
Molecular Mechanisms and Translational Therapies for Human Epidermal Receptor 2 Positive Breast Cancer

 ,
,
Abstract
:1. Introduction
2. HER2 Biology and Its Role in Breast Cancer
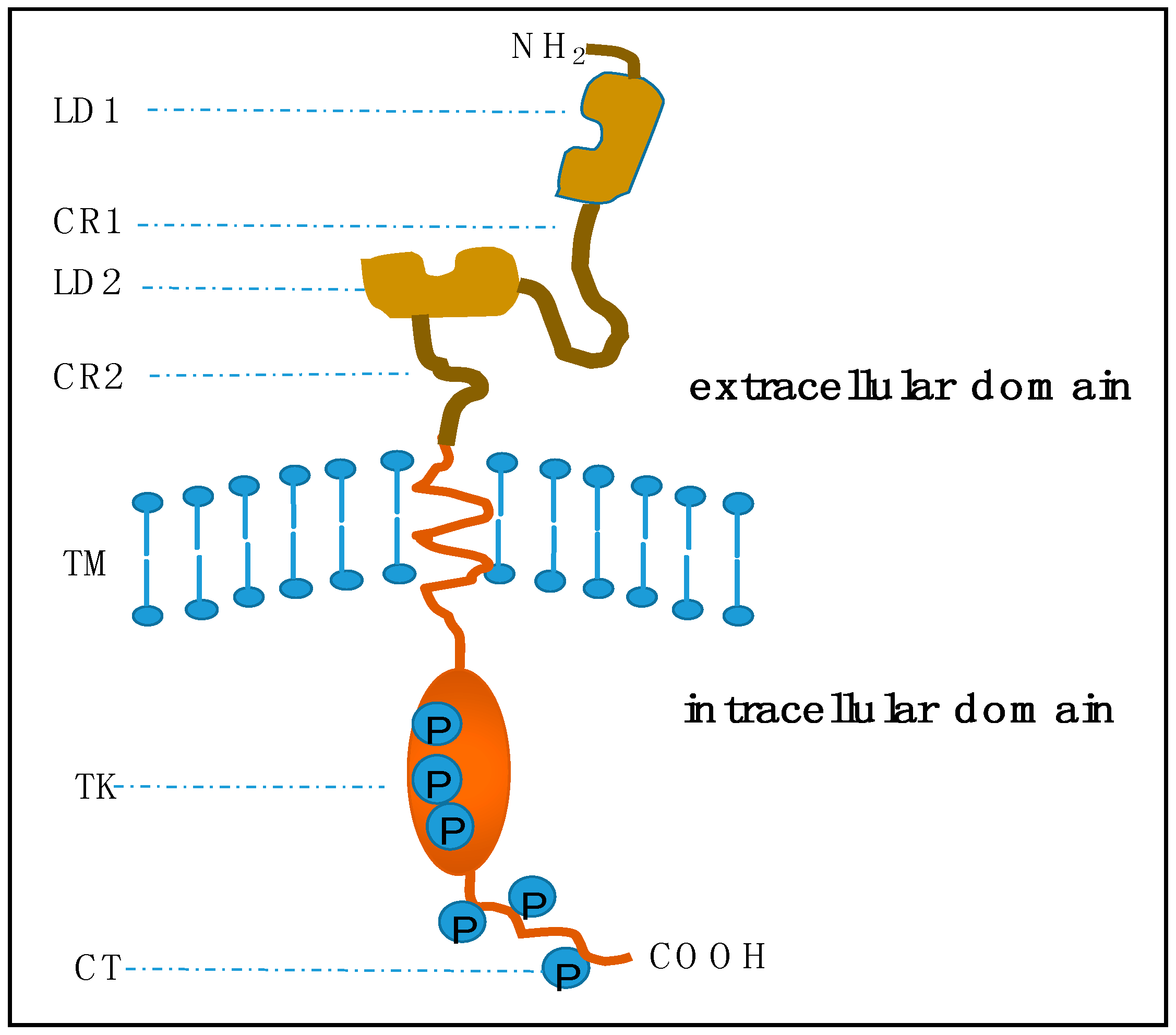
2.1. Structure of HER2 and Its Physiological Role in Signaling Pathways
2.1.1. Structure of HER2
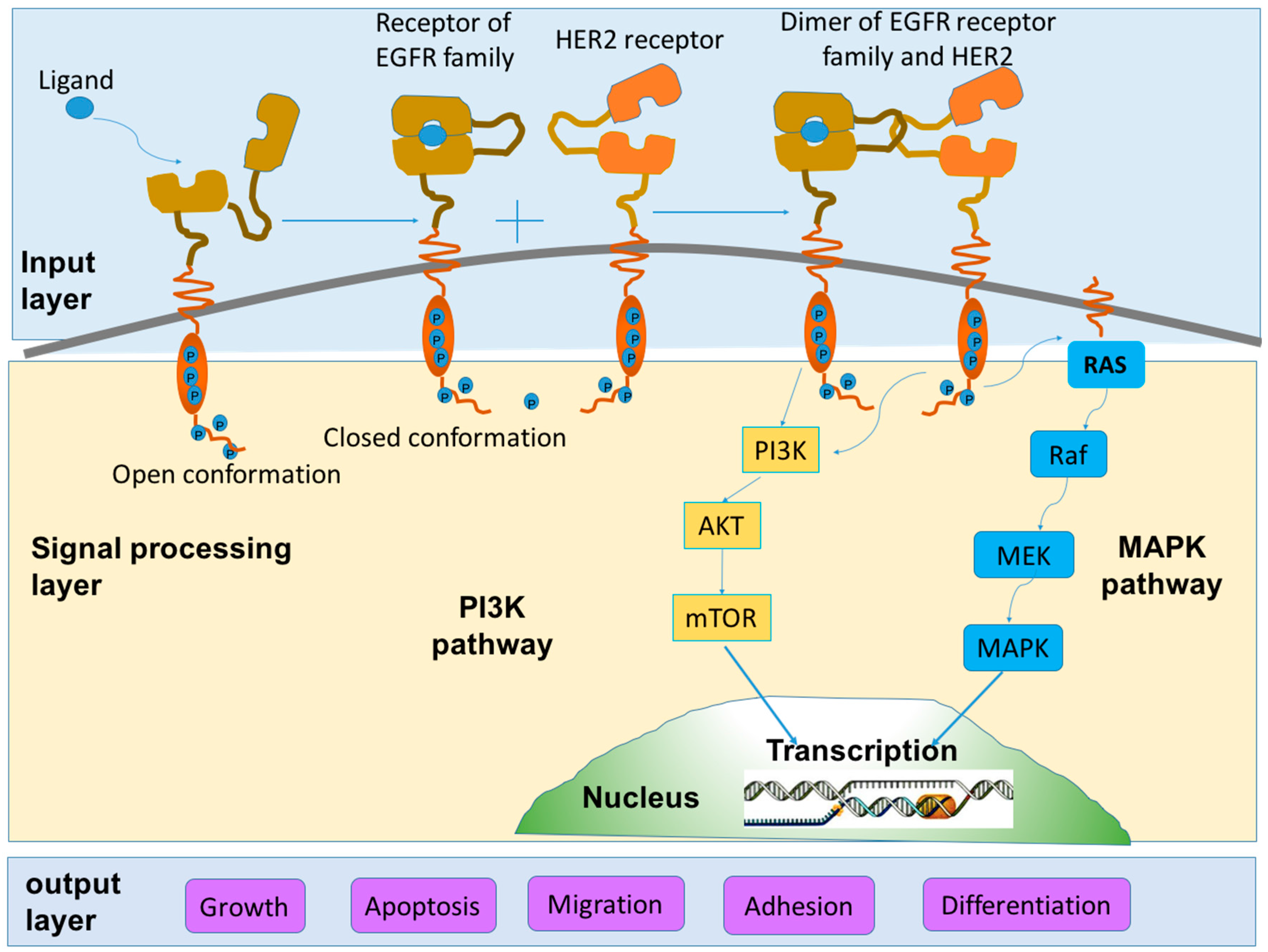
2.1.2. Role in Signaling Pathways
2.2. Tumorigenic Action of HER2
2.2.1. HER2 and Breast Cancer
2.2.2. HER2 and Other Cancers
3. Diagnosis of HER2+ BC
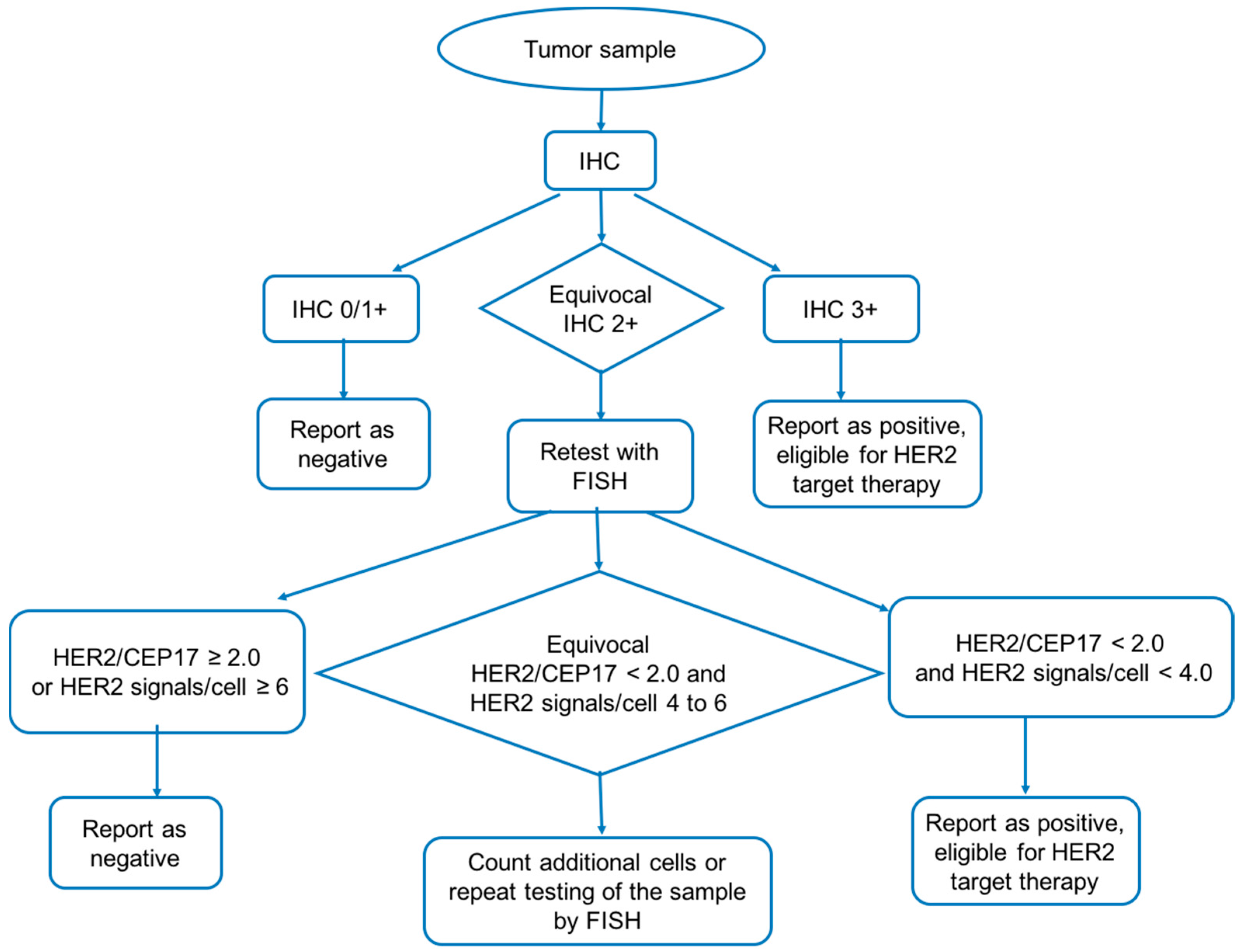
3.1. Immunohistochemistry
3.2. Fluorescent In Situ hybridization (FISH)
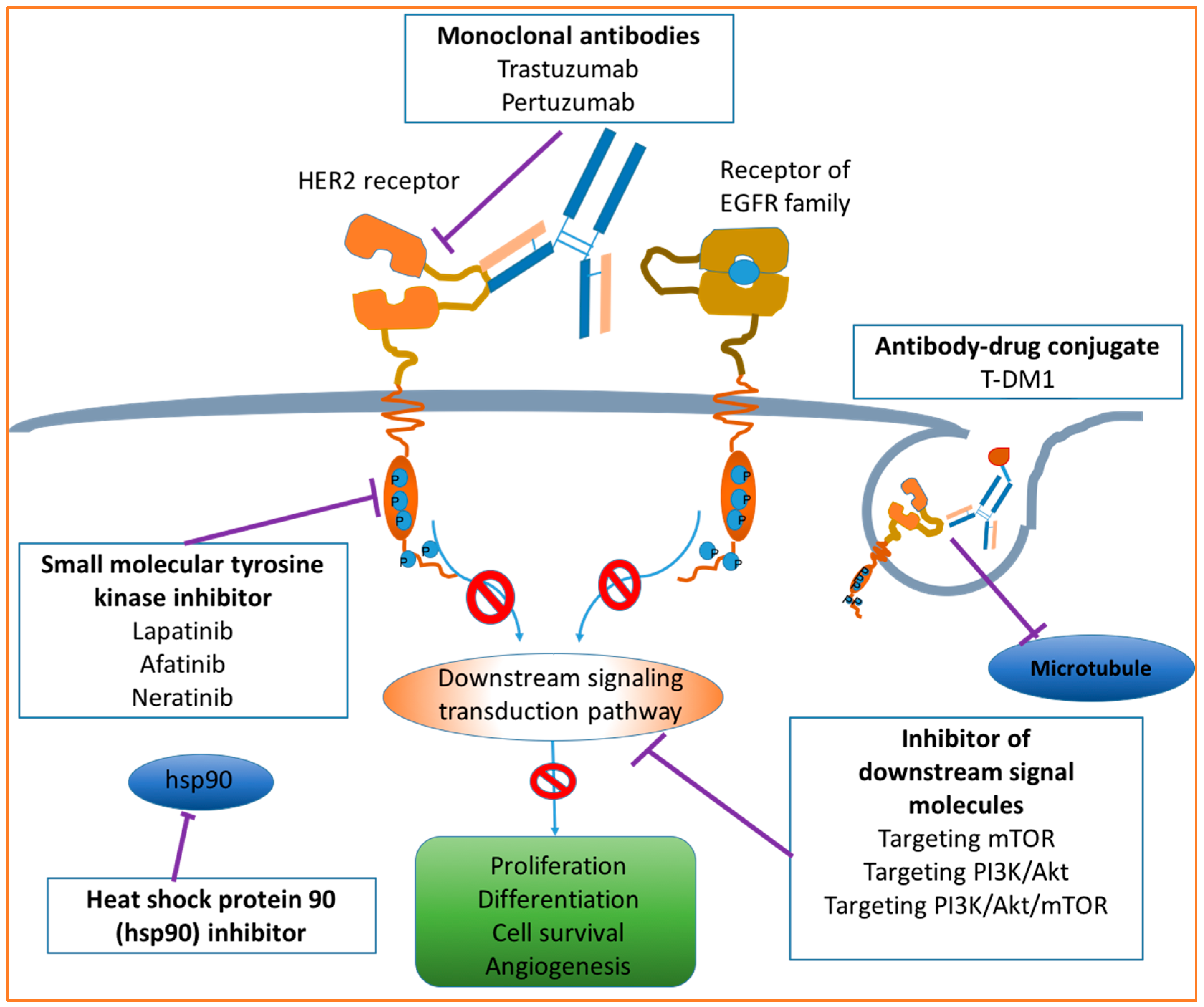
4. Drugs Targeting HER2
4.1. Monoclonal Antibodies
4.1.1. Trastuzumab
Mechanism of Action
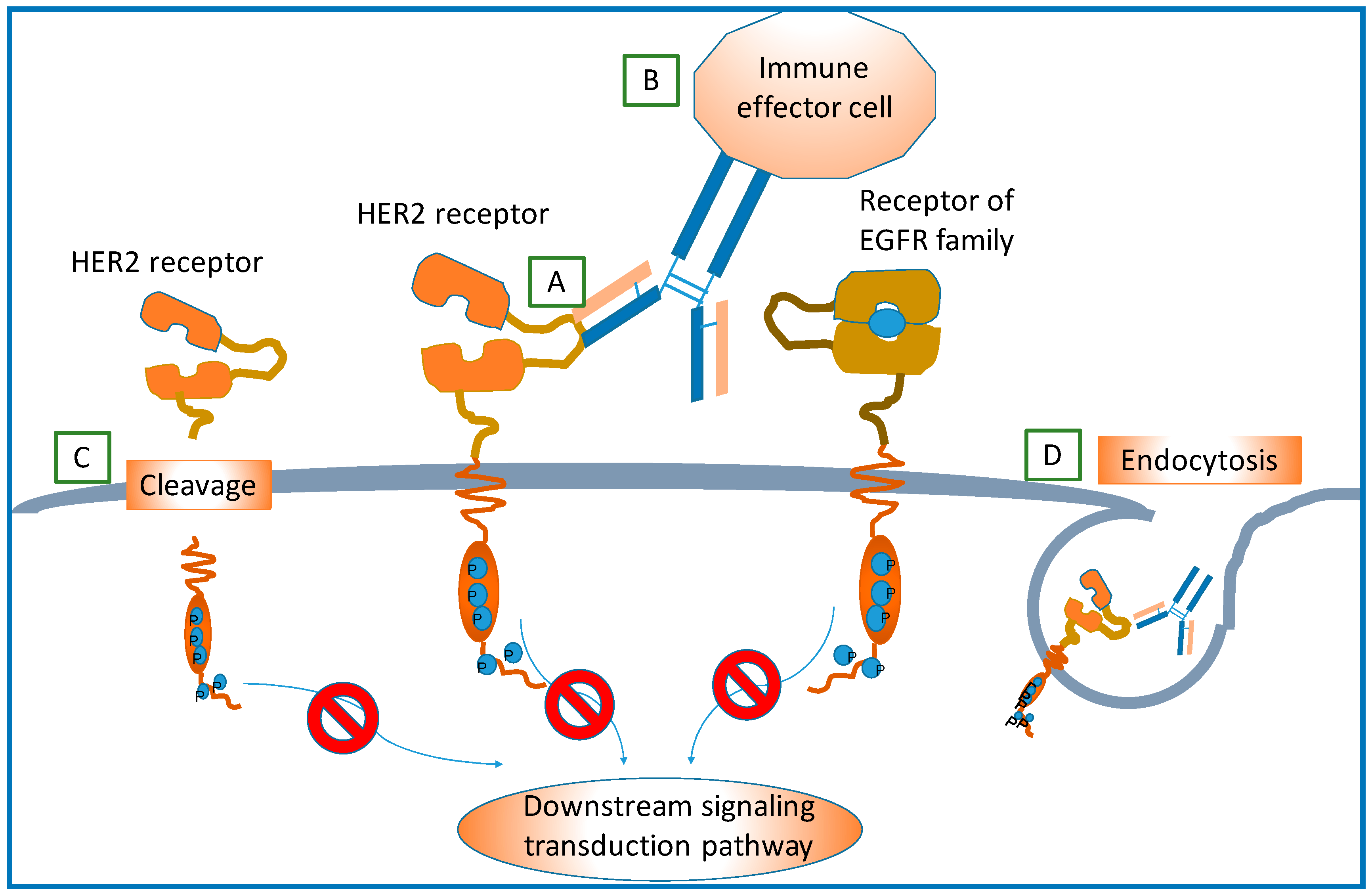
Mechanisms of Resistance
4.1.2. Pertuzumab
4.1.3. Antibody Drugs for HER2+ BC in Ongoing Clinical Trials
4.2. Small Molecule Tyrosine Kinase Inhibitors
4.2.1. Lapatinib
4.2.2. Afatinib
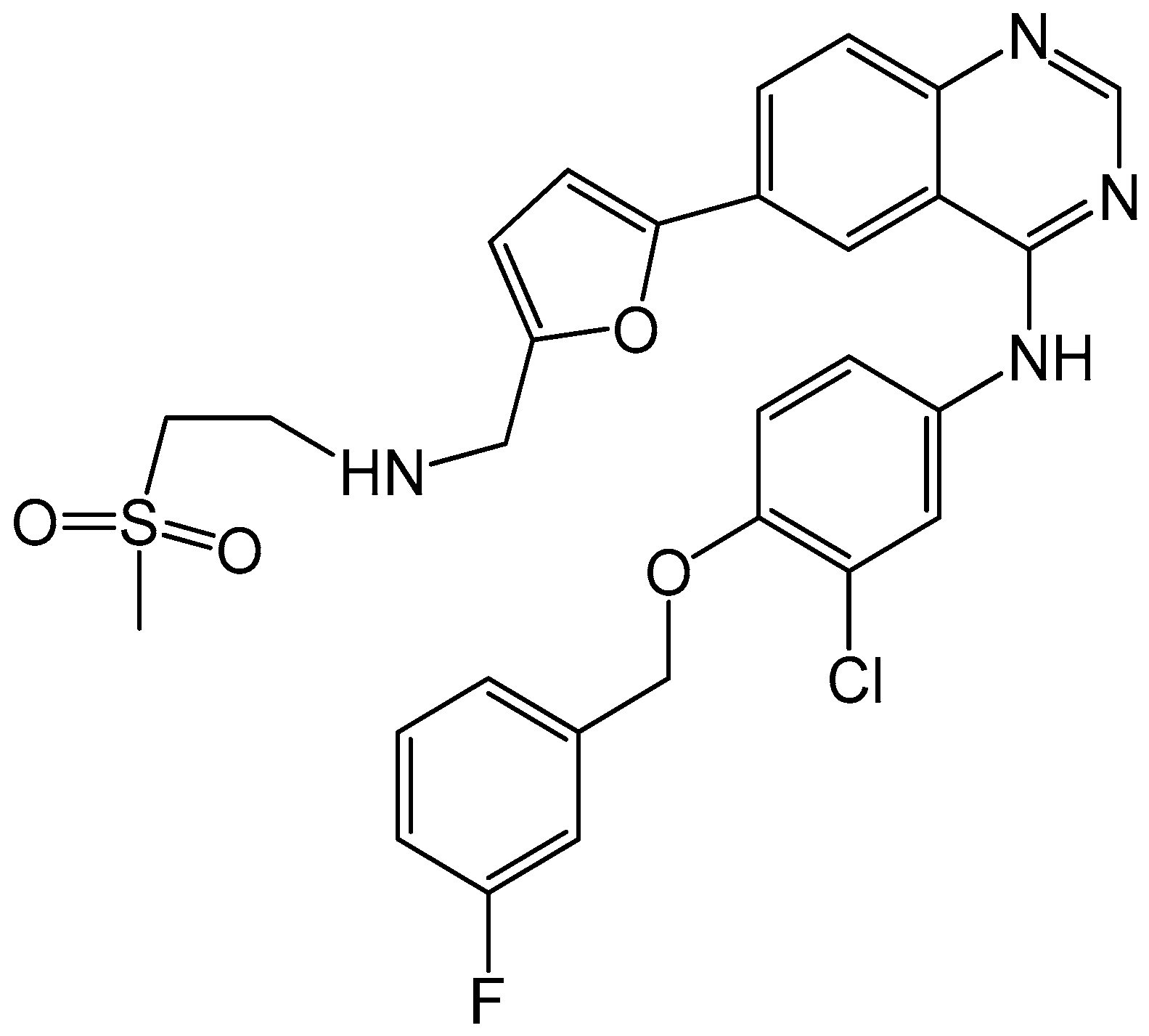
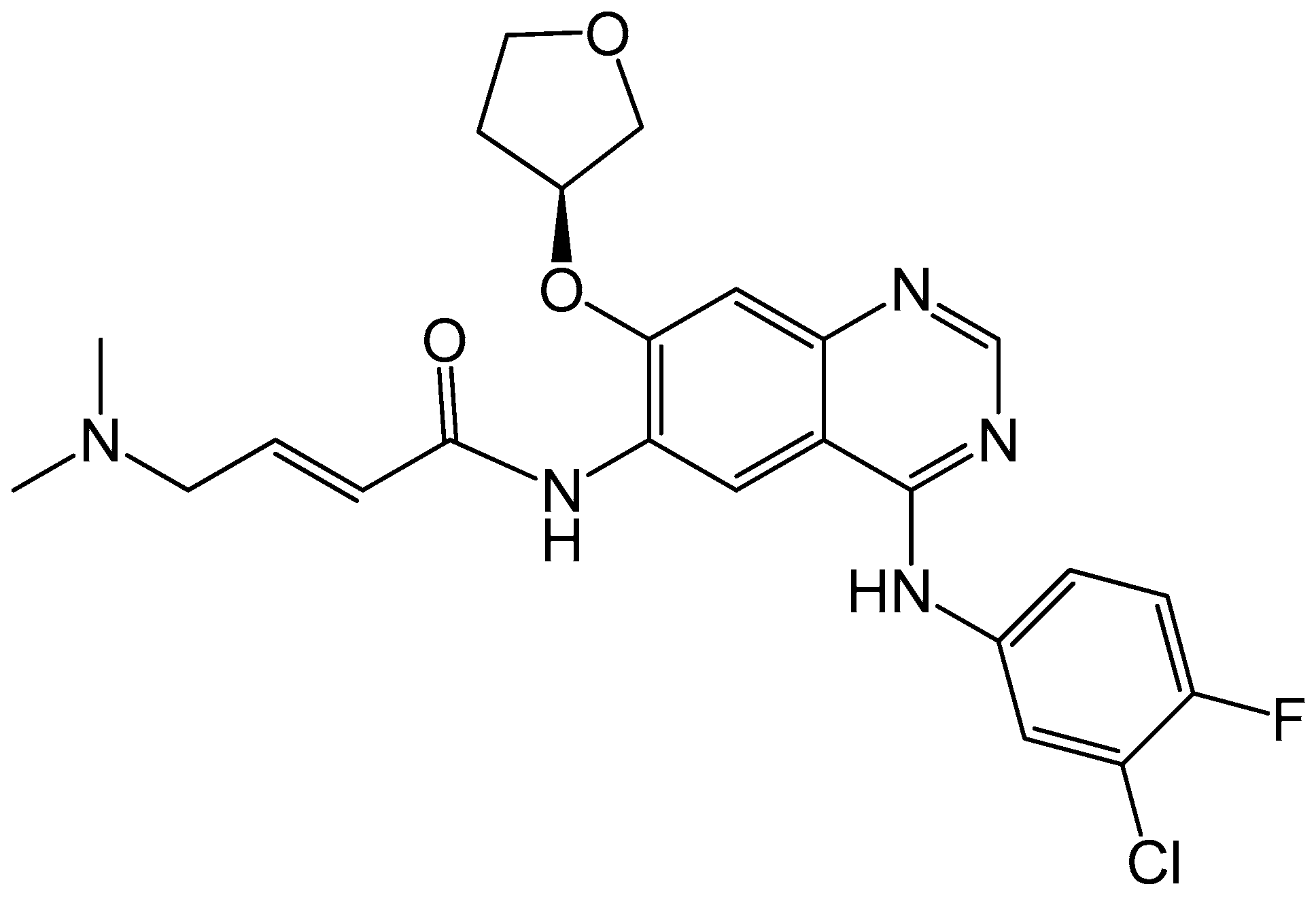
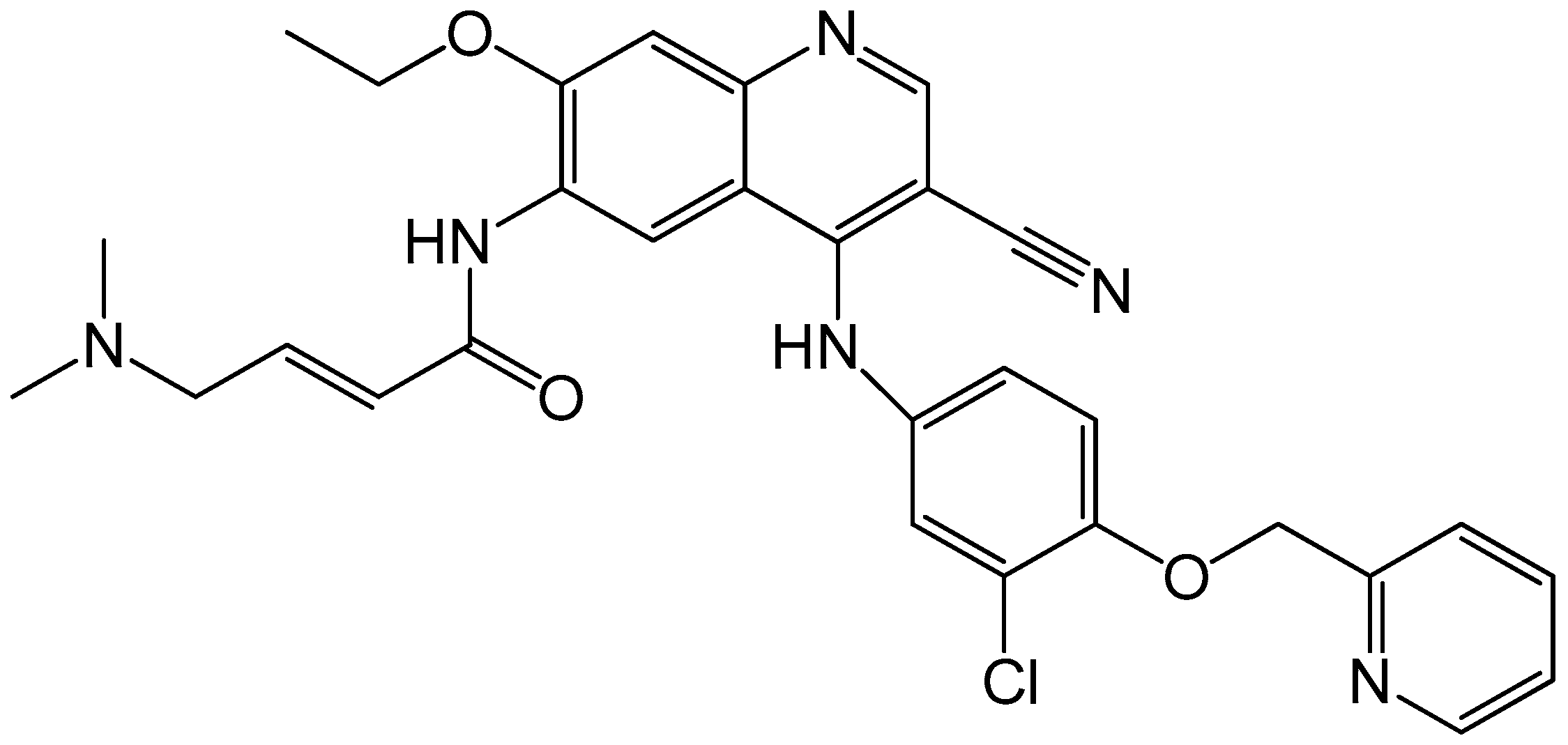
4.2.3. Neratinib
4.3. Antibody–Drug Conjugates
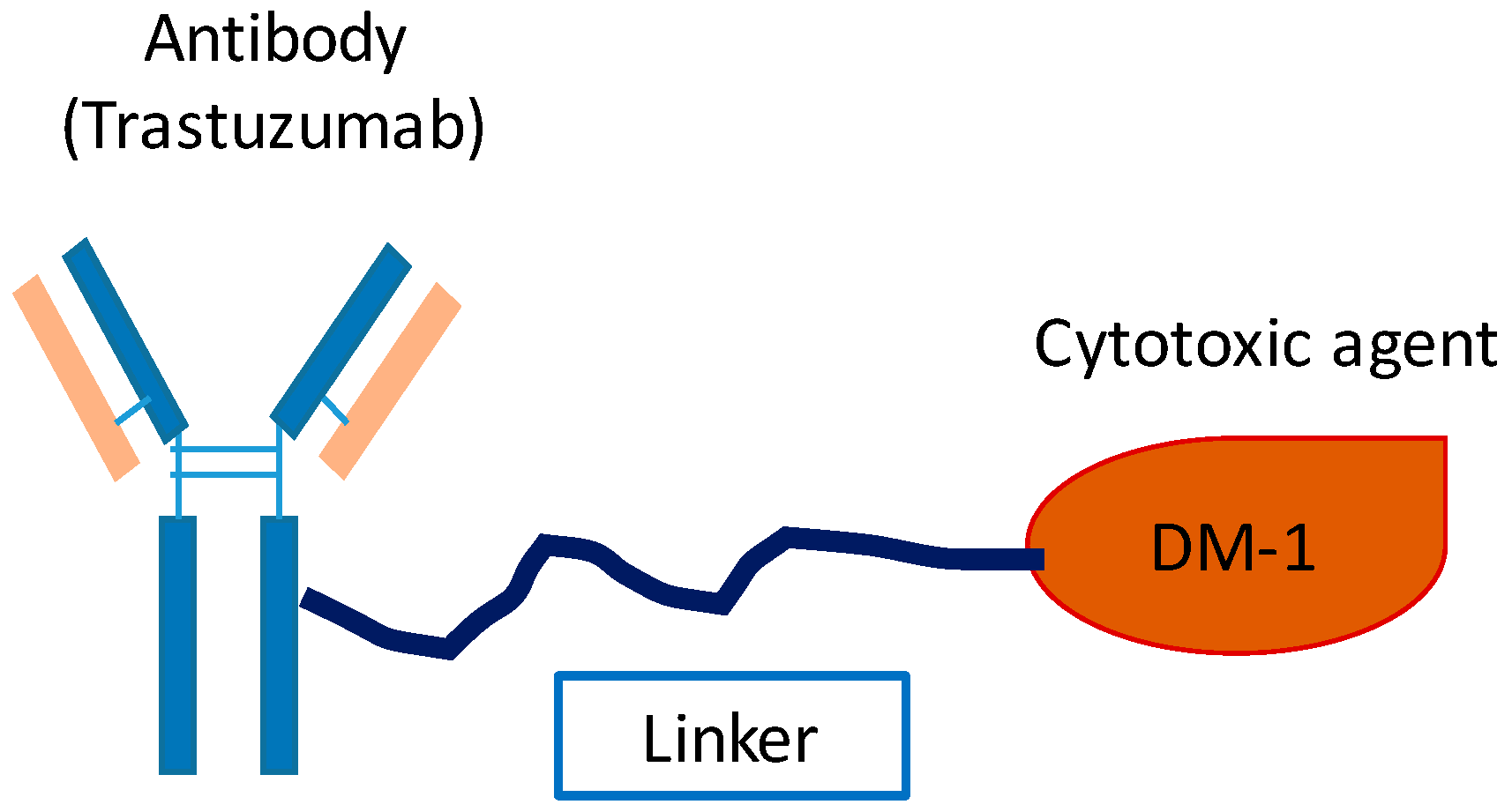
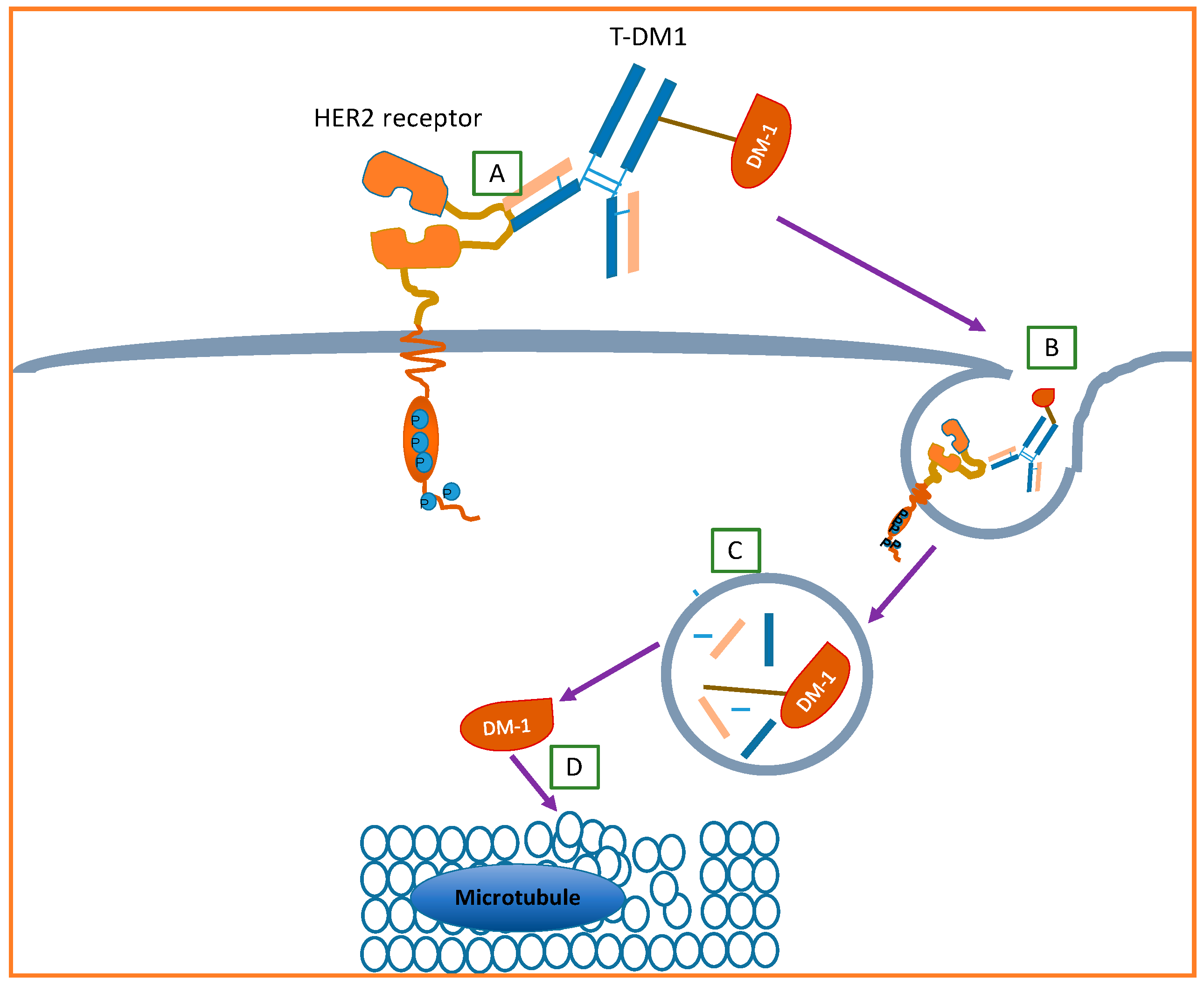
4.3.1. Ado–Trastuzumab Emtansine
4.3.2. A Novel Biparatopic ADC for HER2+ BC
4.4. Other Emerging HER2 Targeted Agents
4.5. Combination Therapy
5. Anti-HER2 Molecular Therapy for Breast Cancer with HER2 Somatic Mutations
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Centers for Disease Control and Prevention. Cancer Among Women. Available online: http://www.cdc.gov/cancer/dcpc/data/women.htm (accessed on 10 September 2016).
- Breastcancer.org. U.S. Breast Cancer Statistics. Available online: http://www.breastcancer.org/symptoms/understand_bc/statistics (accessed on 10 September 2016).
- Orphanos, G.; Kountourakis, P. Targeting the HER2 receptor in metastatic breast cancer. Hematol. Oncol. Stem Cell Ther. 2012, 5, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER−2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J. Studies of the HER−2/neu Proto-oncogene in Human Breast Cancer. Cancer Investig. 1990, 8, 253–254. [Google Scholar] [CrossRef]
- Wahler, J.; Suh, N. Targeting HER2 Positive Breast Cancer with Chemopreventive Agents. Curr. Pharmacol. Rep. 2015, 1, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Ménard, S.; Pupa, S.M.; Campiglio, M.; Tagliabue, E. Biologic and therapeutic role of HER2 in cancer. Oncogene 2003, 22, 6570–6578. [Google Scholar] [CrossRef] [PubMed]
- Wesola, M.; Jelen, M. A comparison of IHC and FISH cytogenetic methods in the evaluation of HER2 status in breast cancer. Adv. Clin. Exp. Med. 2015, 24, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of Chemotherapy Plus a Monoclonal Antibody against HER2 for Metastatic Breast Cancer That Overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J. Treatment of HER2-overexpressing breast cancer. Ann. Oncol. 2010, 21, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Hubalek, M.; Brunner, C.; Matthä, K.; Marth, C. Resistance to HER2-targeted therapy: Mechanisms of trastuzumab resistance and possible strategies to overcome unresponsiveness to treatment. Wien. Med. Wochenschr. 2010, 160, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Incorvati, J.A.; Shah, S.; Mu, Y.; Lu, J. Targeted therapy for HER2 positive breast cancer. J. Hematol. Oncol. 