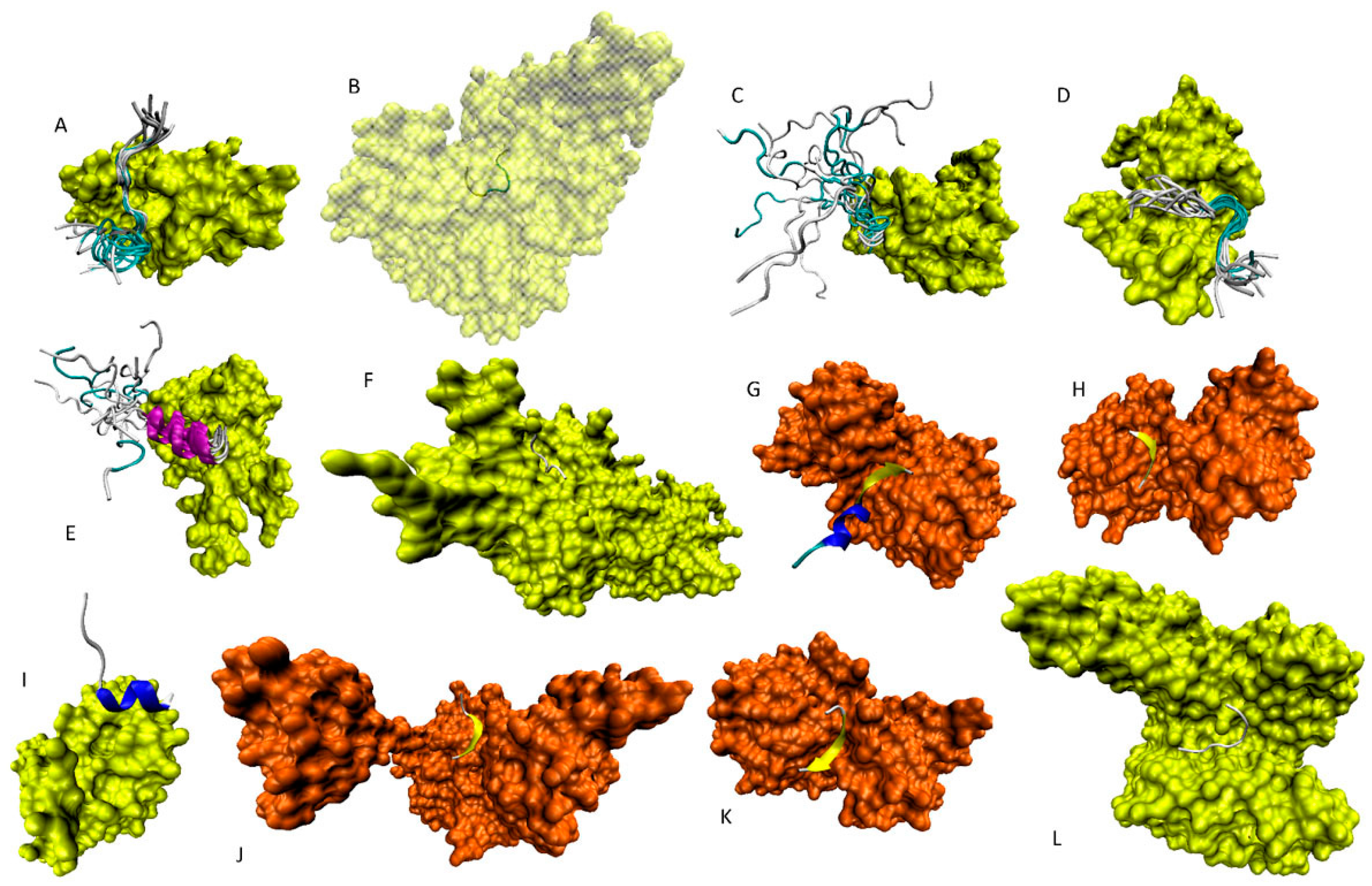
6. Functioning Proteoforms of p53
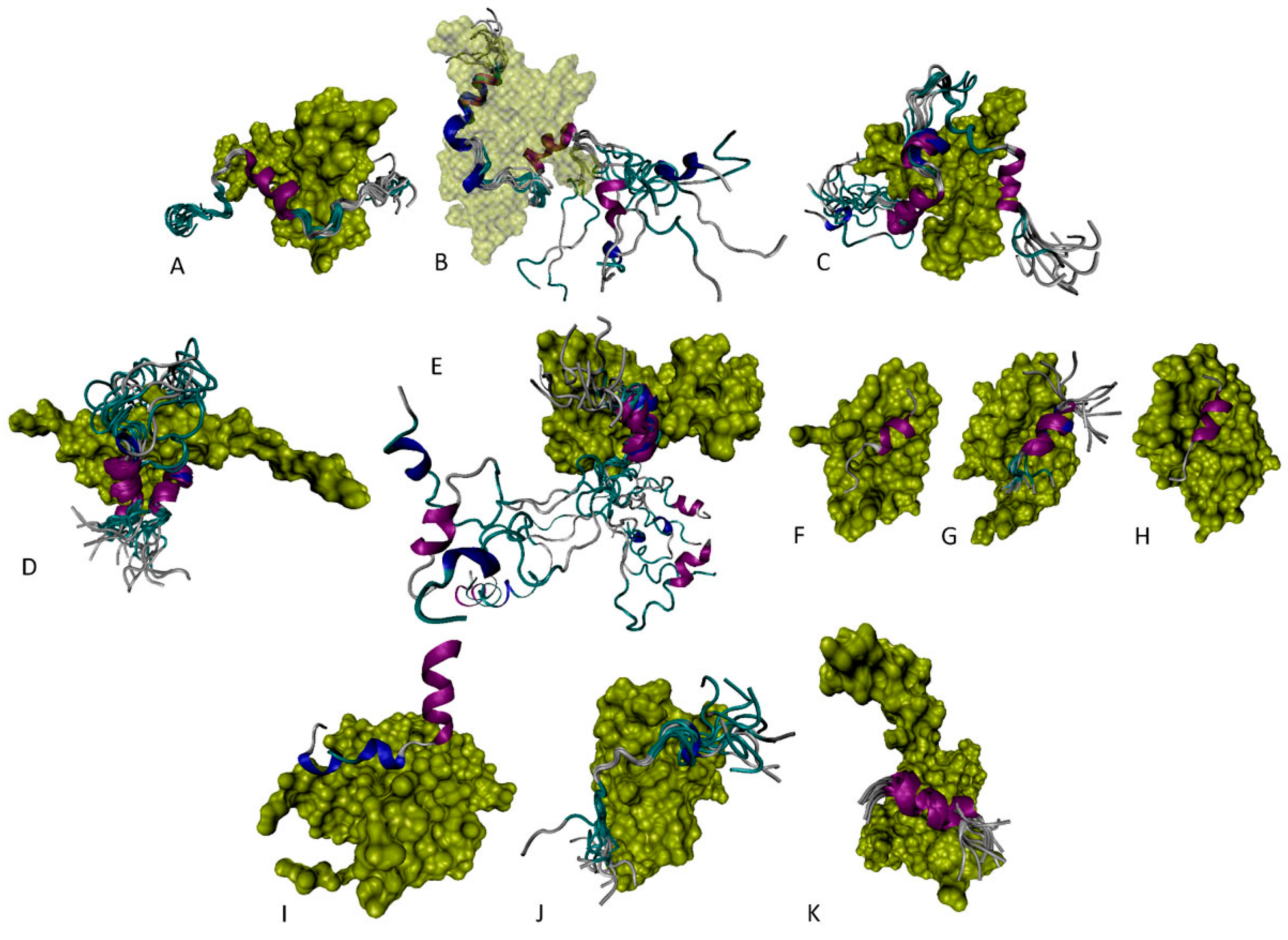
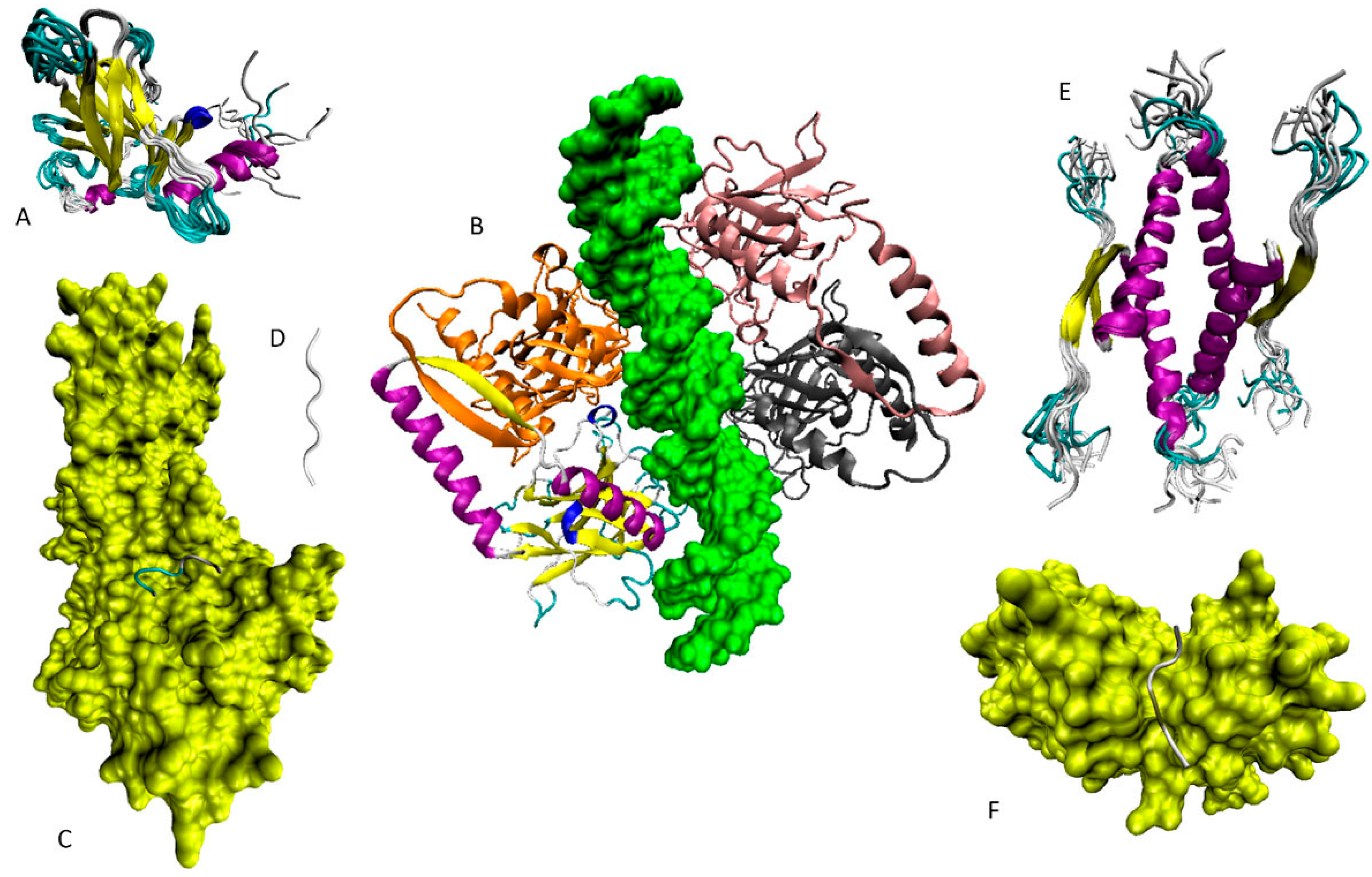
6.1. N-Terminal Proteoforms
6.2. DBD/OD Proteoforms
6.3. C-Terminal Proteoforms
6.4. Tetrameric Proteoforms
7. Inducible/Modified Proteoforms of p53
7.1. Alternative Splicing-Induced p53 Proteoforms
7.2. PTM-Induced p53 Proteoforms
7.3. Mutation-Induced p53 Proteoforms
- (1)
- Almost all residues of p53 are subject to disease-related mutations;
- (2)
- The ordered central part of this protein has more mutations than its disordered tails;
- (3)
- The heterogeneity of missense mutations is also higher for the central region of p53;
- (4)
- Both order-promoting and disorder-promoting residues can be efficiently mutated. In fact, numbers of mutations found for the order-promoting Cys176 and Tyr220 (343 and 324, respectively) are not too different from the corresponding values reported for the disorder-promoting residues Arg213 and Pro278 (327 and 268, respectively). Also, the hottest of p53 hotspots (i.e., residues that have >500 mutations each) are the disorder-promoting residues Arg248, Arg273, Arg175, Gly245, Arg282, and Arg249, with mutations reported 1544, 1425, 1092, 718, 616, and 573 times, respectively;
- (5)
- The abundance and diversity of mutations for a given residue depend mostly on its position within the p53 sequence and not so much on its physico-chemical nature;
- (6)
- Mutations tend to “like” disorder-based binding regions, but are preferentially excluded from the majority of the PTM sites.
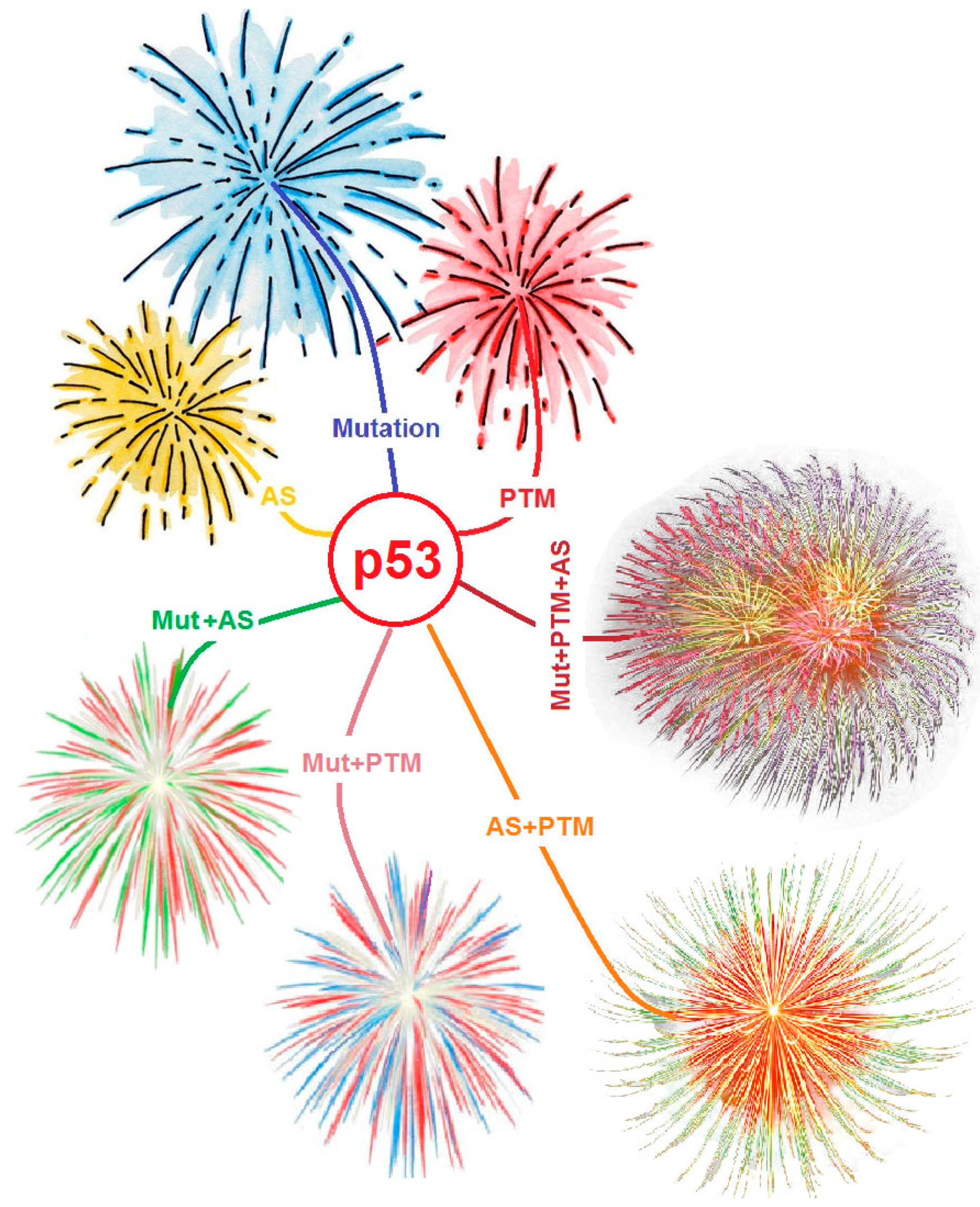
7.4. The More the Merrier: Complex p53 Proteoforms Created by Mutations, PTMs, and Alternative Splicing
8. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Fischer, E. Einfluss der configuration auf die wirkung der enzyme. Ber. Dtsch. Chem. Ges. 1894, 27, 2985–2993. [Google Scholar] [CrossRef]
- Uversky, V.N. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002, 11, 739–756. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. What does it mean to be natively unfolded? Eur. J. Biochem. 2002, 269, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef]
- Tompa, P. The functional benefits of protein disorder. J. Mol. Struct. THEOCHEM 2003, 666, 361–371. [Google Scholar] [CrossRef]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Williams, R.W.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N. Archaic chaos: Intrinsically disordered proteins in archaea. BMC Syst. Biol. 2010, 4, S1. [Google Scholar] [CrossRef] [PubMed]
- Van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing protein intrinsic disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunker, A.K.; Garner, E.; Guilliot, S.; Romero, P.; Albrecht, K.; Hart, J.; Obradovic, Z.; Kissinger, C.; Villafranca, J.E. Protein disorder and the evolution of molecular recognition: Theory, predictions and observations. Pac. Symp. Biocomput. 1998, 473–484. [Google Scholar]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Daughdrill, G.W.; Pielak, G.J.; Uversky, V.N.; Cortese, M.S.; Dunker, A.K. Natively disordered proteins. In Handbook of Protein Folding; Buchner, J., Kiefhaber, T., Eds.; Wiley-VCH, Verlag GmbH & Co., KGaA: Weinheim, Germany, 2005; pp. 271–353. [Google Scholar]
- Uversky, V.N. Unusual biophysics of intrinsically disordered proteins. Biochim. Biophys. Acta 2013, 1834, 932–951. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Dunker, A.K. The case for intrinsically disordered proteins playing contributory roles in molecular recognition without a stable 3D structure. F1000 Biol. Rep. 2013, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Yan, J.; Fan, X.; Mizianty, M.J.; Xue, B.; Wang, K.; Hu, G.; Uversky, V.N.; Kurgan, L. Exceptionally abundant exceptions: Comprehensive characterization of intrinsic disorder in all domains of life. Cell. Mol. Life Sci. 2015, 72, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Na, I.; Redmon, D.; Kopa, M.; Qin, Y.; Xue, B.; Uversky, V.N. Ordered disorder of the astrocytic dystrophin-associated protein complex in the norm and pathology. PLoS ONE 2013, 8, e73476. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Obradovic, Z.; Romero, P.; Garner, E.C.; Brown, C.J. Intrinsic protein disorder in complete genomes. Genome Inform. Ser. Workshop Genome Inform. 2000, 11, 161–171. [Google Scholar] [PubMed]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradovic, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Brown, C.J.; Obradovic, Z. Identification and functions of usefully disordered proteins. Adv. Protein Chem. 2002, 62, 25–49. [Google Scholar] [PubMed]
- Dyson, H.J.; Wright, P.E. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 2002, 12, 54–60. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Obradovic, Z. The protein trinity—Linking function and disorder. Nat. Biotechnol. 2001, 19, 805–806. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Protein folding revisited. A polypeptide chain at the folding-misfolding-nonfolding cross-roads: Which way to go? Cell. Mol. Life Sci. 2003, 60, 1852–1871. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. A decade and a half of protein intrinsic disorder: Biology still waits for physics. Protein Sci. 2013, 22, 693–724. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Vucetic, S.; Iakoucheva, L.M.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N.; Obradovic, Z. Functional anthology of intrinsic disorder. 1. Biological processes and functions of proteins with long disordered regions. J. Proteome Res. 2007, 6, 1882–1898. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Silman, I.; Uversky, V.N.; Sussman, J.L. Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 2008, 18, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Uversky, V.N. Signal transduction via unstructured protein conduits. Nat. Chem. Biol. 2008, 4, 229–230. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, C.J.; Meng, J.; Yang, J.Y.; Yang, M.Q.; Uversky, V.N.; Dunker, A.K. Flexible nets: Disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genom. 2008, 9, S1. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005, 272, 5129–5148. [Google Scholar] [CrossRef] [PubMed]
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.D.; Obradovic, Z.; Dunker, A.K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002, 323, 573–584. [Google Scholar] [CrossRef]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: Introducing the D2 concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Dave, V.; Iakoucheva, L.M.; Malaney, P.; Metallo, S.J.; Pathak, R.R.; Joerger, A.C. Pathological unfoldomics of uncontrolled chaos: Intrinsically disordered proteins and human diseases. Chem. Rev. 2014, 114, 6844–6879. [Google Scholar] [CrossRef] [PubMed]
