1. Introduction
G protein-coupled receptors (GPCRs) form a large protein superfamily comprising nearly 800 members in humans [
1]. GPCRs are mainly located in the plasma membrane, where they detect various extracellular physicochemical signals from inside the body or from external environments such as neurotransmitters, hormones, light, and odorous chemicals during neurological, cardiovascular, sensory and reproductive signaling processes via activation of respective target G protein α subunits (Gαs) and their effector proteins for intracellular signals. GPCR signaling systems are involved in many diseases, and some are major therapeutic targets [
2]. Due to their abundance and variability, GPCR signaling is highly diverse in terms of ligands, ligand-binding sites, GPCR-specific Gα subunits, and downstream effector proteins. In contrast, the intramolecular interactions underpinning the structural rearrangements of activated GPCRs [
3,
4] and the essential and extensive interactions between activated GPCR and Gα [
5,
6] are conserved, at least for class A GPCR signaling. Activation of a specific Gα appears to be mediated by the formation of initial transient and type-specific interactions with activated GPCRs prior to the formation of more stable interactions. This initial transient process can be a potential target for specific GPCR-regulated signaling pathways.
Recently, we found that the second residue of the amphipathic helix 8 in the C-terminal domain of the murine olfactory receptor S6 (
mOR-S6), a GPCR superfamily member, is responsible for initial transient and specific interactions with chimeric Gα
15_olf, but not with Gα
15 [
7]. Our mutagenesis analysis also indicates that the hydrophobic core that is buried between the amphipathic helix 8 and transmembrane domains 1–2 (TM1–2) of
mOR-S6 are important for activation of both Gα
15_olf and Gα
15. In many GPCRs, helix 8 plays several key roles in protein/lipid interaction [
8,
9], receptor internalization [
10], dimerization of receptors [
11], and coupling with G proteins [
12]. By comparing several GPCRs, this review focuses on the functional roles of the C-terminal amphipathic helix 8 of olfactory receptors (ORs) and other GPCRs.
3. Structural Features of Helix 8
The most important structural feature of helix 8 is an amphipathic short α-helix. In rhodopsin (Rhod), helix 8 acts as a membrane surface recognition domain, and adopts a helical structure only in the presence of membranes or membrane mimetics [
9]. Helix 8 begins after a short linker following TM7, at the end of which the conserved NPxxY motif is located, as shown in a selection of class A GPCR sequences (
Figure 1) [
3,
4,
5,
6,
7]. The short linker between TM7 and helix 8 is also important, as described in
Section 5 below. Crystal structures revealed that helix 8 lies parallel to the membrane in both inactive and active states in the β
2 adrenergic receptor (β
2AdR) and in Rhod [
5,
13,
14,
15]. Moreover, in the inactive state of these GPCRs, the third residue (Phe) of helix 8 interacts with the Tyr residue of the NPxxY motif in TM7 [