2013, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Tsang, R.Y.; Finn, R.S. Beyond trastuzumab: Novel therapeutic strategies in HER2-positive metastatic breast cancer. Br. J. Cancer 2012, 106, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Blanquisett, A.; Touya, D.; StrassER−Weippl, K.; Ruiz, R.; St. Louis, J.; Goss, P. Current and emerging therapies of HER2-positive metastatic breast cancer. Breast 2016, 29, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.; Mahato, R.; Cheng, K. The role of HER2 in cancer therapy and targeted drug delivery. J. Control. Release 2010, 146, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Moasser, M.M. The oncogene HER2: Its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 2007, 26, 6469–6487. [Google Scholar] [CrossRef] [PubMed]
- Graus-Porta, D.; Beerli, R.R.; Daly, J.M.; Hynes, N.E. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 1997, 16, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Tzahar, E.; Waterman, H.; Chen, X.; Levkowitz, G.; Karunagaran, D.; Lavi, S.; Ratzkin, B.J.; Yarden, Y. A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol. Cell. Biol. 1996, 16, 5276–5287. [Google Scholar] [CrossRef] [PubMed]
- Issekutz, A.C.; Quinn, P.J.; Lang, B.; Ramsey, S.; Huber, A.M.; Rowter, D.; Karkada, M.; Issekutz, T.B. Coexpression of chemokine receptors CCR5, CXCR3, and CCR4 and ligands for P- and E-selectin on T lymphocytes of patients with juvenile idiopathic arthritis. Arthritis Rheum. 2011, 63, 3467–3476. [Google Scholar] [CrossRef] [PubMed]
- Vijapurkar, U.; Kim, M.S.; Koland, J.G. Roles of mitogen-activated protein kinase and phosphoinositide 3′-kinase in ErbB2/ErbB3 coreceptor-mediated heregulin signaling. Exp. Cell Res. 2003, 284, 291–302. [Google Scholar] [CrossRef]
- Esteva, F.J.; Pusztai, L. Optimizing outcomes in HER2-positive breast cancer: The molecular rationale. Oncology 2005, 19, 5–16. [Google Scholar] [PubMed]
- Kanavos, P. The rising burden of cancer in the developing world. Ann. Oncol. 2006, 17, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Tzahar, E.; Yarden, Y. The ErbB-2/HER2 oncogenic receptor of adenocarcinomas: From orphanhood to multiple stromal ligands. Biochim. Biophys. Acta Rev. Cancer 1998, 1377, M25–M37. [Google Scholar] [CrossRef]
- Karunagaran, D.; Tzahar, E.; Beerli, R.R.; Chen, X.; Graus-Porta, D.; Ratzkin, B.J.; Seger, R.; Hynes, N.E.; Yarden, Y. ErbB-2 is a common auxiliary subunit of NDF and EGF receptors: Implications for breast cancer. EMBO J. 1996, 15, 254–264. [Google Scholar] [PubMed]
- Neve, R.M.; Lane, H.A.; Hynes, N.E. The role of overexpressed HER2 in transformation. Ann. Oncol. 2001, 12 (Suppl. S1), S9–S13. [Google Scholar] [CrossRef] [PubMed]
- Burstein, H.J. The Distinctive Nature of HER2-Positive Breast Cancers—NEJM. N. Engl. J. Med. 2005, 353, 1652–1654. [Google Scholar] [CrossRef] [PubMed]
- Venter, D.; Kumar, S.; Tuzi, N.; Gullick, W. Overexpression of the c-ErbB-2 oncoprotein in human breast carcinomas: Immunohistological assessment correlates with gene amplification. Lancet 1987, 330, 69–72. [Google Scholar] [CrossRef]