- Beadle, G.W.; Tatum, E.L. Genetic control of biochemical reactions in neurospora. Proc. Natl. Acad. Sci. USA 1941, 27, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Bussard, A.E. A scientific revolution? The prion anomaly may challenge the central dogma of molecular biology. EMBO Rep. 2005, 6, 691–694. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Uhlen, M.; Bjorling, E.; Agaton, C.; Szigyarto, C.A.; Amini, B.; Andersen, E.; Andersson, A.C.; Angelidou, P.; Asplund, A.; Asplund, C.; et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol. Cell. Proteom. 2005, 4, 1920–1932. [Google Scholar] [CrossRef] [PubMed]
- Farrah, T.; Deutsch, E.W.; Omenn, G.S.; Sun, Z.; Watts, J.D.; Yamamoto, T.; Shteynberg, D.; Harris, M.M.; Moritz, R.L. State of the human proteome in 2013 as viewed through peptideatlas: Comparing the kidney, urine, and plasma proteomes for the biology- and disease-driven human proteome project. J. Proteome Res. 2014, 13, 60–75. [Google Scholar] [CrossRef] [PubMed]
- Farrah, T.; Deutsch, E.W.; Hoopmann, M.R.; Hallows, J.L.; Sun, Z.; Huang, C.Y.; Moritz, R.L. The state of the human proteome in 2012 as viewed through peptideatlas. J. Proteome Res. 2013, 12, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.J.; Ray, S.; Srivastava, S. The quest of the human proteome and the missing proteins: Digging deeper. OMICS J. Integr. Biol. 2015, 19, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S.; et al. A draft map of the human proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Schluter, H.; Apweiler, R.; Holzhutter, H.G.; Jungblut, P.R. Finding one’s way in proteomics: A protein species nomenclature. Chem. Cent. J. 2009, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.M.; Kelleher, N.L. Consortium for Top Down Proteomics. Proteoform: A single term describing protein complexity. Nat. Methods 2013, 10, 186–187. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. (Intrinsically disordered) Splice variants in the proteome: Implications for novel drug discovery. Genes Genom. 2016, 38, 577–594. [Google Scholar] [CrossRef]
- Iakoucheva, L.M.; Radivojac, P.; Brown, C.J.; O’Connor, T.R.; Sikes, J.G.; Obradovic, Z.; Dunker, A.K. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004, 32, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Pejaver, V.; Hsu, W.L.; Xin, F.; Dunker, A.K.; Uversky, V.N.; Radivojac, P. The structural and functional signatures of proteins that undergo multiple events of post-translational modification. Protein Sci. 2014, 23, 1077–1093. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.R.; Zaidi, S.; Fang, Y.Y.; Uversky, V.N.; Radivojac, P.; Oldfield, C.J.; Cortese, M.S.; Sickmeier, M.; LeGall, T.; Obradovic, Z.; et al. Alternative splicing in concert with protein intrinsic disorder enables increased functional diversity in multicellular organisms. Proc. Natl. Acad. Sci. USA 2006, 103, 8390–8395. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, C.J.; Cheng, Y.; Cortese, M.S.; Romero, P.; Uversky, V.N.; Dunker, A.K. Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry 2005, 44, 12454–12470. [Google Scholar] [CrossRef] [PubMed]
- Radivojac, P.; Iakoucheva, L.M.; Oldfield, C.J.; Obradovic, Z.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder and functional proteomics. Biophys. J. 2007, 92, 1439–1456. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim. Biophys. Acta 2010, 1804, 1231–1264. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Multitude of binding modes attainable by intrinsically disordered proteins: A portrait gallery of disorder-based complexes. Chem. Soc. Rev. 2011, 40, 1623–1634. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Disordered competitive recruiter: Fast and foldable. J. Mol. Biol. 2012, 418, 267–268. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.; Oldfield, C.J.; Radivojac, P.; Vacic, V.; Cortese, M.S.; Dunker, A.K.; Uversky, V.N. Analysis of molecular recognition features (MoRFS). J. Mol. Biol. 2006, 362, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- Vacic, V.; Oldfield, C.J.; Mohan, A.; Radivojac, P.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Characterization of molecular recognition features, morfs, and their binding partners. J. Proteome Res. 2007, 6, 2351–2366. [Google Scholar] [CrossRef] [PubMed]
- Koshland, D.E. Application of a theory of enzyme specificity to protein synthesis. Proc. Natl. Acad. Sci. USA 1958, 44, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Villa, J.; Strajbl, M.; Glennon, T.M.; Sham, Y.Y.; Chu, Z.T.; Warshel, A. How important are entropic contributions to enzyme catalysis? Proc. Natl. Acad. Sci. USA 2000, 97, 11899–11904. [Google Scholar] [CrossRef] [PubMed]
- Eisenmesser, E.Z.; Bosco, D.A.; Akke, M.; Kern, D. Enzyme dynamics during catalysis. Science 2002, 295, 1520–1523. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, M.J.; Scrutton, N.S. A new conceptual framework for enzyme catalysis. Hydrogen tunnelling coupled to enzyme dynamics in flavoprotein and quinoprotein enzymes. Eur. J. Biochem. 2002, 269, 3096–3102. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, P.T.; Benkovic, S.J. Preorganization and protein dynamics in enzyme catalysis. Chem. Rec. 2002, 2, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.K.; Billeter, S.R.; Rajagopalan, P.T.; Benkovic, S.J.; Hammes-Schiffer, S. Network of coupled promoting motions in enzyme catalysis. Proc. Natl. Acad. Sci. USA 2002, 99, 2794–2799. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.K.; Geist, A.; Gorin, A. Protein dynamics and enzymatic catalysis: Investigating the peptidyl-prolyl cis-trans isomerization activity of cyclophilin A. Biochemistry 2004, 43, 10605–10618. [Google Scholar] [CrossRef] [PubMed]
- Tousignant, A.; Pelletier, J.N. Protein motions promote catalysis. Chem. Biol. 2004, 11, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.K. Role of protein dynamics in reaction rate enhancement by enzymes. J. Am. Chem. Soc. 2005, 127, 15248–15256. [Google Scholar] [CrossRef] [PubMed]
- Eisenmesser, E.Z.; Millet, O.; Labeikovsky, W.; Korzhnev, D.M.; Wolf-Watz, M.; Bosco, D.A.; Skalicky, J.J.; Kay, L.E.; Kern, D. Intrinsic dynamics of an enzyme underlies catalysis. Nature 2005, 438, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.W.; Bahar, I. Coupling between catalytic site and collective dynamics: A requirement for mechanochemical activity of enzymes. Structure 2005, 13, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.H.; Parson, W.W.; Warshel, A. Dynamical contributions to enzyme catalysis: Critical tests of a popular hypothesis. Chem. Rev. 2006, 106, 1737–1756. [Google Scholar] [CrossRef] [PubMed]
- Frauenfelder, H.; Chen, G.; Berendzen, J.; Fenimore, P.W.; Jansson, H.; McMahon, B.H.; Stroe, I.R.; Swenson, J.; Young, R.D. A unified model of protein dynamics. Proc. Natl. Acad. Sci. USA 2009, 106, 5129–5134. [Google Scholar] [CrossRef] [PubMed]
- Austin, R.H.; Beeson, K.W.; Eisenstein, L.; Frauenfelder, H.; Gunsalus, I.C. Dynamics of ligand binding to myoglobin. Biochemistry 1975, 14, 5355–5373. [Google Scholar] [CrossRef] [PubMed]