5,
15], and mutation within this motif causes a significant reduction in signaling activity [
15,
16]. The third residue of helix 8 is commonly hydrophobic (Phe, Ile, Val, etc.) in mammalian class A GPCRs, including ORs that comprise the largest superfamily [
17]. In the inactive state of GPCR, the Tyr-Phe/Ile/Val intramolecular interaction forms part of the hydrophobic core between helix 8 and TM1–2 [
5,
15], which stabilizes the orientation and position of the N-terminal region of helix 8 [
7].
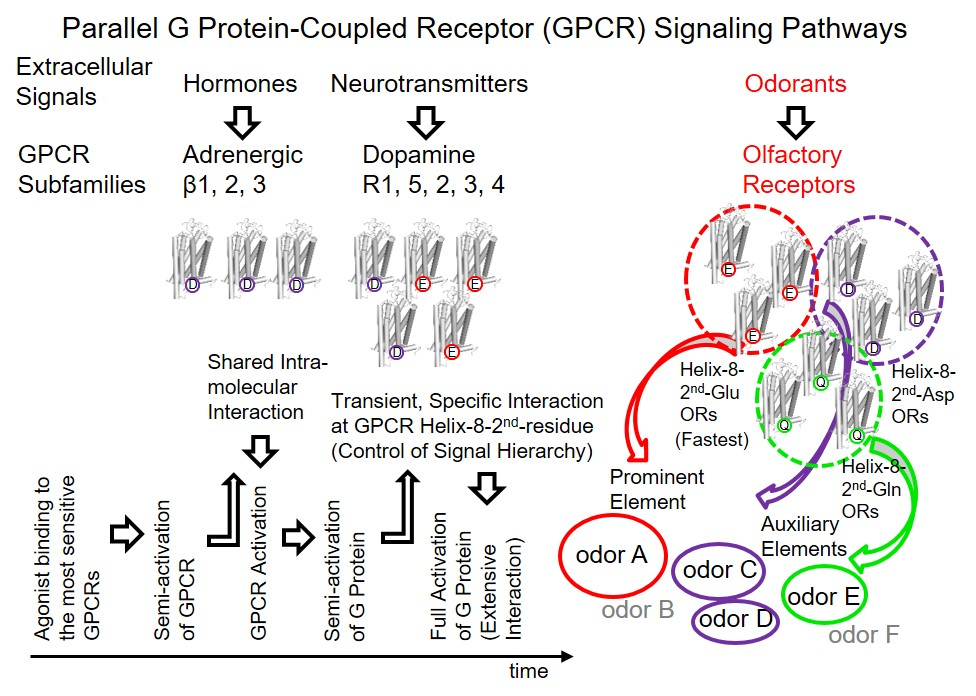
To investigate the structural details of
mOR-S6 using alanine scanning mutagenesis, a model for the 3D structure was constructed by homology modeling (
Figure 2) [
7] with crystal structures of active Rhod (the Protein Data Bank (PDB) id 3PQR) and β
2AdR (PDB id 3SN6) as templates. The best model was chosen based on the discrete optimized protein energy (DOPE) score (statistical score derived from atom pairing frequencies in the PDB) using MODELLER 9.1 [
18], and the model was further validated by PROCHECK (ver. 3.5) [
19]. In both Rhod and β
2AdR structures, the C-terminal amphipathic helix 8 is stabilized by a hydrophobic core on the intracellular side of the membrane [
12,
13]. Similarly, in our homology model, the hydrophobic core of both the N-terminal linker (Thr
300) and helix 8 (Ile
303, Leu
307, Val
308, Leu
310 and Phe
311) of
mOR-S6 are surrounded by TM1 (Phe
44, Leu
48, and Thr
52), IL1 (Leu
59) and TM2 (Tyr
64). The hydrophobic residues of helix 8 can be categorized into two groups: The first group contains Thr
300, Ile
303, and Leu
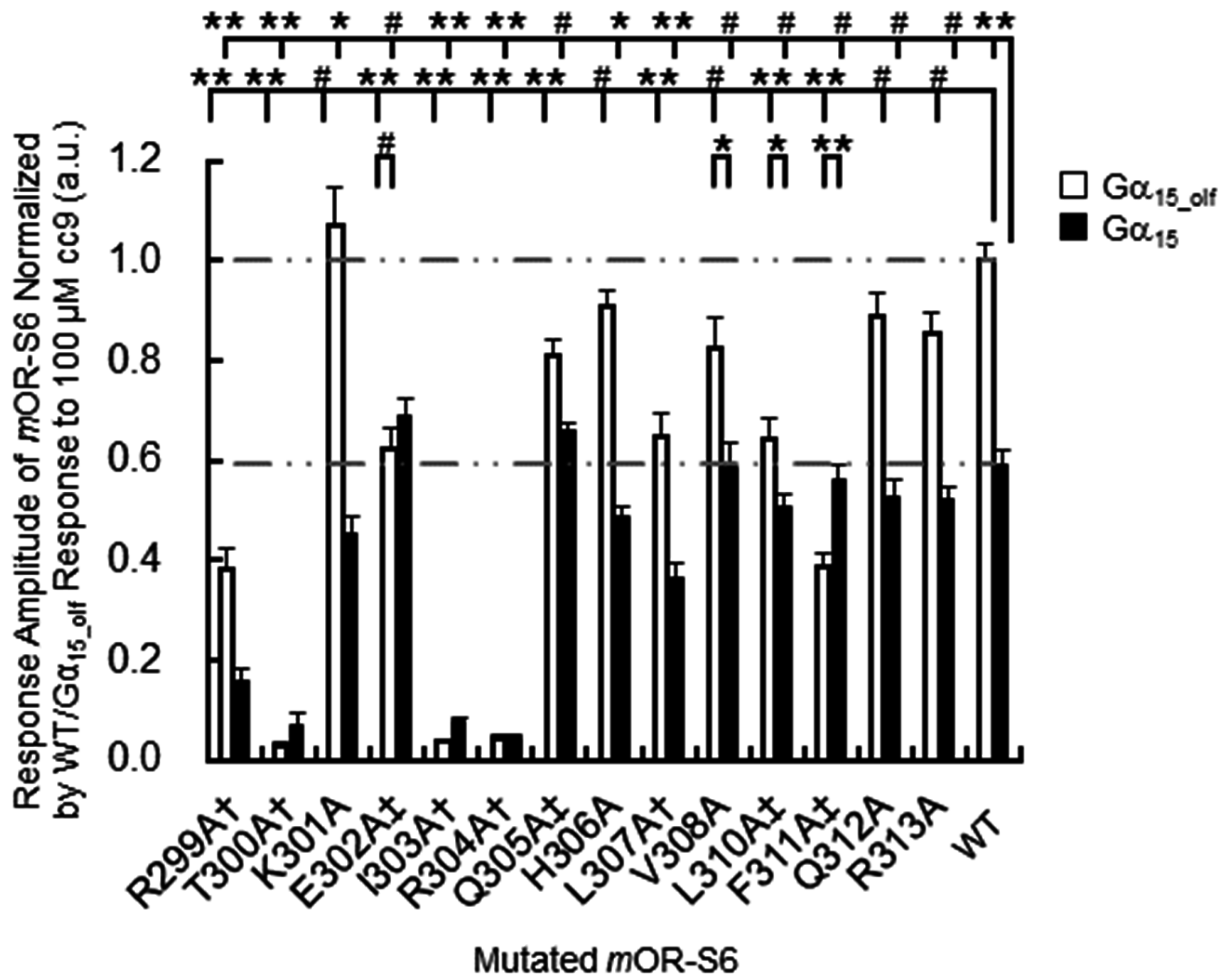
307, which are located at the N-terminal side and the middle region of helix 8. These residues play a crucial role in appropriately positioning helix 8, and mutation of these residues likely disrupt the hydrophobic core and prevent activation of Gα. Indeed, in our alanine-scanning mutagenesis analysis of helix 8, mutation of I303A in
mOR-S6, equivalent to Phe
332 in β
2AdR, caused a drastic decrease in agonist-induced Ca
2+ responses in HEK293 cells (
Figure 3) [
7]. This result indicates that disruption of the hydrophobic interactions between Ile
303 and Thr
300 (N-terminal linker), Leu
307 (helix 8) and Leu
59 (IL1) lead to impaired activation of Gα by
mOR-S6. The third residue of helix 8, Ile
303 in
mOR-S6, appears to be essential for stabilizing the structure of helix 8, and it is also essential for Gα activation along with Thr
300, the last residue of the N-terminal linker region. Alanine mutations T300A and L307A led to drastic and moderate decreases in Ca
2+ responses, respectively, since the effect of mutating the N-terminus was greater than that of the middle region (
Figure 2 and
Figure 3) [
7]. Scanning mutagenesis of the M1 muscarinic acetylcholine receptor (M1R, specific to G
q, a member of the G
q/11) similarly indicated that hydrophobic core residues are functionally important [
20].