- Natali, P.G.; Nicotra, M.R.; Bigotti, A.; Venturo, I.; Slamon, D.J.; Fendly, B.M.; Ullrich, A. Expression of the p185 encoded by HER2 oncogene in normal and transformed human tissues. Int. J. Cancer 1990, 45, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Thor, B.A.D.; Liu, S.; Edgerton, S.; Ii, D.M.; Kasowitz, K.M.; Benz, C.C.; Stern, D.F.; Digiovanna, M.P. Activation (Tyrosine Phosphorylation) of ErbB-2 (HER-2/neu): A Study of Incidence and Correlation With Outcome in Breast Cancer. J. Clin. Oncol. 2016, 18, 3230–3239. [Google Scholar]
- Yu, D.; Hung, M.C. Overexpression of ErbB2 in cancer and ErbB2-targeting strategies. Oncogene 2000, 19, 6115–6121. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Yu, D. Molecular mechanisms of ErbB2-mediated breast cancer chemoresistance. Adv. Exp. Med. Biol. 2007, 608, 119–129. [Google Scholar] [PubMed]
- Sakai, K.; Mori, S.; Kawamoto, T.; Taniguchi, S.; Kobori, O.; Morioka, Y.; Kuroki, T.; Kano, K. Expression of epidermal growth factor receptors on normal human gastric epithelia and gastric carcinomas. J. Natl. Cancer Inst. 1986, 77, 1047–1052. [Google Scholar] [PubMed]
- Gomez-Martín, C.; Lopez-Rios, F.; Aparicio, J.; Barriuso, J.; García-Carbonero, R.; Pazo, R.; Rivera, F.; Salgado, M.; Salud, A.; Vázquez-Sequeiros, E.; et al. A critical review of HER2-positive gastric cancer evaluation and treatment: From trastuzumab, and beyond. Cancer Lett. 2014, 351, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Rü Schoff, J.; Hanna, W.; Bilous, M.; Hofmann, M.; Osamura, R.Y.; Penault-Llorca, F.; Van De Vijver, M.; Viale, G. HER2 testing in gastric cancer: A practical approach. Mod. Pathol. 2012, 25, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Gravalos, C.; Jimeno, A. HER2 in gastric cancer: A new prognostic factor and a novel therapeutic target. Ann. Oncol. 2008, 19, 1523–1529. [Google Scholar] [CrossRef] [PubMed]
- Chua, T.C.; Merrett, N.D. Clinicopathologic factors associated with HER2-positive gastric cancer and its impact on survival outcomes—A systematic review. Int. J. Cancer 2012, 130, 2845–2856. [Google Scholar] [CrossRef] [PubMed]
- Uchino, S.; Tsuda, H.; Maruyama, K. Overexpression of c-erB-2 Protein in Gastric Cancer. Its Correlation with Long-term Survival of Patients. Cancer 1993, 72, 3179–3184. [Google Scholar] [CrossRef]
- Pils, D.; Pinter, A.; Reibenwein, J.; Alfanz, A.; Horak, P.; Schmid, B.C.; Hefler, L.; Horvat, R.; Reinthaller, A.; Zeillinger, R.; et al. In ovarian cancer the prognostic influence of HER2/neu is not dependent on the CXCR4/SDF-1 signalling pathway. Br. J. Cancer 2007, 96, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Bookman, M.A.; Darcy, K.M.; Clarke-Pearson, D.; Boothby, R.A.; Horowitz, I.R. Evaluation of monoclonal humanized anti-HER2 antibody, trastuzumab, in patients with recurrent or refractory ovarian or primary peritoneal carcinoma with overexpression of HER2: A phase II trial of the Gynecologic Oncology Group. J. Clin. Oncol. 2003, 21, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Steffensen, K.D.; Waldstrøm, M.; Jeppesen, U.; Jakobsen, E.; Brandslund, I.; Jakobsen, A. The prognostic importance of cyclooxygenase 2 and HER2 expression in epithelial ovarian cancer. Int. J. Gynecol. Cancer 2007, 17, 798–807. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I. HER2 in prostate cancer—A viable target or innocent bystander? J. Natl. Cancer Inst. 2000, 92, 1866–1868. [Google Scholar] [CrossRef] [PubMed]