- Frauenfelder, H.; Petsko, G.A.; Tsernoglou, D. Temperature-dependent X-ray diffraction as a probe of protein structural dynamics. Nature 1979, 280, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Artymiuk, P.J.; Blake, C.C.; Grace, D.E.; Oatley, S.J.; Phillips, D.C.; Sternberg, M.J. Crystallographic studies of the dynamic properties of lysozyme. Nature 1979, 280, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Frauenfelder, H.; Petsko, G.A. Structural dynamics of liganded myoglobin. Biophys. J. 1980, 32, 465–483. [Google Scholar] [CrossRef]
- Beece, D.; Eisenstein, L.; Frauenfelder, H.; Good, D.; Marden, M.C.; Reinisch, L.; Reynolds, A.H.; Sorensen, L.B.; Yue, K.T. Solvent viscosity and protein dynamics. Biochemistry 1980, 19, 5147–5157. [Google Scholar] [CrossRef] [PubMed]
- Parak, F.; Frolov, E.N.; Mossbauer, R.L.; Goldanskii, V.I. Dynamics of metmyoglobin crystals investigated by nuclear gamma resonance absorption. J. Mol. Biol. 1981, 145, 825–833. [Google Scholar] [CrossRef]
- Hartmann, H.; Parak, F.; Steigemann, W.; Petsko, G.A.; Ponzi, D.R.; Frauenfelder, H. Conformational substates in a protein: Structure and dynamics of metmyoglobin at 80 K. Proc. Natl. Acad. Sci. USA 1982, 79, 4967–4971. [Google Scholar] [CrossRef] [PubMed]
- Burger, V.M.; Gurry, T.; Stultz, C.M. Intrinsically disordered proteins: Where computation meets experiment. Polymers 2014, 6, 2684–2719. [Google Scholar] [CrossRef]
- Leopold, P.E.; Montal, M.; Onuchic, J.N. Protein folding funnels: A kinetic approach to the sequence-structure relationship. Proc. Natl. Acad. Sci. USA 1992, 89, 8721–8725. [Google Scholar] [CrossRef] [PubMed]
- Onuchic, J.N.; Wolynes, P.G. Theory of protein folding. Curr. Opin. Struct. Biol. 2004, 14, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Socci, N.D.; Onuchic, J.N.; Wolynes, P.G. Protein folding mechanisms and the multidimensional folding funnel. Proteins 1998, 32, 136–158. [Google Scholar] [CrossRef]
- Onuchic, J.N.; Luthey-Schulten, Z.; Wolynes, P.G. Theory of protein folding: The energy landscape perspective. Annu. Rev. Phys. Chem. 1997, 48, 545–600. [Google Scholar] [CrossRef] [PubMed]
- Onuchic, J.N.; Socci, N.D.; Luthey-Schulten, Z.; Wolynes, P.G. Protein folding funnels: The nature of the transition state ensemble. Fold Des. 1996, 1, 441–450. [Google Scholar] [CrossRef]
- Turoverov, K.K.; Kuznetsova, I.M.; Uversky, V.N. The protein kingdom extended: Ordered and intrinsically disordered proteins, their folding, supramolecular complex formation, and aggregation. Prog. Biophys. Mol. Biol. 2010, 102, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. Unstructural biology coming of age. Curr. Opin. Struct. Biol. 2011, 21, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, S.B.; Trach, S.O. Estimation of macromolecule concentrations and excluded volume effects for the cytoplasm of Escherichia coli. J. Mol. Biol. 1991, 222, 599–620. [Google Scholar] [CrossRef]
- Van den Berg, B.; Ellis, R.J.; Dobson, C.M. Effects of macromolecular crowding on protein folding and aggregation. EMBO J. 1999, 18, 6927–6933. [Google Scholar] [CrossRef] [PubMed]
- Rivas, G.; Ferrone, F.; Herzfeld, J. Life in a crowded world. EMBO Rep. 2004, 5, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.J.; Minton, A.P. Cell biology: Join the crowd. Nature 2003, 425, 27–28. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, S.B.; Minton, A.P. Macromolecular crowding: Biochemical, biophysical, and physiological consequences. Annu. Rev. Biophys. Biomol. Struct. 1993, 22, 27–65. [Google Scholar] [CrossRef] [PubMed]
- Fulton, A.B. How crowded is the cytoplasm? Cell 1982, 30, 345–347. [Google Scholar] [CrossRef]
- Minton, A.P. Influence of excluded volume upon macromolecular structure and associations in ‘crowded’ media. Curr. Opin. Biotechnol. 1997, 8, 65–69. [Google Scholar] [CrossRef]
- Ellis, R.J. Macromolecular crowding: Obvious but underappreciated. Trends Biochem. Sci. 2001, 26, 597–604. [Google Scholar] [CrossRef]
- Minton, A.P. Protein folding: Thickening the broth. Curr. Biol. 2000, 10, R97–R99. [Google Scholar] [CrossRef]
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Miyashita, T.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293–299. [Google Scholar] [PubMed]
- Soussi, T. The p53 tumor suppressor gene: From molecular biology to clinical investigation. Ann. N. Y. Acad. Sci. 2000, 910, 121–137. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the light: The growing complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, J.C. p53 family isoforms. Curr. Pharm. Biotechnol. 2007, 8, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.W.; Appella, E. Signaling to the p53 tumor suppressor through pathways activated by genotoxic and nongenotoxic stress. In Handbook of Cell Signaling; Bradshaw, R.A., Dennis, E.A., Eds.; Academic Press: New York, NY, USA, 2004; pp. 237–247. [Google Scholar]
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; et al. The string database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011, 39, D561–D568. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Oliner, J.D.; Pietenpol, J.A.; Thiagalingam, S.; Gyuris, J.; Kinzler, K.W.; Vogelstein, B. Oncoprotein Mdm2 conceals the activation domain of tumour suppressor p53. Nature 1993, 362, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Chene, P. Inhibition of the p53-Mdm2 interaction: Targeting a protein-protein interface. Mol. Cancer Res. 2004, 2, 20–28. [Google Scholar] [PubMed]
- Momand, J.; Zambetti, G.P.; Olson, D.C.; George, D.; Levine, A.J. The Mdm2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992, 69, 1237–1245. [Google Scholar] [CrossRef]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein Mdm2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef]
- Roth, J.; Dobbelstein, M.; Freedman, D.A.; Shenk, T.; Levine, A.J. Nucleo-cytoplasmic shuttling of the HDM2 oncoprotein regulates the levels of the p53 protein via a pathway used by the human immunodeficiency virus REV protein. EMBO J. 1998, 17, 554–564. [Google Scholar] [CrossRef] [PubMed]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the Mdm2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Mok, K.H.; Muhandiram, R.; Park, K.H.; Suk, J.E.; Kim, D.H.; Chang, J.; Sung, Y.C.; Choi, K.Y.; Han, K.H. Local structural elements in the mostly unstructured transcriptional activation domain of human p53. J. Biol. Chem. 2000, 275, 29426–29432. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. Structural biology of the tumor suppressor p53. Annu. Rev. Biochem. 2008, 77, 557–582. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Oldfield, C.J.; Midic, U.; Xie, H.; Xue, B.; Vucetic, S.; Iakoucheva, L.M.; Obradovic, Z.; Dunker, A.K. Unfoldomics of human diseases: Linking protein intrinsic disorder with diseases. BMC Genom. 2009, 10, S7. [Google Scholar] [CrossRef] [PubMed]