In order to identify GPCR residues responsible for specific interaction with Gα, we investigated the kinetics of agonist-induced cellular Ca
2+ responses of
mOR-S6 by comparing chimeric Gα
15_olf with Gα
15. The chimeric Gα
15_olf has the Gα
olf (a member of the G
s class) C-terminal
376KQYE motif instead of the corresponding
369DEIN sequence present in Gα
15 (a member of the G
q/11 class), and this change improves the rapidity of the response (2.2-fold shorter Ca
2+ response onset latency) as well as the response amplitude (1.7-fold), compared with Gα
15 [
7,
23]. As expected, EC
50 values for the most potent agonist of
mOR-S6 showed no significant difference between Gα
15_olf and Gα
15 [
23]. These results indicate that the observed improvements in kinetics are likely attributable to specific interactions at the C-terminal region of Gα
15_olf with ORs. In β
2AdR, Arg
131 of the DRY motif is packed against the fourth residue (Tyr
391) of the C-terminal region of Gα
s [
5]. This intimate interaction between
mOR-S6 DRY-motif Arg
126 and Gα
15_olf C-terminal fourth Tyr
371 is believed to be responsible for the rapid and robust responses of ORs with Gα
15_olf. During the initial interaction step, conformational heterogeneity of TM7 in agonist-bound β
2AdR [
24] may facilitate interactions between the Gα C-terminal domain and the GPCR TM7 helix 8. Further kinetic analysis to unequivocally define the residues responsible for the specific interactions between GPCRs and G proteins are discussed in
Section 5 below.
The second group of
mOR-S6 helix 8 hydrophobic residues includes Leu
310 and Phe
311, located at the C-terminal interface between helix 8 and TM1. Alanine mutants L310A and F311A of
mOR-S6 caused moderate and dramatic decreases in Ca
2+ responses with Gα
15_olf, respectively, compared with
mOR-S6 (
Figure 3). Weakening of the hydrophobic core in the vicinity of the helix 8 C-terminal region likely increases helix 8 flexibility and destabilizes its structure. This suggests that activation of Gα
15_olf requires a solid and stable helix 8, whereas activation of Gα
15 does not have this requirement. In total, seven and five of helix-8 alanine mutants reduced the signaling of
mOR-S6 via Gα
15_olf and Gα
15, respectively (
Figure 3). Immunostaining of N-terminal rhodopsin-tagged
mOR-S6 with anti-rhodopsin antibody confirmed that all
mOR-S6 mutants were efficiently expressed and membrane-localized [
7].
4. Shared Features of Different GPCR–G Protein Interactions in Inactive and Active States
In the intra- and inter-molecular interactions of GPCRs that occur during activation and inactivation, the relative position of conserved motifs and residues is clearly important. Key residues controlling GPCR–G protein coupling are believed to be located at the intracellular end of TM5, the N-terminal region of intracellular loop 3 (IL3), the junction of TM3 (including the DRY motif) and IL2, the C-terminal TM6, and the junction of TM7 and helix 8 [
4,
5,
6,
21,
22]. Comparison of structural differences between inactive and active states of class A GPCRs indicates shared features in the structural rearrangement of activated GPCRs (
Figure 1) [
3,
4]. In β
2AdR (specific to G
s, a member of the G
s class), Rhod (specific to G
t, a member of the G
i/o class), μ-opioid receptor (µOR, specific to G
i, a member of the G
i/o class) and M2 muscarinic receptor (M2R, specific to G
i), Arg3.50 of the DRY motif interacts with either Glu6.30 or Thr6.34 in the inactive states (except for M2R and β
2AdR), but similarly with Tyr5.58 in all active states (
x.yz numbers follow the Ballesteros-Weinstein numbering method for GPCRs [
25]) (
Figure 1, Arg
131–none → Tyr
219 in β
2AdR; Arg
135–Glu
247 → Tyr
223 in Rhod; Arg
165–Thr
279 → Tyr
252 in µOR) [
3]. Tyr7.53 of the NPxxY motif and the adjacent Cys/Ile/Ala7.54 also interact with the third (Phe8.50) and fourth (Arg/Lys8.51) residues of helix 8 respectively, in the inactive states [
4,
14,
15], whereas Tyr7.53 interacts with Tyr5.58 and Leu3.43 through a water-mediated polar network in active state structures (
Figure 1, Tyr
326–Phe
332 → Tyr
219 + Leu
124 and Cys
327–Arg
333 → none in β
2AdR; Tyr
306–Phe
313 → Tyr
223 + Leu
128 and Ile
307–Arg
314 → none in Rhod; Tyr
336–Phe
343 → Tyr
252 + Leu
158 and Ala
337–Lys
344 → none in µOR) [
3]. In our scanning mutagenesis analysis, mutation of the
mOR-S6 helix-8 fourth residue (R8.51A) resulted in a drastic decrease in Ca
2+ responses for both Gα
15_olf and Gα
15 (
Figure 3) [
7]. This is consistent with previous reports that β
2AdR Arg8.51
333 [
6] and β
1 adrenergic receptor Arg8.51
384 [
12] are essential for coupling with G proteins. Notably, recent analysis has indicated that Ile/Leu/Met3.46 interacts with Leu/Val/Ile6.37 in inactive states, but with Tyr7.53 in active states (
Figure 1, Ile
127–Leu
275 → Tyr
326 in β
2AdR; Leu
131–Val
254 → Tyr
306 in Rhod; Met
161–Val
282 → Tyr
336 in µOR) [
4].