- Signoretti, S.; Montironi, R.; Manola, J.; Altimari, A.; Tam, C.; Bubley, G.; Balk, S.; Thomas, G.; Kaplan, I.; Hlatky, L.; et al. HER−2-neu expression and progression toward androgen independence in human prostate cancer. J. Natl. Cancer Inst. 2000, 92, 1918–1925. [Google Scholar] [CrossRef] [PubMed]
- Bilous, M.; Dowsett, M.; Hanna, W.; Isola, J.; Lebeau, A.; Moreno, A.; Penault-Llorca, F.; Rüschoff, J.; Tomasic, G.; van de Vijver, M. Current perspectives on HER2 testing: A review of national testing guidelines. Mod. Pathol. 2003, 16, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Sáez, A.; Andreu, F.J.; Seguí, M.A.; Baré, M.L.; Fernández, S.; Dinarés, C.; Rey, M. HER-2 gene amplification by chromogenic in situ hybridisation (CISH) compared with fluorescence in situ hybridisation (FISH) in breast cancer−A study of two hundred cases. Breast 2006, 15, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Dong, Y.; Yoshimoto, N.; Asano, T.; Hato, Y.; Yamashita, H.; Sato, S.; Takahashi, S.; Fujii, Y.; Toyama, T. HER2 mutation status in Japanese HER2-negative breast cancer patients. Jpn. J. Clin. Oncol. 2014, 44, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Moelans, C.B.; de Weger, R.A.; van der Wall, E.; van Diest, P.J. Current technologies for HER2 testing in breast cancer. Crit. Rev. Oncol. Hematol. 2011, 80, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Hicks, D.G.; Tubbs, R.R. Assessment of the HER2 status in breast cancer by fluorescence in situ hybridization: A technical review with interpretive guidelines. Hum. Pathol. 2005, 36, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.S.; Fletcher, J.A.; Linette, G.P.; Stec, J.; Clark, E.; Ayers, M.; Symmans, W.F.; Pusztai, L.; Bloom, K.J. The HER−2/neu gene and protein in breast cancer 2003: Biomarker and target of therapy. Oncologist 2003, 8, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Nitta, H.; Kelly, B.D.; Allred, C.; Jewell, S.; Banks, P.; Dennis, E.; Grogan, T.M. The assessment of HER2 status in breast cancer: The past, the present, and the future. Pathol. Int. 2016, 66, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Hicks, D.G.; Kulkarni, S. HER2+ breast cancer: Review of biologic relevance and optimal use of diagnostic tools. Am. J. Clin. Pathol. 2008, 129, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Dowsett, M.; Cooke, T.; Ellis, I.; Gusterson, B.; Mallon, E.; Walker, R. Assessment of HER2 status in breast cancer: Why, when and how? Eur. J. Cancer 2000, 36, 170–176. [Google Scholar] [CrossRef]
- Krishnamurti, U. HER2 in Breast Cancer: A review and update. Adv. Anat. Pathol. 2014, 21, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Wolff, A.C.; Hammond, M.E.H.; Hicks, D.G.; Dowsett, M.; Mcshane, L.M.; Allison, K.H.; Allred, D.C.; Bartlett, J.M.S.; Bilous, M.; Fitzgibbons, P.; et al. Recommendations for Human Epidermal Growth Factor Receptor 2 Testing in Breast Cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Update. J. Clin. Oncol. 2013, 31, 3997–4014. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.L.; Cobleigh, M.A.; Tripathy, D.; Gutheil, J.C.; Harris, L.N.; Fehrenbacher, L.; Slamon, D.J.; Murphy, M.; Novotny, W.F.; Burchmore, M.; et al. Efficacy and Safety of Trastuzumab as a Single Agent in First-Line Treatment of HER2-Overexpressing Metastatic Breast Cancer. J. Clin. Oncol. 2003, 20, 719–726. [Google Scholar] [CrossRef]