- Soussi, T.; Beroud, C. Assessing TP53 status in human tumours to evaluate clinical outcome. Nat. Rev. Cancer 2001, 1, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Oates, M.E.; Romero, P.; Ishida, T.; Ghalwash, M.; Mizianty, M.J.; Xue, B.; Dosztanyi, Z.; Uversky, V.N.; Obradovic, Z.; Kurgan, L.; et al. D(2)P(2): Database of disordered protein predictions. Nucleic Acids Res. 2013, 41, D508–D516. [Google Scholar] [CrossRef] [PubMed]
- Dosztanyi, Z.; Csizmok, V.; Tompa, P.; Simon, I. Iupred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Ishida, T.; Kinoshita, K. Prdos: Prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 2007, 35, W460–W464. [Google Scholar] [CrossRef] [PubMed]
- Obradovic, Z.; Peng, K.; Vucetic, S.; Radivojac, P.; Dunker, A.K. Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins 2005, 61, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Radivojac, P.; Vucetic, S.; Dunker, A.K.; Obradovic, Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinform. 2006, 7, 208. [Google Scholar] [CrossRef] [PubMed]
- Walsh, I.; Martin, A.J.; di Domenico, T.; Tosatto, S.C. Espritz: Accurate and fast prediction of protein disorder. Bioinformatics 2012, 28, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Tidow, H.; Melero, R.; Mylonas, E.; Freund, S.M.; Grossmann, J.G.; Carazo, J.M.; Svergun, D.I.; Valle, M.; Fersht, A.R. Quaternary structures of tumor suppressor p53 and a specific p53 DNA complex. Proc. Natl. Acad. Sci. USA 2007, 104, 12324–12329. [Google Scholar] [CrossRef] [PubMed]
- Wells, M.; Tidow, H.; Rutherford, T.J.; Markwick, P.; Jensen, M.R.; Mylonas, E.; Svergun, D.I.; Blackledge, M.; Fersht, A.R. Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proc. Natl. Acad. Sci. USA 2008, 105, 5762–5767. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Jenkins, L.M.; Durell, S.R.; Hayashi, R.; Mazur, S.J.; Cherry, S.; Tropea, J.E.; Miller, M.; Wlodawer, A.; Appella, E.; et al. Structural basis for p300 TAZ2-p53 TAD1 binding and modulation by phosphorylation. Structure 2009, 17, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Krois, A.S.; Ferreon, J.C.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Recognition of the disordered p53 transactivation domain by the transcriptional adapter Zinc finger domains of CREB-binding protein. Proc. Natl. Acad. Sci. USA 2016, 113, E1853–E1862. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Structure of the p53 transactivation domain in complex with the nuclear receptor coactivator binding domain of CREB binding protein. Biochemistry 2010, 49, 9964–9971. [Google Scholar] [CrossRef] [PubMed]
- Rowell, J.P.; Simpson, K.L.; Stott, K.; Watson, M.; Thomas, J.O. HMGB1-facilitated p53 DNA binding occurs via HMG-BOX/p53 transactivation domain interaction, regulated by the acidic tail. Structure 2012, 20, 2014–2024. [Google Scholar] [CrossRef] [PubMed]
- Grace, C.R.; Ban, D.; Min, J.; Mayasundari, A.; Min, L.; Finch, K.E.; Griffiths, L.; Bharatham, N.; Bashford, D.; Kiplin Guy, R.; et al. Monitoring ligand-induced protein ordering in drug discovery. J. Mol. Biol. 2016, 428, 1290–1303. [Google Scholar] [CrossRef] [PubMed]
- Popowicz, G.M.; Czarna, A.; Holak, T.A. Structure of the human mdmx protein bound to the p53 tumor suppressor transactivation domain. Cell Cycle 2008, 7, 2441–2443. [Google Scholar] [CrossRef] [PubMed]
- Bochkareva, E.; Kaustov, L.; Ayed, A.; Yi, G.S.; Lu, Y.; Pineda-Lucena, A.; Liao, J.C.; Okorokov, A.L.; Milner, J.; Arrowsmith, C.H.; et al. Single-stranded DNA mimicry in the p53 transactivation domain interaction with replication protein A. Proc. Natl. Acad. Sci. USA 2005, 102, 15412–15417. [Google Scholar] [CrossRef] [PubMed]
- Okuda, M.; Nishimura, Y. Extended string binding mode of the phosphorylated transactivation domain of tumor suppressor p53. J. Am. Chem. Soc. 2014, 136, 14143–14152. [Google Scholar] [CrossRef] [PubMed]
- Di Lello, P.; Jenkins, L.M.; Jones, T.N.; Nguyen, B.D.; Hara, T.; Yamaguchi, H.; Dikeakos, J.D.; Appella, E.; Legault, P.; Omichinski, J.G. Structure of the TFB1/p53 complex: Insights into the interaction between the p62/TFB1 subunit of tfiih and the activation domain of p53. Mol. Cell 2006, 22, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Canadillas, J.M.; Tidow, H.; Freund, S.M.; Rutherford, T.J.; Ang, H.C.; Fersht, A.R. Solution structure of p53 core domain: Structural basis for its instability. Proc. Natl. Acad. Sci. USA 2006, 103, 2109–2114. [Google Scholar] [CrossRef] [PubMed]
- Emamzadah, S.; Tropia, L.; Vincenti, I.; Falquet, B.; Halazonetis, T.D. Reversal of the DNA-binding-induced loop L1 conformational switch in an engineered human p53 protein. J. Mol. Biol. 2014, 426, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.J.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal-structure of a p53 tumor-suppressor DNA complex—Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Joo, W.S.; Jeffrey, P.D.; Cantor, S.B.; Finnin, M.S.; Livingston, D.M.; Pavletich, N.P. Structure of the 53BP1 BRCT region bound to p53 and its comparison to the BRCAL1 BRCT structure. Genes Dev. 2002, 16, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Gorina, S.; Pavletich, N.P. Structure of the p53 tumor suppressor bound to the ankyrin and SH3 domains of 53BP2. Science 1996, 274, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Lilyestrom, W.; Klein, M.G.; Zhang, R.G.; Joachimiak, A.; Chen, X.J.S. Crystal structure of SV40 large T-antigen bound to p53: Interplay between a viral oncoprotein and a cellular tumor suppressor. Genes Dev. 2006, 20, 2373–2382. [Google Scholar] [CrossRef] [PubMed]
- Schimpl, M.; Borodkin, V.S.; Gray, L.J.; van Aalten, D.M. Synergy of peptide and sugar in O-glcnacase substrate recognition. Chem. Biol. 2012, 19, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Soragni, A.; Janzen, D.M.; Johnson, L.M.; Lindgren, A.G.; Thai-Quynh Nguyen, A.; Tiourin, E.; Soriaga, A.B.; Lu, J.; Jiang, L.; Faull, K.F.; et al. A designed inhibitor of p53 aggregation rescues p53 tumor suppression in ovarian carcinomas. Cancer Cell 2016, 29, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Clore, G.M.; Omichinski, J.G.; Sakaguchi, K.; Zambrano, N.; Sakamoto, H.; Appella, E.; Gronenborn, A.M. High-resolution structure of the oligomerization domain of p53 by multidimensional NMR. Science 1994, 265, 386–391. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.; Saridakis, V.; Sarkari, F.; Duan, S.; Wu, T.; Arrowsmith, C.H.; Frappier, L. Molecular recognition of p53 and Mdm2 by USP7/HAUSP. Nat. Struct. Mol. Biol. 2006, 13, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Tong, Q.; Cui, G.; Botuyan, M.V.; Rothbart, S.B.; Hayashi, R.; Musselman, C.A.; Singh, N.; Appella, E.; Strahl, B.D.; Mer, G.; et al. Structural plasticity of methyllysine recognition by the tandem Tudor domain of 53BP1. Structure 2015, 23, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, L.; Zhang, H.; Luo, X.; Dai, J.; Zhou, S.; Gu, J.; Zhu, J.; Atadja, P.; Lu, C.; et al. Structure of human SMYD2 protein reveals the basis of p53 tumor suppressor methylation. J. Biol. Chem. 2011, 286, 38725–38737. [Google Scholar] [CrossRef] [PubMed]