A key feature of β
2AdR activation is the ~14 Å outward movement of the intracellular portion of TM6, creating a cavity large enough to accommodate the C-terminus of Gα [
5,
26]. The active state of β
2AdR is stabilized by extensive interactions with Gα [
5]. In an atomic resolution structure of the β
2AdR-G
s complex, the essential and stable interface buried between activated β
2AdR and Gα
s is formed by IL2, TM5 and TM6 of β
2AdR and by helix-α5, the αN-β1 junction, the top of strand β3 strand, and helix-α4 of Gα
s [
5]. Among the most conserved amino acids, β
2AdR Arg
131 (TM3 DRY motif) is packed against both Gα
s Tyr
391 (helix-α5, fourth residue from the C-terminus of Gα
s) and β
2AdR Tyr
326 (TM7 NPxxY motif) [
5]. β
2AdR Leu
275 (TM6) also interacts with Gα
s Leu
393 (the penultimate residue for the C-terminus) [
5,
26]. In addition, β
2AdR Phe
139 (IL2) docks into a hydrophobic pocket formed by Gα
s His
41 (β1-strand), Val
217 (β3-strand), Phe
376 (helix-α5), Cys
379 (helix-α5), Arg
380 (helix-α5) and Ile
383 (helix-α5) [
5]. The position of Phe
139 (IL2) is stabilized by interactions between Asp
130 (DRY motif) and Tyr
141 (IL2) [
5]. Notably, the residue corresponding to Phe
139 is a Phe or Leu in almost all G
s-coupled GPCRs [
5]. In the crystal structure of Rhod in complex with the Gα
t C-terminal peptide (GαCT2), the Rhod D(E)RY motif Arg
135 forms a hydrogen bond to the backbone carbonyl oxygen at the fourth residue from the C-terminus of GαCT2 in the C347V mutant [
22], which is similar to the packing of the β
2AdR DRY motif Arg
131 against Gα
s Tyr
391. However, rather than Arg
135, Rhod D(E)RY motif Glu
134 binds to NPxxY motif Asn
302 via a water-mediated polar network [
22].
The results of a solution-state nuclear magnetic resonance (NMR) study raised the possibility that the propagation of conformational changes in GPCRs occurs via initial interactions between GPCR helix 8 and the associated G protein [
27]. Using C
13-dimethylated µOR, NMR spectroscopy revealed that the agonist-induced spectral changes in helix 8 (Lys8.51
344) and IC1 (Lys
98, Lys
100) were larger than those of TM6 (Lys6.24
269/Lys6.26
271) and TM5 (Lys5.66
260). Interestingly, the presence of both an agonist and a Gα
i-mimetic nanobody resulted in a complete loss in the intensity of peaks corresponding to helix 8 and the IC1 Lys residue, and a drastic reduction in the intensity of the TM6 Lys peak, with a concomitant appearance of new intense peak. The spectral shift in the TM6 Lys peak presumably reflects the >10 Å outward movement of TM6 in the active state. Sharp, narrow intense peaks for TM6 and TM5 Lys residues indicate a relatively stable conformation for these features, while broad and irregular peaks for helix 8 and IC1 suggest that two or more conformations of helix 8 and IC1 exchange on a low ms time scale in µOR in the presence of agonist alone [
27]. In contrast to the relatively stable positions of TM6 and TM5, helix 8 is likely to be more flexible and thus able to adopt the required conformations for forming specific interactions with Gα C-terminal residues, as will be described in the next section.
5. Helix 8 N-Terminal Residues of GPCRs Are Responsible for Rapid Kinetics Associated with Specific G Protein Activation
Establishing which residues of GPCRs are responsible for the specific interactions with G proteins has received considerable attention. Chimeric mutants of Rhod in which
300NKQ is replaced with the
330SPD sequence of β
2AdR (the middle of which is the first amino acid of helix 8) displayed a dramatic decrease in the ability to activate the target G
t [
28,
29]. Furthermore, we examined the contribution made by each residue of helix 8 of
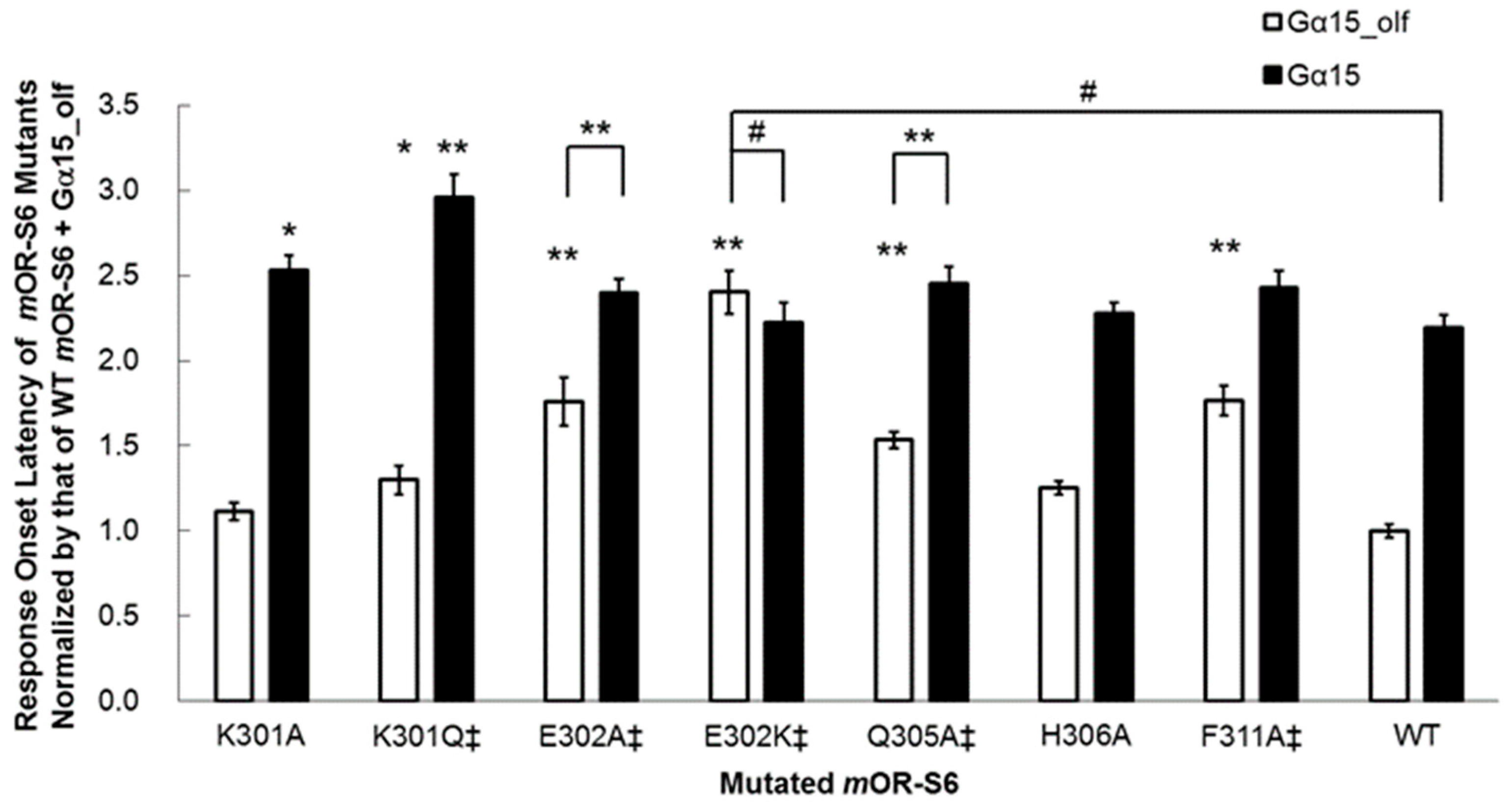
mOR-S6 to the response kinetics using alanine-scanning mutagenesis. Four mutations (E302A, Q305A, L310A, and F311A) caused a decrease in agonist-induced Ca
2+ responses in HEK293 cells via Gα
15_olf, but not via Gα
15 (
Figure 3) [
7]. Of these four residues, only mutation of Glu
302 to alanine resulted in no significant difference in the amplitude of the Ca
2+ response between Gα
15_olf and Gα
15, but a significant difference in response onset latency was still apparent (