- Buzdar, A.U.; Ibrahim, N.K.; Francis, D.; Booser, D.J.; Thomas, E.S.; Theriault, R.L.; Pusztai, L.; Green, M.C.; Arun, B.K.; Giordano, S.H.; et al. Significantly higher pathologic complete remission rate after neoadjuvant therapy with trastuzumab, paclitaxel, and epirubicin chemotherapy: Results of a randomized trial in human epidermal growth factor receptor 2-positive operable breast cancer. J. Clin. Oncol. 2005, 23, 3676–3685. [Google Scholar] [CrossRef] [PubMed]
- Dahabreh, I.J.; Linardou, H.; Siannis, F.; Fountzilas, G.; Murray, S. Trastuzumab in the adjuvant treatment of early-stage breast cancer: A systematic review and meta-analysis of randomized controlled trials. Oncologist 2008, 13, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Fishbane, S.; Schiller, B.; Locatelli, F.; Covic, A.C.; Provenzano, R.; Wiecek, A.; Levin, N.W.; Kaplan, M.; Macdougall, I.C.; Francisco, C.; et al. Peginesatide in patients with anemia undergoing hemodialysis. N. Engl. J. Med. 2013, 368, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Hudis, C.A. Trastuzumab—Mechanism of Action and Use in Clinical Practice. N. Engl. J. Med. 2007, 357, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.; Claret, F.X. Trastuzumab: Updated mechanisms of action and resistance in breast cancer. Front. Oncol. 2012, 2, 62. [Google Scholar] [CrossRef] [PubMed]
- Valabrega, G.; Montemurro, F.; Aglietta, M. Trastuzumab: Mechanism of action, resistance and future perspectives in HER2-overexpressing breast cancer. Ann. Oncol. 2007, 18, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.L.A.; Lee, S.-C. Mechanisms of Resistance to Trastuzumab and Novel Therapeutic Strategies in HER2-Positive Breast Cancer. Int. J. Breast Cancer 2012, 2012, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, J. Mechanisms of resistance to trastuzumab: An updated review. Chin. Ger. J. Clin. Oncol. 2010, 9, 660–665. [Google Scholar] [CrossRef]
- Rexer, B.N.; Arteaga, C.L. Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: Mechanisms and clinical implications. Crit. Rev. Oncog. 2012, 17, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Nady, P.; Friedlander, E.; Tanner, M.; Kapanen, A.I.; Carraway, K.L.; Isola, J.; Jovin, T.M. Decreased availability and lack of activation of ErbB2 in JIMT-1; a herceptin-resistant, MUC4-overexpressing breast cancer cell line. Cancer Res. 2005, 65, 473–482. [Google Scholar]
- Nahta, R. Molecular Mechanisms of Trastuzumab-Based Treatment in HER2-Overexpressing Breast Cancer. ISRN Oncol. 2012, 2012, 428062. [Google Scholar] [CrossRef] [PubMed]
- Gagliato, D.D.M.; Jardim, D.L.F.; Marchesi, M.S.P.; Hortobagyi, G.N. Mechanisms of resistance and sensitivity to anti-HER2 therapies in HER2+ breast cancer. Oncotarget 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Wilks, S.T. Potential of overcoming resistance to HER2-targeted therapies through the PI3K/Akt/mTOR pathway. Breast 2015, 24, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Lan, K.H.; Zhou, X.; Tan, M.; Esteva, F.J.; Sahin, A.A.; Klos, K.S.; Li, P.; Monia, B.P.; Nguyen, N.T.; et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 2004, 6, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Saez, R.; Molina, M.A.; Ramsey, E.E.; Rojo, F.; Keenan, E.J.; Albanell, J.; Lluch, A.; Garcia-Conde, J.; Baselga, J.; Clinton, G.M. p95HER-2 predicts worse outcome in patients with HER-2-positive breast cancer. Clin. Cancer Res. 2006, 12, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Valabrega, G.; Montemurro, F.; Sarotto, I.; Petrelli, A.; Rubini, P.; Tacchetti, C.; Aglietta, M.; Comoglio, P.M.; Giordano, S. TGFα expression impairs Trastuzumab-induced HER2 downregulation. Oncogene 2005, 24, 3002–3010. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zi, X.; Zhao, Y.; Mascarenhas, D.; Pollak, M. Insulin-like growth factor-I receptor signaling and resistance to Trastuzumab (Herceptin). J. Natl. Cancer Inst. 2001, 93, 1852–1857. [Google Scholar] [CrossRef] [PubMed]
- Franklin, M.C.; Carey, K.D.; Vajdos, F.F.; Leahy, D.J.; De Vos, A.M.; Sliwkowski, M.X. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell 2004, 5, 317–328. [Google Scholar] [CrossRef]
- Aspenstro, P.; Na, I.; Lee, V.M.; Kypta, R.; Gumbiner, B.M. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 2003, 421, 756–760. [Google Scholar]
- Agus, D.B.; Gordon, M.S.; Taylor, C.; Natale, R.B.; Karlan, B.; Mendelson, D.S.; Press, M.F.; Allison, D.E.; Sliwkowski, M.X.; Lieberman, G.; et al. Phase I clinical study of pertuzumab, a novel HER dimerization inhibitor, in patients with advanced cancer. J. Clin. Oncol. 2005, 23, 2534–2543. [Google Scholar] [CrossRef] [PubMed]
- Hubalek, M.; Brantner, C.; Marth, C. Role of pertuzumab in the treatment of HER2-positive breast cancer. Breast Cancer Targets Ther. 2012, 4, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Baselga, J.; Kim, S.-B.; Ro, J.; Semiglazov, V.; Campone, M.; Ciruelos, E.; Ferrero, J.-M.; Schneeweiss, A.; Heeson, S.; et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N. Engl. J. Med. 2015, 372, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Vahdat, L.T. Novel data in metastatic breast cancer. Clin. Adv. Hematol. Oncol. 2013, 11, 13–15. [Google Scholar] [PubMed]
- Tripathy, D.; Pegram, M.D.; Stanford, C.A. Recent Developments in the Treatment of HER2-Positive Breast Cancer. Am. J. Hematol. Oncol. 2016, 12, 26–32. [Google Scholar]
- Richards, D.; Braiteh, F.; Anthony, S.; Edenfield, W.; Hellerstedt, B.; Raju, R.; Conkling, P. A Phase 1 Study of MM-111; a Bispecific HER2/HER3 Antibody Fusion Protein, Combined with Multiple Treatment Regimens in Patients with Advanced HER2 Positive Solid Tumors. Ann. Oncol. 2012, 23, 170. [Google Scholar]
- McDonagh, C.F.; Huhalov, A.; Harms, B.D.; Adams, S.; Paragas, V.; Oyama, S.; Zhang, B.; Luus, L.; Overland, R.; Nguyen, S.; et al. Antitumor Activity of a Novel Bispecific Antibody That Targets the ErbB2/ErbB3 Oncogenic Unit and Inhibits Heregulin-Induced Activation of ErbB3. Mol. Cancer Ther. 2012, 11, 582–593. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.R.; Leary, A. Lapatinib: A novel EGFR/HER2 tyrosine kinase inhibitor for cancer. Drugs Today 2006, 42, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Nahta, R.; Yu, D.; Hung, M.-C.; Hortobagyi, G.N.; Esteva, F.J. Mechanisms of Disease: Understanding resistance to HER2-targeted therapy in human breast cancer. Nat. Clin. Pract. Oncol. 2006, 3, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Wood, E.R. A Unique Structure for Epidermal Growth Factor Receptor Bound to GW572016 (Lapatinib): Relationships among Protein Conformation, Inhibitor Off-Rate, and Receptor Activity in Tumor Cells. Cancer Res. 2004, 64, 6652–6659. [Google Scholar] [CrossRef] [PubMed]