- Mujtaba, S.; He, Y.; Zeng, L.; Yan, S.; Plotnikova, O.; Sachchidanand; Sanchez, R.; Zeleznik-Le, N.J.; Ronai, Z.; Zhou, M.M. Structural mechanism of the bromodomain of the coactivator CBP in p53 transcriptional activation. Mol. Cell 2004, 13, 251–263. [Google Scholar] [CrossRef]
- Chuikov, S.; Kurash, J.K.; Wilson, J.R.; Xiao, B.; Justin, N.; Ivanov, G.S.; McKinney, K.; Tempst, P.; Prives, C.; Gamblin, S.J.; et al. Regulation of p53 activity through lysine methylation. Nature 2004, 432, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Rustandi, R.R.; Baldisseri, D.M.; Weber, D.J. Structure of the negative regulatory domain of p53 bound to S100B (ββ). Nat. Struct. Biol. 2000, 7, 570–574. [Google Scholar] [PubMed]
- Avalos, J.L.; Bever, K.M.; Wolberger, C. Mechanism of sirtuin inhibition by nicotinamide: Altering the NAD(+) cosubstrate specificity of a SIR2 enzyme. Mol. Cell 2005, 17, 855–868. [Google Scholar] [CrossRef] [PubMed]
- Tong, Q.; Mazur, S.J.; Rincon-Arano, H.; Rothbart, S.B.; Kuznetsov, D.M.; Cui, G.; Liu, W.H.; Gete, Y.; Klein, B.J.; Jenkins, L.; et al. An acetyl-methyl switch drives a conformational change in p53. Structure 2015, 23, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.; Fischer, F.; Nguyen, G.T.; Lakshminarasimhan, M.; Schutkowski, M.; Weyand, M.; Steegborn, C. EX-527 inhibits sirtuins by exploiting their unique NAD+-dependent deacetylation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, E2772–E2781. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Case, A.W.; Riera, T.V.; Considine, T.; Lee, J.E.; Hamuro, Y.; Zhao, H.; Jiang, Y.; Sweitzer, S.M.; Pietrak, B.; et al. Crystallographic structure of a small molecule SIRT1 activator-enzyme complex. Nat. Commun. 2015, 6, 7645. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, B.; Mondry, J.; Thiel, P.; Weyand, M.; Ottmann, C. Structure of the p53 C-terminus bound to 14-3-3: Implications for stabilization of the p53 tetramer. FEBS Lett. 2010, 584, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Balagurumoorthy, P.; Sakamoto, H.; Lewis, M.S.; Zambrano, N.; Clore, G.M.; Gronenborn, A.M.; Appella, E.; Harrington, R.E. Four p53 DNA-binding domain peptides bind natural p53-response elements and bend the DNA. Proc. Natl. Acad. Sci. USA 1995, 92, 8591–8595. [Google Scholar] [CrossRef] [PubMed]
- Kamada, R.; Toguchi, Y.; Nomura, T.; Imagawa, T.; Sakaguchi, K. Tetramer formation of tumor suppressor protein p53: Structure, function, and applications. Biopolymers 2016, 106, 598–612. [Google Scholar] [CrossRef] [PubMed]
- Pietsch, E.C.; Leu, J.I.; Frank, A.; Dumont, P.; George, D.L.; Murphy, M.E. The tetramerization domain of p53 is required for efficient bak oligomerization. Cancer Biol. Ther. 2007, 6, 1576–1583. [Google Scholar] [CrossRef] [PubMed]
- Van Dieck, J.; Fernandez-Fernandez, M.R.; Veprintsev, D.B.; Fersht, A.R. Modulation of the oligomerization state of p53 by differential binding of proteins of the S100 family to p53 monomers and tetramers. J. Biol. Chem. 2009, 284, 13804–13811. [Google Scholar] [CrossRef] [PubMed]
- Chene, P. The role of tetramerization in p53 function. Oncogene 2001, 20, 2611–2617. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Li, H.H.; Bula, C.M.; Liu, X. Stimulation of p53 DNA binding by C-ABL requires the p53 C terminus and tetramerization. Mol. Cell. Biol. 2000, 20, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Delphin, C.; Ronjat, M.; Deloulme, J.C.; Garin, G.; Debussche, L.; Higashimoto, Y.; Sakaguchi, K.; Baudier, J. Calcium-dependent interaction of S100B with the C-terminal domain of the tumor suppressor p53. J. Biol. Chem. 1999, 274, 10539–10544. [Google Scholar] [CrossRef] [PubMed]
- Stommel, J.M.; Marchenko, N.D.; Jimenez, G.S.; Moll, U.M.; Hope, T.J.; Wahl, G.M. A leucine-rich nuclear export signal in the p53 tetramerization domain: Regulation of subcellular localization and p53 activity by NES masking. EMBO J. 1999, 18, 1660–1672. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; An, S.S. Role of p53 isoforms and aggregations in cancer. Medicine 2016, 95, e3993. [Google Scholar] [CrossRef] [PubMed]
- Okumura, N.; Yoshida, H.; Kitagishi, Y.; Nishimura, Y.; Matsuda, S. Alternative splicings on p53, BRCA1 and PTEN genes involved in breast cancer. Biochem. Biophys. Res. Commun. 2011, 413, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, J.C.; Fernandes, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005, 19, 2122–2137. [Google Scholar] [CrossRef] [PubMed]
- Marcel, V.; Petit, I.; Murray-Zmijewski, F.; Goullet de Rugy, T.; Fernandes, K.; Meuray, V.; Diot, A.; Lane, D.P.; Aberdam, D.; Bourdon, J.C. Diverse p63 and p73 isoforms regulate Δ133p53 expression through modulation of the internal TP53 promoter activity. Cell Death Differ. 2012, 19, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Bernard, H.; Garmy-Susini, B.; Ainaoui, N.; van den Berghe, L.; Peurichard, A.; Javerzat, S.; Bikfalvi, A.; Lane, D.P.; Bourdon, J.C.; Prats, A.C. The p53 isoform, delta133p53alpha, stimulates angiogenesis and tumour progression. Oncogene 2013, 32, 2150–2160. [Google Scholar] [CrossRef] [PubMed]
- Olivares-Illana, V.; Fahraeus, R. p53 isoforms gain functions. Oncogene 2010, 29, 5113–5119. [Google Scholar] [CrossRef] [PubMed]
- Surget, S.; Khoury, M.P.; Bourdon, J.C. Uncovering the role of p53 splice variants in human malignancy: A clinical perspective. Onco Targets Ther. 2013, 7, 57–68. [Google Scholar] [PubMed]
- Yin, Y.; Stephen, C.W.; Luciani, M.G.; Fahraeus, R. P53 stability and activity is regulated by Mdm2-mediated induction of alternative p53 translation products. Nat. Cell Biol. 2002, 4, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.J.; Hrstka, R.; Candeias, M.; Bourougaa, K.; Vojtesek, B.; Fahraeus, R. Stress-dependent changes in the properties of p53 complexes by the alternative translation product p53/47. Cell Cycle 2008, 7, 950–959. [Google Scholar] [CrossRef] [PubMed]
- Bourougaa, K.; Naski, N.; Boularan, C.; Mlynarczyk, C.; Candeias, M.M.; Marullo, S.; Fahraeus, R. Endoplasmic reticulum stress induces G2 cell-cycle arrest via mrna translation of the p53 isoform p53/47. Mol. Cell 2010, 38, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Courtois, S.; Verhaegh, G.; North, S.; Luciani, M.G.; Lassus, P.; Hibner, U.; Oren, M.; Hainaut, P. Deltan-p53, a natural isoform of p53 lacking the first transactivation domain, counteracts growth suppression by wild-type p53. Oncogene 2002, 21, 6722–6728. [Google Scholar] [CrossRef] [PubMed]
- Holmila, R.; Fouquet, C.; Cadranel, J.; Zalcman, G.; Soussi, T. Splice mutations in the p53 gene: Case report and review of the literature. Hum. Mutat. 2003, 21, 101–102. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, A.; He, M.; Schmidt, C.A.; Huhn, D.; Liu, E.T. Genetic alterations in the p53 gene in the blast crisis of chronic myelogenous leukemia: Analysis by polymerase chain reaction based techniques. Leukemia 1993, 7, 593–600. [Google Scholar] [PubMed]