Figure 4) [
7]. Interestingly, this second residue of helix 8 is negatively charged (Glu or Asp) or uncharged, but is polar (Gln) in the OR family (
Table 1).
We examined the effect of introducing a positively charged residue in the E302K mutant, and the improved kinetics of the onset latency and amplitude with Gα
15_olf were completely abolished in this variant, which showed no significant differences from wild-type
mOR-S6 with Gα
15 (
Figure 4) [
7]. These results suggest that the N-terminal acidic residue of helix 8 of an OR is responsible for rapid activation of Gα
15_olf. In the crystal structure of the opsin–Gα
t C-terminal peptide (GαCT) complex, the second residue of helix 8 (Gln
312) interacts with the sixth residue from the C-terminus of Gα
t (Lys
345) and the opsin helix-8 N-terminal linker residue Asn
310, in addition to the interaction between the opsin D(E)RY motif Arg
135 and the fourth residue from the C-terminus of Gα
t (Cys
347) (
Figure 1) [
30]. Molecular modeling revealed differences between intermediary (R*–G
t(GDP) complex) and stable (R*–G
t(empty) complex) interactions [
31]. Specifically, the second residue of helix 8 of Rhod (Gln
312) interacts in an intermediate manner with the fourth residue from the C-terminus of Gα
t (Cys
347), but then stably interacts with the sixth residue from the C-terminus (Lys
345). However, in the crystal structures of the Rhod–GαCT2 complex and the stable β
2AdR–G
s complex, no such interaction between the second residue of helix 8 and Gα was observed. This difference is likely attributable to the C347V mutation of GαCT2 and the stable (i.e., not intermediate) active state of β
2AdR, respectively. Taken together, these observations indicate that the initial transient and specific interaction between the second residue of
mOR-S6 helix 8 (Glu
302; Gln
312 in opsin) and the sixth residue from the C-terminus of Gα
15_olf (Lys
369; Lys
345 in Gα
t) likely facilitates the rapid formation of the active state in the OR–Gα
15_olf complex, but not in the OR–Gα
15 complex. If this is the case, the question arises as to which residues of
mOR-S6 initially interact with Gα
15.
Notably, the KE301-302EK double mutant of
mOR-S6 exhibits an impaired Ca
2+ response via both Gα
15_olf and Gα
15 (our unpublished data). Moreover, mutation of the first residue of helix 8 of
mOR-S6 (Lys
301), which is conserved in the OR family, resulted in mutants that displayed a complicated behavior [
7]. The K301A mutation resulted in a significant, but not drastic, decrease in Ca
2+ responses via Gα
15 but no change in the responses via Gα
15_olf (
Figure 3). Meanwhile, the K301A mutation delayed the onset latency, consistent with the decreased response via Gα
15 but no change via Gα
15_olf (
Figure 4). Mutation to an uncharged polar residue (K301Q) resulted in similar changes to K301A in terms of response kinetics. However, in contrast to the KE301-302EK double mutant and the K301A/Q mutants, the K301E single mutant with a negatively charged residue, displayed a drastic and selective decrease in response to Gα
15, but not for Gα
15_olf. These results raised the possibility that Lys
301 may attract a negatively charged region of Gα
15, but not necessarily for Gα
15_olf. Based on sequence differences between Gα
15_olf 369KQYE and Gα
15 369DEIN, Gα
15 Asp
369 and/or Glu
370 might be involved in such an initial attraction. In the M3 muscarinic acetylcholine receptor (M3R, specific to G
q/11), an agonist-induced increase in disulfide cross-linking of the first residue of helix 8 (via the K548C mutant) and the α4/β6 loop of G
q (via the D321C mutant) was observed, and was greatly reduced by the pretreatment of membranes with the inverse agonist, atropine [
32]. This indicates an interaction between M3R helix 8 Lys
548 and G
q α4/β6 loop Asp
321. Similarly,
mOR-S6 helix 8 Lys
301 may interact with Gα
15 α4/β6 loop Asp
328 with slower response kinetics than the inter-helix interaction between
mOR-S6 and Gα
15_olf, while the K301E mutation may impair the interaction with Gα
15 and hence decrease its activation. Thus, kinetic analysis is very useful for evaluating specific interactions between GPCRs and G proteins.