- Gomez, H.L.; Doval, D.C.; Chavez, M.A.; Ang, P.C.-S.; Aziz, Z.; Nag, S.; Ng, C.; Franco, S.X.; Chow, L.W.C.; Arbushites, M.C.; et al. Efficacy and safety of lapatinib as first-line therapy for ErbB2-amplified locally advanced or metastatic breast cancer. J. Clin. Oncol. 2008, 26, 2999–3005. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Mullin, R.J.; Keith, B.R.; Liu, L.-H.; Ma, H.; Rusnak, D.W.; Owens, G.; Alligood, K.J.; Spector, N.L. Anti-tumor activity of GW572016: A dual tyrosine kinase inhibitor blocks EGF activation of EGFR/ErbB2 and downstream Erk1/2 and AKT pathways. Oncogene 2002, 21, 6255–6263. [Google Scholar] [CrossRef] [PubMed]
- Konecny, G.E.; Pegram, M.D.; Venkatesan, N.; Finn, R.; Yang, G.; Rahmeh, M.; Untch, M.; Rusnak, D.W.; Spehar, G.; Mullin, R.J.; et al. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER−2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 2006, 66, 1630–1639. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Gerard, C.M.; Liu, L.; Baudson, N.M.; Ory, T.L.; Spector, N.L. Combining lapatinib (GW572016), a small molecule inhibitor of ErbB1 and ErbB2 tyrosine kinases, with therapeutic anti-ErbB2 antibodies enhances apoptosis of ErbB2-overexpressing breast cancer cells. Oncogene 2005, 24, 6213–6221. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.U.; Guo, H.; Yap, J.T.; Mayer, I.A.; Falkson, C.I.; Hobday, T.J.; Dees, E.C.; Richardson, A.L.; Nanda, R.; Rimawi, M.F.; et al. Phase II Study of Lapatinib in Combination with Trastuzumab in Patients with Human Epidermal Growth Factor Receptor 2-Positive Metastatic Breast Cancer: Clinical Outcomes and Predictive Value of Early [18F]Fluorodeoxyglucose Positron Emission Tomography I. J. Clin. Oncol. 2015, 33, 2623–2631. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.U.; Winer, E.P.; Wheatley, D.; Carey, L.A.; Houston, S.; Mendelson, D.; Munster, P.; Frakes, L.; Kelly, S.; Garcia, A.A.; et al. A phase II study of afatinib (BIBW 2992), an irreversible ErbB family blocker, in patients with HER2-positive metastatic breast cancer progressing after trastuzumab. Breast Cancer Res. Treat. 2012, 133, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Burstein, H.J.; Sun, Y.; Dirix, L.Y.; Jiang, Z.; Paridaens, R.; Tan, A.R.; Awada, A.; Ranade, A.; Jiao, S.; Schwartz, G.; et al. Neratinib, an irreversible ErbB receptor tyrosine kinase inhibitor, in patients with advanced ErbB2-positive breast cancer. J. Clin. Oncol. 2010, 28, 1301–1307. [Google Scholar] [CrossRef] [PubMed]
- Amiri-Kordestani, L.; Blumenthal, G.M.; Xu, Q.C.; Zhang, L.; Tang, S.W.; Ha, L.; Weinberg, W.C.; Chi, B.; Candau-Chacon, R.; Hughes, P.; et al. FDA Approval: Ado-trastuzumab Emtansine for the Treatment of Patients with HER2-Positive Metastatic Breast Cancer. Clin. Cancer Res. 2014, 20, 4436–4442. [Google Scholar] [CrossRef] [PubMed]
- Esteva, F.J.; Miller, K.D.; Teicher, B.A. What Can We Learn about Antibody-Drug Conjugates from the T-DM1 Experience? In 2015 Educational Book; ASCO: Alexandria, VA, USA, 2015; pp. 117–125. [Google Scholar]
- Martínez, M.T.; Pérez-Fidalgo, J.A.; Martín-Martorell, P.; Cejalvo, J.M.; Pons, V.; Bermejo, B.; Martín, M.; Albanell, J.; Lluch, A. Treatment of HER2 positive advanced breast cancer with T-DM1: A review of the literature. Crit. Rev. Oncol. Hematol. 2016, 97, 96–106. [Google Scholar]