- Marcel, V.; Perrier, S.; Aoubala, M.; Ageorges, S.; Groves, M.J.; Diot, A.; Fernandes, K.; Tauro, S.; Bourdon, J.C. Delta160p53 is a novel N-terminal p53 isoform encoded by Δ133p53 transcript. FEBS Lett. 2010, 584, 4463–4468. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, I.; Marcel, V.; Olivier, M.; Oren, M.; Rotter, V.; Hainaut, P. Understanding wild-type and mutant p53 activities in human cancer: New landmarks on the way to targeted therapies. Cancer Gene Ther. 2011, 18, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Marcel, V.; Khoury, M.P.; Fernandes, K.; Diot, A.; Lane, D.P.; Bourdon, J.C. Detecting p53 isoforms at protein level. Methods Mol. Biol. 2013, 962, 15–29. [Google Scholar] [PubMed]
- Khoury, M.P.; Bourdon, J.C. p53 isoforms: An intracellular microprocessor? Genes Cancer 2011, 2, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Khoury, M.P.; Bourdon, J.C. The isoforms of the p53 protein. Cold Spring Harb. Perspect. Biol. 2010, 2, a000927. [Google Scholar] [CrossRef] [PubMed]
- Nutthasirikul, N.; Limpaiboon, T.; Leelayuwat, C.; Patrakitkomjorn, S.; Jearanaikoon, P. Ratio disruption of the 133p53 and TAp53 isoform equilibrium correlates with poor clinical outcome in intrahepatic cholangiocarcinoma. Int. J. Oncol. 2013, 42, 1181–1188. [Google Scholar] [PubMed]
- Takahashi, R.; Giannini, C.; Sarkaria, J.N.; Schroeder, M.; Rogers, J.; Mastroeni, D.; Scrable, H. p53 isoform profiling in glioblastoma and injured brain. Oncogene 2013, 32, 3165–3174. [Google Scholar] [CrossRef] [PubMed]
- Hofstetter, G.; Berger, A.; Fiegl, H.; Slade, N.; Zoric, A.; Holzer, B.; Schuster, E.; Mobus, V.J.; Reimer, D.; Daxenbichler, G.; et al. Alternative splicing of p53 and p73: The novel p53 splice variant p53Δ is an independent prognostic marker in ovarian cancer. Oncogene 2010, 29, 1997–2004. [Google Scholar] [CrossRef] [PubMed]
- Marabese, M.; Marchini, S.; Marrazzo, E.; Mariani, P.; Cattaneo, D.; Fossati, R.; Compagnoni, A.; Signorelli, M.; Moll, U.M.; Codegoni, A.M.; et al. Expression levels of p53 and p73 isoforms in stage I and stage III ovarian cancer. Eur. J. Cancer 2008, 44, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Boldrup, L.; Bourdon, J.C.; Coates, P.J.; Sjostrom, B.; Nylander, K. Expression of p53 isoforms in squamous cell carcinoma of the head and neck. Eur. J. Cancer 2007, 43, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Anensen, N.; Oyan, A.M.; Bourdon, J.C.; Kalland, K.H.; Bruserud, O.; Gjertsen, B.T. A distinct p53 protein isoform signature reflects the onset of induction chemotherapy for acute myeloid leukemia. Clin. Cancer Res. 2006, 12, 3985–3992. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, J.C.; Khoury, M.P.; Diot, A.; Baker, L.; Fernandes, K.; Aoubala, M.; Quinlan, P.; Purdie, C.A.; Jordan, L.B.; Prats, A.C.; et al. p53 mutant breast cancer patients expressing p53γ have as good a prognosis as wild-type p53 breast cancer patients. Breast Cancer Res. 2011, 13, R7. [Google Scholar] [CrossRef] [PubMed]
- Avery-Kiejda, K.A.; Zhang, X.D.; Adams, L.J.; Scott, R.J.; Vojtesek, B.; Lane, D.P.; Hersey, P. Small molecular weight variants of p53 are expressed in human melanoma cells and are induced by the DNA-damaging agent cisplatin. Clin. Cancer Res. 2008, 14, 1659–1668. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, L.; Segun, A.D.; Mersch, S.; Blasberg, N.; Grinstein, E.; Wai, D.; Anlauf, M.; Gabbert, H.E.; Mahotka, C.; Heikaus, S. Regulation of p53 isoform expression in renal cell carcinoma. Front. Biosci. 2010, 2, 1042–1053. [Google Scholar]
- Song, W.; Huo, S.W.; Lu, J.J.; Liu, Z.; Fang, X.L.; Jin, X.B.; Yuan, M.Z. Expression of p53 isoforms in renal cell carcinoma. Chin. Med. J. 2009, 122, 921–926. [Google Scholar] [PubMed]
- Li, X.; Romero, P.; Rani, M.; Dunker, A.K.; Obradovic, Z. Predicting protein disorder for N-, C-, and internal regions. Genome Inform. Workshop Genome Inform. 1999, 10, 30–40. [Google Scholar]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. Pondr-fit: A meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 2010, 1804, 996–1010. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Vucetic, S.; Radivojac, P.; Brown, C.J.; Dunker, A.K.; Obradovic, Z. Optimizing long intrinsic disorder predictors with protein evolutionary information. J. Bioinform. Comput. Biol. 2005, 3, 35–60. [Google Scholar] [CrossRef] [PubMed]
- Meszaros, B.; Simon, I.; Dosztanyi, Z. Prediction of protein binding regions in disordered proteins. PLoS Comput. Biol. 2009, 5, e1000376. [Google Scholar] [CrossRef] [PubMed]
- Dosztanyi, Z.; Meszaros, B.; Simon, I. Anchor: Web server for predicting protein binding regions in disordered proteins. Bioinformatics 2009, 25, 2745–2746. [Google Scholar] [CrossRef] [PubMed]
- Buljan, M.; Chalancon, G.; Eustermann, S.; Wagner, G.P.; Fuxreiter, M.; Bateman, A.; Babu, M.M. Tissue-specific splicing of disordered segments that embed binding motifs rewires protein interaction networks. Mol. Cell 2012, 46, 871–883. [Google Scholar] [CrossRef] [PubMed]
- Buljan, M.; Chalancon, G.; Dunker, A.K.; Bateman, A.; Balaji, S.; Fuxreiter, M.; Babu, M.M. Alternative splicing of intrinsically disordered regions and rewiring of protein interactions. Curr. Opin. Struct. Biol. 2013, 23, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Wrecked regulation of intrinsically disordered proteins in diseases: Pathogenicity of deregulated regulators. Front. Mol. Biosci. 2014, 1, 6. [Google Scholar] [CrossRef] [PubMed]
- Meek, D.W.; Anderson, C.W. Posttranslational modification of p53: Cooperative integrators of function. Cold Spring Harb. Perspect. Biol. 2009, 1, a000950. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Gu, W. p53 post-translational modification: Deregulated in tumorigenesis. Trends Mol. Med. 2010, 16, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, Y.; Li, L.; McNutt, M.A.; Wu, L.; Lu, S.; Yu, Y.; Zhou, W.; Feng, J.; Chai, G.; et al. An ATM- and Rad3-related (ATR) signaling pathway and a phosphorylation-acetylation cascade are involved in activation of p53/p21Waf1/Cip1 in response to 5-Aza-2′-deoxycytidine treatment. J. Biol. Chem. 2008, 283, 2564–2574. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Yamaguchi, H.; Higashimoto, Y.; Chao, C.; Xu, Y.; Fornace, A.J., Jr.; Appella, E.; Anderson, C.W. Phosphorylation site interdependence of human p53 post-translational modifications in response to stress. J. Biol. Chem. 2003, 278, 37536–37544. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Goodarzi, A.A.; Higashimoto, Y.; Noda, Y.; Lees-Miller, S.P.; Appella, E.; Anderson, C.W. ATM mediates phosphorylation at multiple p53 sites, including Ser(46), in response to ionizing radiation. J. Biol. Chem. 2002, 277, 12491–12494. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, K.; Saito, S.; Higashimoto, Y.; Roy, S.; Anderson, C.W.; Appella, E. Damage-mediated phosphorylation of human p53 threonine 18 through a cascade mediated by a casein 1-like kinase. Effect on Mdm2 binding. J. Biol. Chem. 2000, 275, 9278–9283. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, K.; Herrera, J.E.; Saito, S.; Miki, T.; Bustin, M.; Vassilev, A.; Anderson, C.W.; Appella, E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 1998, 12, 2831–2841. [Google Scholar] [CrossRef] [PubMed]