As described above, transient interactions between the second residue of
mOR-S6 helix 8 (Glu
302) and the sixth residue from the C-terminus of Gα
15_olf (Lys
369) likely facilitate the rapid formation of a more stable and active OR–Gα
15_olf complex, resulting in a rapid and robust Ca
2+ response. If this is the case, the question arises as to exactly how
mOR-S6 helix 8 accommodates the Gα α5 C-terminal region between TM3 and TM5 in the stable and active state. Considering the simplest case of β
2AdR (
mOR-S6) and its relative movement toward Gα, the C-terminus of Gα α5 may forward towards the N-terminal region of β
2AdR (
mOR-S6) helix 8 under TM domain assembly from the intracellular spacing between TM3 and TM5. This relative movement is likely the trigger for an outward movement of the intracellular portion of TM6 that resides on the front side of the N-terminus of helix 8 and may be ready to move following rearrangement of the TM domains upon agonist binding to β
2AdR. A forward movement of the C-terminal region of Gα α5 would then promote its docking onto the N-terminus of β
2AdR (
mOR-S6) helix 8, resulting in the formation of a specific interaction between the sixth residue from the C-terminus of Gα
s (Arg
389 in helix-α5; Lys
369 of Gα
15_olf) and the second residue of β
2AdR helix 8 (Asp
331; Glu
302 in
mOR-S6) at the corner of helix 8 and the membrane surface, rather than at the open surface of helix 8 (the first residue of this region,
Figure 2). This step also facilitates the breakage of the interaction between the NPxxY motif Tyr7.58
326 (Tyr7.58
296 of
mOR-S6) and the third residue of helix 8 (Phe8.50
332; Ile8.50
304? of
mOR-S6), which is caused by the outward movement of the adjacent Asp
331 due to the forward momentum of the transiently interacting C-terminal region of Gα. This presumably results in the movement of helix 8 and Gα C-terminus being pushed back towards TM3 through intra-TM interactions that underpin the elastic properties. This likely results in intimate interactions between β
2AdR TM3 DRY-motif Arg
131 and both the fourth residue from the C-terminus of Gα
s (Tyr
391) and β
2AdR NPxxY motif Tyr
326, which stabilizes the active state of the ternary complex [
5]. These proposed transient perturbations of helix 8 are consistent with the moderately dynamic conformational changes observed for C
13-dimethylated µOR [
27].
This model also explains the greater selective decrease in the Ca
2+ response for Gα
15_olf observed in
mOR-S6 F311A compared with the L310A mutant, since weakening of the hydrophobic core at the C-terminus of helix 8 likely increases its flexibility and destabilizes the position of Glu
302 between the membrane and the open surface to a greater extent than disruption of the hydrophobic core within the middle of helix 8. This model, therefore, offers a possible explanation for the rapid formation of a more stable ternary GPCR–G protein complex. Truncated mutants provide further support that helix 8 is essential in GPCR signaling [
7,
33].
Notably, molecular dynamics simulation and mutagenesis studies of the cannabinoid 1 (CB1, specific to G
i) receptor suggested that Arg
400 (the penultimate amino acid of the N-terminal linker) interacts with the penultimate residue of Gα
i (Leu
353) [
34]. The penultimate Leu
393 point mutation to Ala in Gα
s also reduced the activity of both β
2AdR and luteinizing hormone receptor (LHR) [
35]. Furthermore, in our scanning mutagenesis analysis, two mutations of this CB1 Arg
400 equivalent (R299A and R299E), respectively, markedly reduced and completely ablated the Ca
2+ response [
7]. These results suggest that the penultimate residue of the N-terminal linker between helix 8 and TM7 might be responsible for the recruitment of the G protein C-terminus.
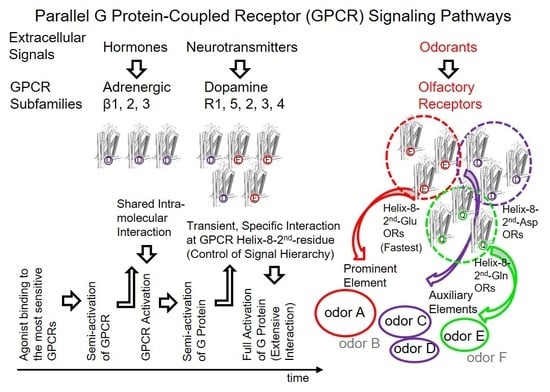
6. The Second Residue of Helix 8 Partially Governs the Hierarchy of GPCR-Associated Information in Parallel GPCR Signaling Pathways
The replacement of Gα
15 369DEIN with Gα
olf 376KQYE improved the response kinetics of
mOR-S6 via the chimeric Gα
15_olf by shortening the onset latency 2.2-fold [
7,
23], but replacement of
mOR-S6 Glu
302 with Arg
302 completely eliminated the effect of this mutation on
mOR-S6-mediated Ca
2+ responses [
7]. These findings clearly indicate that the second residue of helix 8 is a major determinant of the initial specific interaction with the target G protein that are essential for a rapid and robust response, rather than with Arg or Ala at the second position of helix 8 in ORs or non-target G proteins. In the olfactory system, the second residue of helix 8 appears to govern the sensory processing hierarchy of elemental odors that are represented in the third-order neurons of olfactory pathways [
36,
37,
38,
39].
We proposed a mechanism for supersensitive odor discrimination wherein signals from the helix 8 second residue Glu of dorsal ORs determines the most prominent elemental odor of a given odorant [
36]. In odor detection/discrimination behavioral assays, wild-type mice can discriminate similar odors of enantiomeric pairs at sub-ppq (<10
−15) level, which equates to supersensitivity for enantiomer detection, whereas transgenic mice in which all dorsal ORs are ablated display a >10
10-fold reduction in enantiomer discrimination sensitivity, although the supersensitive detection capability for (−)-enantiomers is retained [
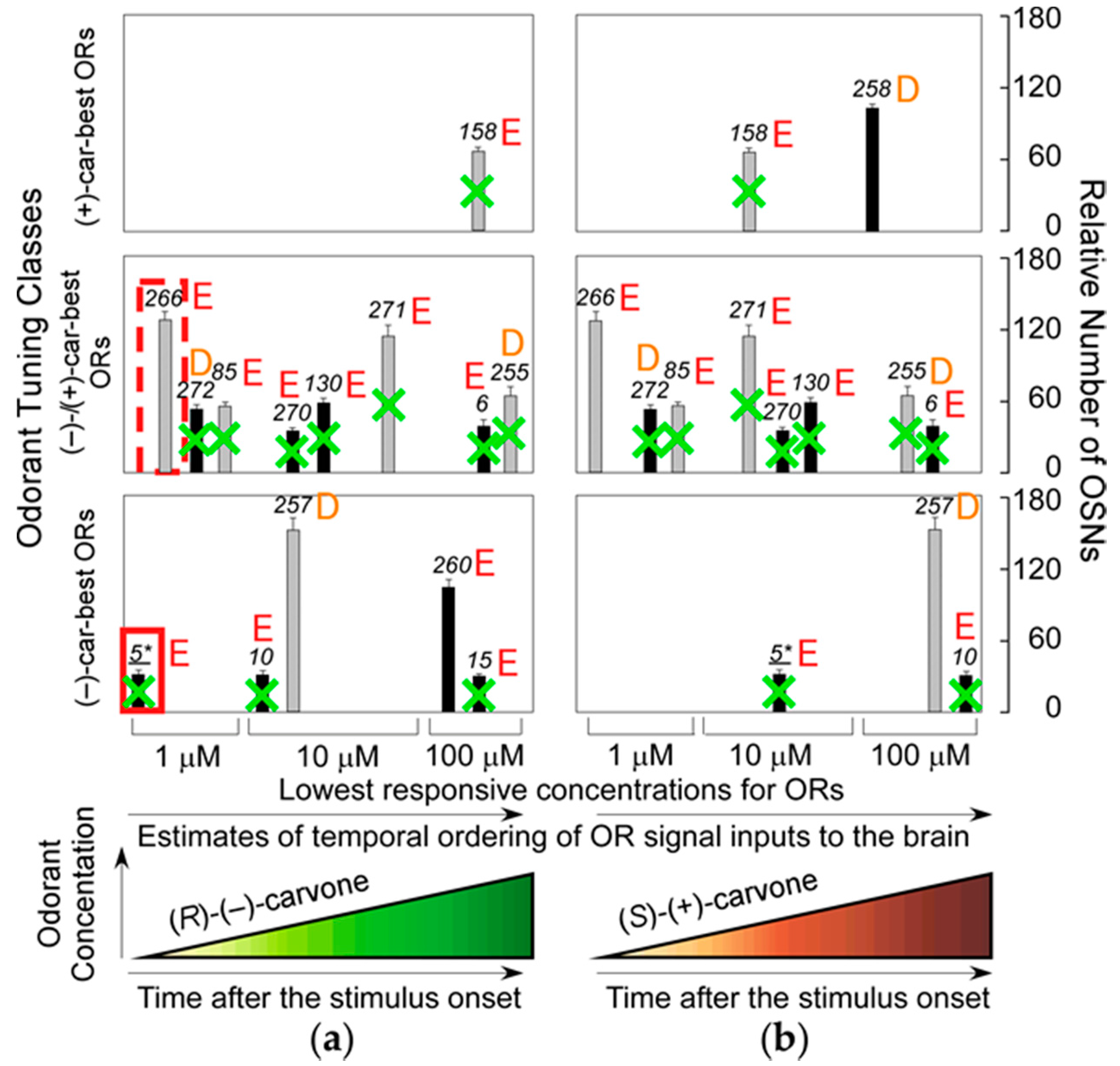
37]. This result indicates that the most sensitive ORs that enable the transgenic mice to detect (−)-enantiomers but not (+)-enantiomers at sub-ppq level do not allow the mice to discriminate (−) from (+)-enantiomers with supersensitivity (odor discrimination paradox), and suggests that some of the most sensitive ORs ablated may enhance characteristic elemental odors in wild-type mice. Among the ablated dorsal ORs with a Glu in the second position of helix 8,
mOR-
car-c5 is one of the most sensitive and specific for (
R)-(−)-carvone (
Figure 5). These results indicate that the highly sensitive helix-8-second-Glu dorsal ORs play a critical role in hierarchical elemental odor coding by summating synchronized inputs from cognate ORs to third-order neurons for elemental odors through feedforward inhibition [
37].
The hierarchical odor-coding hypothesis was first proposed following receptor code analysis for carvone enantiomers [
38]. This odor-decoding model considers that the olfactory system can extract sensory information by summating signals from multiple receptors in the third-order neurons of olfactory pathways via input synchronization through feedforward inhibition of the pyramidal cells in the anterior piriform cortex (aPC), the second olfactory center [
36,
37,
39]. This sensory strategy is analogous to that in vision, wherein the four elemental colors (red, green, yellow and blue) are primarily extracted by the third-order neurons (ganglion cells) or the higher visual pathway through summation of synchronized inputs from one or two types of receptors following inhibition by signals from M-cone and S-cone photoreceptors. Elemental colors allow us to perceive all visible hues in different weighted combinations, and similarly, elemental odors likely allow us to discriminate various odors in different weighted combinations.
Olfactory feedforward inhibition is activated in the rostro-ventral portion of the aPC (aPC
vr) [
40]. Notably, in insects, input synchronization via inhibition is also important for discrimination of similar odors [
41]. Furthermore, mutual inhibition between different odors was previously examined in a mixture of rose and fox-unique TMT odors in mice [
42]. A rose-odor-induced decrease was apparent in cells positive for the TMT odor in the aPC
vr, and this was accompanied by a subsequent decrease in the TMT-induced stress response. This suggests that signals from ORs activated by the co-applied rose odor weakens the feedforward inhibition from ORs for TMT and thus weakens the subsequent signal integration of cognate TMT ORs. Compared to the sum of the responses to the two individual odors, the total number of cells positive for the mixture of TMT and rose odor in the dorsal part of the anterior piriform cortex also decreased, suggesting a decrease in the perceived intensity of the TMT odor [
42]. In contrast to the rose odor, caraway odor did not alleviate the TMT-induced stress response, suggesting a hierarchy of elemental odors in the order rose > TMT > caraway, at least under the experimental conditions employed. Signals from helix-8-second-Glu dorsal ORs are likely associated with the most prominent (upper level) signaling for a given odor (the most prominent elemental odor), whereas other ORs are presumably related to lower levels (auxiliary) of the odor (weaker elemental odors).