- Dumaz, N.; Milne, D.M.; Meek, D.W. Protein kinase CK1 is a p53-threonine 18 kinase which requires prior phosphorylation of serine 15. FEBS Lett. 1999, 463, 312–316. [Google Scholar] [CrossRef]
- Jenkins, L.M.; Yamaguchi, H.; Hayashi, R.; Cherry, S.; Tropea, J.E.; Miller, M.; Wlodawer, A.; Appella, E.; Mazur, S.J. Two distinct motifs within the p53 transactivation domain bind to the TAZ2 domain of p300 and are differentially affected by phosphorylation. Biochemistry 2009, 48, 1244–1255. [Google Scholar] [CrossRef] [PubMed]
- Polley, S.; Guha, S.; Roy, N.S.; Kar, S.; Sakaguchi, K.; Chuman, Y.; Swaminathan, V.; Kundu, T.; Roy, S. Differential recognition of phosphorylated transactivation domains of p53 by different p300 domains. J. Mol. Biol. 2008, 376, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Dumaz, N.; Meek, D.W. Serine15 phosphorylation stimulates p53 transactivation but does not directly influence interaction with HDM2. EMBO J. 1999, 18, 7002–7010. [Google Scholar] [CrossRef] [PubMed]
- Lambert, P.F.; Kashanchi, F.; Radonovich, M.F.; Shiekhattar, R.; Brady, J.N. Phosphorylation of p53 serine 15 increases interaction with CBP. J. Biol. Chem. 1998, 273, 33048–33053. [Google Scholar] [CrossRef] [PubMed]
- Teufel, D.P.; Bycroft, M.; Fersht, A.R. Regulation by phosphorylation of the relative affinities of the N-terminal transactivation domains of p53 for p300 domains and Mdm2. Oncogene 2009, 28, 2112–2118. [Google Scholar] [CrossRef] [PubMed]
- Shieh, S.Y.; Taya, Y.; Prives, C. DNA damage-inducible phosphorylation of p53 at n-terminal sites including a novel site, Ser20, requires tetramerization. EMBO J. 1999, 18, 1815–1823. [Google Scholar] [CrossRef] [PubMed]
- Chehab, N.H.; Malikzay, A.; Stavridi, E.S.; Halazonetis, T.D. Phosphorylation of Ser-20 mediates stabilization of human p53 in response to DNA damage. Proc. Natl. Acad. Sci. USA 1999, 96, 13777–13782. [Google Scholar] [CrossRef] [PubMed]
- Unger, T.; Juven-Gershon, T.; Moallem, E.; Berger, M.; Vogt Sionov, R.; Lozano, G.; Oren, M.; Haupt, Y. Critical role for Ser20 of human p53 in the negative regulation of p53 by Mdm2. EMBO J. 1999, 18, 1805–1814. [Google Scholar] [CrossRef] [PubMed]
- Dumaz, N.; Milne, D.M.; Jardine, L.J.; Meek, D.W. Critical roles for the serine 20, but not the serine 15, phosphorylation site and for the polyproline domain in regulating p53 turnover. Biochem. J. 2001, 359, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Schon, O.; Friedler, A.; Bycroft, M.; Freund, S.M.; Fersht, A.R. Molecular mechanism of the interaction between Mdm2 and p53. J. Mol. Biol. 2002, 323, 491–501. [Google Scholar] [CrossRef]
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation is indispensable for p53 activation. Cell 2008, 133, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.; Vousden, K.H. Modifications of p53: Competing for the lysines. Curr. Opin. Genet. Dev. 2009, 19, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.L.; Gu, W. Ubiquitination, phosphorylation and acetylation: The molecular basis for p53 regulation. Curr. Opin. Cell Biol. 2003, 15, 164–171. [Google Scholar] [CrossRef]
- Nguyen, T.A.; Menendez, D.; Resnick, M.A.; Anderson, C.W. Mutant TP53 posttranslational modifications: Challenges and opportunities. Hum. Mutat. 2014, 35, 738–755. [Google Scholar] [CrossRef] [PubMed]
- Hock, A.; Vousden, K.H. Regulation of the p53 pathway by ubiquitin and related proteins. Int. J. Biochem. Cell Biol. 2010, 42, 1618–1621. [Google Scholar] [CrossRef] [PubMed]
- Itahana, Y.; Ke, H.; Zhang, Y. p53 oligomerization is essential for its C-terminal lysine acetylation. J. Biol. Chem. 2009, 284, 5158–5164. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.; Laister, R.C.; Lemak, A.; Wu, B.; Tai, E.; Duan, S.; Lukin, J.; Sunnerhagen, M.; Srisailam, S.; Karra, M.; et al. Molecular basis of PIRH2-mediated p53 ubiquitylation. Nat. Struct. Mol. Biol. 2008, 15, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Hjerpe, R.; Aillet, F.; Lopitz-Otsoa, F.; Lang, V.; Torres-Ramos, M.; Farras, R.; Hay, R.T.; Rodriguez, M.S. Oligomerization conditions Mdm2-mediated efficient p53 polyubiquitylation but not its proteasomal degradation. Int. J. Biochem. Cell Biol. 2010, 42, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.; Manzl, C.; Strasser, A.; Villunger, A. How important are post-translational modifications in p53 for selectivity in target-gene transcription and tumour suppression? Cell Death Differ. 2007, 14, 1561–1575. [Google Scholar] [CrossRef] [PubMed]
- Toledo, F.; Wahl, G.M. Regulating the p53 pathway: In vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 2006, 6, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Bode, A.M.; Dong, Z. Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 2004, 4, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Appella, E.; Anderson, C.W. Post-translational modifications and activation of p53 by genotoxic stresses. Eur. J. Biochem. 2001, 268, 2764–2772. [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.A.; Espinosa, J.M. The impact of post-transcriptional regulation in the p53 network. Brief. Funct. Genom. 2013, 12, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.; Zhu, W.G. Surf the post-translational modification network of p53 regulation. Int. J. Biol. Sci. 2012, 8, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, L.M.; Durell, S.R.; Mazur, S.J.; Appella, E. p53 N-terminal phosphorylation: A defining layer of complex regulation. Carcinogenesis 2012, 33, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Momand, J.; Finlay, C.A. The p53 tumour suppressor gene. Nature 1991, 351, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Murphy, M.E. Genetic modifiers of the p53 pathway. Cold Spring Harb. Perspect. Med. 2016, 6, a026302. [Google Scholar] [CrossRef] [PubMed]
- Leroy, B.; Fournier, J.L.; Ishioka, C.; Monti, P.; Inga, A.; Fronza, G.; Soussi, T. The TP53 website: An integrative resource centre for the TP53 mutation database and TP53 mutant analysis. Nucleic Acids Res. 2013, 41, D962–D969. [Google Scholar] [CrossRef] [PubMed]
- Petitjean, A.; Mathe, E.; Kato, S.; Ishioka, C.; Tavtigian, S.V.; Hainaut, P.; Olivier, M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum. Mutat. 2007, 28, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed]
- Kamihara, J.; Rana, H.Q.; Garber, J.E. Germline TP53 mutations and the changing landscape of li-fraumeni syndrome. Hum. Mutat. 2014, 35, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, G.; Ballinger, M.L.; Wong, S.; Hewitt, C.; James, P.; Young, M.A.; Cipponi, A.; Pang, T.; Goode, D.L.; Dobrovic, A.; et al. High frequency of germline TP53 mutations in a prospective adult-onset sarcoma cohort. PLoS ONE 2013, 8, e69026. [Google Scholar] [CrossRef] [PubMed]
- Karakas, Z.; Tugcu, D.; Unuvar, A.; Atay, D.; Akcay, A.; Gedik, H.; Kayserili, H.; Dogan, O.; Anak, S.; Devecioglu, O. Li-fraumeni syndrome in a turkish family. Pediatr. Hematol. Oncol. 2010, 27, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Lalloo, F.; Varley, J.; Moran, A.; Ellis, D.; O’Dair, L.; Pharoah, P.; Antoniou, A.; Hartley, R.; Shenton, A.; Seal, S.; et al. BRCA1, BRCA2 and TP53 mutations in very early-onset breast cancer with associated risks to relatives. Eur. J. Cancer 2006, 42, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Achatz, M.I.; Olivier, M.; Le Calvez, F.; Martel-Planche, G.; Lopes, A.; Rossi, B.M.; Ashton-Prolla, P.; Giugliani, R.; Palmero, E.I.; Vargas, F.R.; et al. The TP53 mutation, R337H, is associated with li-fraumeni and li-fraumeni-like syndromes in brazilian families. Cancer Lett. 2007, 245, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, B.C.; Sandrini, R.; Zambetti, G.P.; Pereira, R.M.; Cheng, C.; Liu, W.; Lacerda, L.; Pianovski, M.A.; Michalkiewicz, E.; Jenkins, J.; et al. Penetrance of adrenocortical tumours associated with the germline TP53 R337H mutation. J. Med. Genet. 2006, 43, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Chompret, A.; Brugieres, L.; Ronsin, M.; Gardes, M.; Dessarps-Freichey, F.; Abel, A.; Hua, D.; Ligot, L.; Dondon, M.G.; Bressac-de Paillerets, B.; et al. p53 germline mutations in childhood cancers and cancer risk for carrier individuals. Br. J. Cancer 2000, 82, 1932–1937. [Google Scholar] [PubMed]
- Luca, J.W.; Strong, L.C.; Hansen, M.F. A germline missense mutation R337C in exon 10 of the human p53 gene. Hum. Mutat. 1998, (Suppl. 1), S58–S61. [Google Scholar] [CrossRef] [PubMed]
- Lomax, M.E.; Barnes, D.M.; Gilchrist, R.; Picksley, S.M.; Varley, J.M.; Camplejohn, R.S. Two functional assays employed to detect an unusual mutation in the oligomerisation domain of p53 in a li-fraumeni like family. Oncogene 1997, 14, 1869–1874. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, S.J.; Sakaguchi, K.; Lees-Miller, S.P.; Fiscella, M.; Mercer, W.E.; Anderson, C.W.; Appella, E. Phosphorylation at Ser-15 and Ser-392 in mutant p53 molecules from human tumors is altered compared to wild-type p53. Proc. Natl. Acad. Sci. USA 1993, 90, 5954–5958. [Google Scholar] [CrossRef] [PubMed]
- Latronico, A.C.; Pinto, E.M.; Domenice, S.; Fragoso, M.C.; Martin, R.M.; Zerbini, M.C.; Lucon, A.M.; Mendonca, B.B. An inherited mutation outside the highly conserved DNA-binding domain of the p53 tumor suppressor protein in children and adults with sporadic adrenocortical tumors. J. Clin. Endocrinol. Metab. 2001, 86, 4970–4973. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, R.C.; Sandrini, F.; Figueiredo, B.; Zambetti, G.P.; Michalkiewicz, E.; Lafferty, A.R.; DeLacerda, L.; Rabin, M.; Cadwell, C.; Sampaio, G.; et al. An inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proc. Natl. Acad. Sci. USA 2001, 98, 9330–9335. [Google Scholar] [CrossRef] [PubMed]
- Minamoto, T.; Buschmann, T.; Habelhah, H.; Matusevich, E.; Tahara, H.; Boerresen-Dale, A.L.; Harris, C.; Sidransky, D.; Ronai, Z. Distinct pattern of p53 phosphorylation in human tumors. Oncogene 2001, 20, 3341–3347. [Google Scholar] [CrossRef] [PubMed]
- Warnock, L.J.; Raines, S.A.; Milner, J. Aurora a mediates cross-talk between N- and C-terminal post-translational modifications of p53. Cancer Biol. Ther. 2011, 12, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Han, S.Y.; Liu, W.; Otsuka, K.; Shibata, H.; Kanamaru, R.; Ishioka, C. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 8424–8429. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H.; Norman, J.C. p53 and its mutants in tumor cell migration and invasion. J. Cell Biol. 2011, 192, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Adorno, M.; Cordenonsi, M.; Montagner, M.; Dupont, S.; Wong, C.; Hann, B.; Solari, A.; Bobisse, S.; Rondina, M.B.; Guzzardo, V.; et al. A mutant-p53/smad complex opposes p63 to empower TGFβ-induced metastasis. Cell 2009, 137, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Okaichi, K.; Nose, K.; Kotake, T.; Izumi, N.; Kudo, T. Phosphorylation of p53 modifies sensitivity to ionizing radiation. Anticancer Res. 2011, 31, 2255–2258. [Google Scholar] [PubMed]
- Valenti, F.; Fausti, F.; Biagioni, F.; Shay, T.; Fontemaggi, G.; Domany, E.; Yaffe, M.B.; Strano, S.; Blandino, G.; di Agostino, S. Mutant p53 oncogenic functions are sustained by PIK2 kinase through an autoregulatory feedback loop. Cell Cycle 2011, 10, 4330–4340. [Google Scholar] [CrossRef] [PubMed]
- Zerbini, L.F.; Wang, Y.; Correa, R.G.; Cho, J.Y.; Libermann, T.A. Blockage of NF-κB induces serine 15 phosphorylation of mutant p53 by JNK kinase in prostate cancer cells. Cell Cycle 2005, 4, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, O.C.; Choudhury, S.; Kolukula, V.; Vietsch, E.E.; Catania, J.; Preet, A.; Reynoso, K.; Bargonetti, J.; Wellstein, A.; Albanese, C.; et al. Dietary downregulation of mutant p53 levels via glucose restriction: Mechanisms and implications for tumor therapy. Cell Cycle 2012, 11, 4436–4446. [Google Scholar] [CrossRef] [PubMed]
- Perez, R.E.; Knights, C.D.; Sahu, G.; Catania, J.; Kolukula, V.K.; Stoler, D.; Graessmann, A.; Ogryzko, V.; Pishvaian, M.; Albanese, C.; et al. Restoration of DNA-binding and growth-suppressive activity of mutant forms of p53 via a PCAF-mediated acetylation pathway. J. Cell. Physiol. 2010, 225, 394–405. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Marchenko, N.D.; Schulz, R.; Fischer, V.; Velasco-Hernandez, T.; Talos, F.; Moll, U.M. Functional inactivation of endogenous Mdm2 and chip by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol. Cancer Res. 2011, 9, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Luz, F.A.; Brigido, P.C.; Moraes, A.S.; Silva, M.J. Aberrant splicing in cancer: Mediators of malignant progression through an imperfect splice program shift. Oncology 2016. [Google Scholar] [CrossRef] [PubMed]











© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uversky, V.N. p53 Proteoforms and Intrinsic Disorder: An Illustration of the Protein Structure–Function Continuum Concept. Int. J. Mol. Sci. 2016, 17, 1874. https://doi.org/10.3390/ijms17111874
Uversky VN. p53 Proteoforms and Intrinsic Disorder: An Illustration of the Protein Structure–Function Continuum Concept. International Journal of Molecular Sciences. 2016; 17(11):1874. https://doi.org/10.3390/ijms17111874
Chicago/Turabian StyleUversky, Vladimir N. 2016. "p53 Proteoforms and Intrinsic Disorder: An Illustration of the Protein Structure–Function Continuum Concept" International Journal of Molecular Sciences 17, no. 11: 1874. https://doi.org/10.3390/ijms17111874
APA StyleUversky, V. N. (2016). p53 Proteoforms and Intrinsic Disorder: An Illustration of the Protein Structure–Function Continuum Concept. International Journal of Molecular Sciences, 17(11), 1874. https://doi.org/10.3390/ijms17111874