In this way, helix-8-second-Glu of ORs appear to govern, at least in part, olfactory information processing of hierarchical elemental odors through earlier and more intense signals than those processed by helix-8-second-Ala or Lys ORs in parallel GPCR signaling pathways. Among 374 (52 class I and 322 class II) human ORs, a total of 45% (23% (12/52) in class I and 48% (155/322) in class II) have a Glu at the second position of helix 8, while the 33% and 16% have an Asp and Gln, respectively (
Table 1). Interestingly, Glu and Gln are identical in terms of side-chain size (i.e., they are isosteric). However, although Glu and Asp both have a negative charge, the side chain of Asp is shorter by one carbon atom, and there are no helix-8-second-Asp ORs among human or murine class-I ORs that are expressed in the dorsal zone (dorsal ORs). Moreover, the frequency of helix-8-second-Glu, Asp and Gln ORs are almost identical between human and murine class-I ORs: 23% vs. 24%, 0% vs. 0% and 69% and 67%, respectively (
Table 1). Helix-8 second residues were >90% (39/42 and 204/226 in class I and II, respectively) identical between human and murine ORs (
Table S1). These results suggest that ORs with different residues at the second position of helix 8 play distinct roles in elemental odor representation. As described above, our results suggest that the most sensitive helix-8-second-Glu dorsal ORs emphasize (
R)-(−)-carvone-unique elemental odors in the brain by selective summation of cognate OR signals via synchronized inputs to the third-order neurons through feedforward inhibition driven by signals from the most sensitive helix-8-second-Glu dorsal ORs with a shorter onset latency (one of which is enclosed by the red rectangular in
Figure 5) [
36,
37]. If ORs with longer onset latencies determine the most prominent elemental odors, odor perception must change during development as odor representation in the brain adapts over time.
Furthermore, helix-8-second-Glu ORs accounted for 73% (11/15) of the 15 identified carvone ORs in a single-cell RT-PCR study of 2740 randomly sampled murine olfactory sensory neurons, which is 1.6-fold more than the average number of human helix-8-second-Glu ORs. Along with the absence of any helix-8-second-Asp class-I ORs in both human and mouse, this indicates that helix-8-second-Glu ORs operate as determinants of odor representation. Interestingly, helix-8-second-Gln class-I ORs make up the largest group (67%–69%), which is ca. 3-fold and 10-fold larger than helix-8-second-Glu class-I ORs and helix-8-second-Gln class-II ORs, respectively. Future research should focus on the question of which target-prominent or auxiliary elemental odors different types of ORs contribute to identifying, amplifying or classifying.
7. Potential Roles of Helix 8 in GPCR Membrane Surface Expression, Internalization, Regulation of Phosphorylation and Dimerization
In parallel GPCR signaling pathways, a proper ratio of signaling proteins is likely required for ensuring adequate sensory information processing or systematic functional regulation. Inhibition of GPCRs by phosphorylation of the C-terminal region may disrupt the proper sequence of multistep interactions between GPCRs and their target G proteins, resulting in their removal from the membrane. Non-interactive GPCR mutants must also be removed because they could reduce the total GPCR sensitivity by capturing target agonists, leading to a decrease in the effective agonist concentration. Arrestin-mediated internalization of phosphorylated GPCRs is likely to be one of the regulatory mechanisms employed to maintain the proper sensitized/desensitized GPCR ratio.
In the thyrotropin-releasing hormone receptor (TRHR, specific to G
q/11), agonist-dependent phosphorylation by GPCR kinases occurs in both wild type (>35%) and helix-8-second K326R mutant forms (ca. 40%) but not in the K326Q mutant (ca. 5%) [
43]. In total, 70% of wild-type TRHR was internalized following complex formation with arrestin, but internalization was only 40% for 6K → 6Q mutant (including helix-8-second-Lys). The high internalization rate suggests that wild-type TRHR may be of less importance than the 6K → 6Q TRHR mutant, or overexpressed to a great extent than could be measured accurately. This also suggests that the G protein coupling specificity-determining second residue of helix 8 is also a determinant of receptor phosphorylation and internalization via formation of arrestin-receptor complexes. When Rhod is phosphorylated in the C-terminal region, the conformational dynamics of helix 8 controls binding to arrestin and subsequent arrestin activation during the desensitization process [
44]. Enhancement of agonist-induced receptor internalization by a single mutation in helix 8 has also been reported for the human calcitonin receptor-like receptor [
10].
In melanin-concentrating hormone receptor 1, a proximal dibasic pair of residues in the fourth and fifth positions of helix 8 is required for GPCR cell surface expression and signaling [
45]. Furthermore, in the type 1 angiotensin receptor, helix 8 has been reported to interact with a myriad of proteins, including caveolin, angiotensin II type 1 receptor-associated protein, and γ-aminobutyric acid receptor-associated protein in membrane expression, G proteins, phospholipase C, Jak2, calmodulin and SHP-2 in signaling, and regulators in lateral receptor migration, receptor internalization and nuclear transcription factors [
46]. Helix 8 also plays a key role in protein/lipid interactions [
8,
9] and dimerization of receptors [
11] or heteroreceptors (fibroblast growth factor receptor 1 and 5-hydroxytryptamine 1A receptor that play an enhancing role in hippocampal plasticity) [
47], and is, therefore, responsible for multiple functions in parallel GPCR signaling pathways.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}