- Lewis Phillips, G.D.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blättler, W.A.; Lambert, J.M.; Chari, R.V.J.; Lutz, R.J.; et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar]
- Li, J.Y.; Perry, S.R.; Muniz-Medina, V.; Wang, X.; Wetzel, L.K.; Rebelatto, M.C.; Hinrichs, M.J.M.; Bezabeh, B.Z.; Fleming, R.L.; Dimasi, N.; et al. A Biparatopic HER2-Targeting Antibody-Drug Conjugate Induces Tumor Regression in Primary Models Refractory to or Ineligible for HER2-Targeted Therapy. Cancer Cell 2016, 29, 117–129. [Google Scholar]
- Solit, D.B.; Zheng, F.F.; Drobnjak, M.; Mu, P.N.; Higgins, B.; Verbel, D.; Heller, G.; Tong, W.; Cordon-cardo, C.; Agus, D.B.; et al. 17-Allylamino-17-demethoxygeldanamycin Induces the Degradation of Androgen Receptor and HER−2/neu and Inhibits the Growth of Prostate Cancer Xenografts Advances in Brief 17-Allylamino-17-demethoxygeldanamycin Induces the Degradation of Androgen Receptor. Clin. Cancer Res. 2002, 8, 986–993. [Google Scholar]
- Pick, E.; Kluger, Y.; Giltnane, J.M.; Moeder, C.; Camp, R.L.; Rimm, D.L.; Kluger, H.M. High HSP90 expression is associated with decreased survival in breast cancer. Cancer Res. 2007, 67, 2932–2937. [Google Scholar]
- Press, D. HER2 breast cancer therapies: A review. Biol. Targets Ther. 2009, 289–301. [Google Scholar]
- Huszno, J.; Nowara, E. Current therapeutic strategies of anti-HER2 treatment in advanced breast cancer patients. Współczesna Onkol. 2016, 1, 1–7. [Google Scholar]
- Munagala, R.; Aqil, F.; Gupta, R.C. Promising molecular targeted therapies in breast cancer. Indian J. Pharmacol. 2011, 43, 236–245. [Google Scholar] [PubMed]
- Modi, S.; Stopeck, A.; Linden, H.; Solit, D.; Chandarlapaty, S.; Rosen, N.; D’Andrea, G.; Dickler, M.; Moynahan, M.E.; Sugarman, S.; et al. HSP90 inhibition is effective in breast cancer: A phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin. Cancer Res. 2011, 17, 5132–5139. [Google Scholar] [PubMed]
- Modi, S.; Stopeck, A.T.; Gordon, M.S.; Mendelson, D.; Solit, D.B.; Bagatell, R.; Ma, W.; Wheler, J.; Rosen, N.; Norton, L.; et al. Combination of trastuzumab and tanespimycin (17-AAG, KOS-953) is safe and active in trastuzumab-refractory HER-2-overexpressing breast cancer: A phase I dose-escalation study. J. Clin. Oncol. 2007, 25, 5410–5417. [Google Scholar] [PubMed]
- Nielsen, D.L.; Kümler, I.; Palshof, J.A.E.; Andersson, M. Efficacy of HER2-targeted therapy in metastatic breast cancer. Monoclonal antibodies and tyrosine kinase inhibitors. Breast 2013, 22, 1–12. [Google Scholar] [PubMed]
- Arteaga, C.L.; Sliwkowski, M.X.; Osborne, C.K.; Perez, E.A.; Puglisi, F.; Gianni, L. Treatment of HER2-positive breast cancer: Current status and future perspectives. Nat. Rev. Clin. Oncol. 2011, 9, 16–32. [Google Scholar] [PubMed]
- Cameron, D.A.; Stein, S. Drug Insight: Intracellular inhibitors of HER2—Clinical development of lapatinib in breast cancer. Nat. Clin. Pract. Oncol. 2008, 5, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, K.L.; Burstein, H.J.; Storniolo, A.M.; Rugo, H.; Sledge, G.; Koehler, M.; Ellis, C.; Casey, M.; Vukelja, S.; Bischoff, J.; et al. Randomized study of lapatinib alone or in combination with trastuzumab in women with ErbB2-positive, trastuzumab-refractory metastatic breast cancer. J. Clin. Oncol. 2010, 28, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Bradbury, I.; Eidtmann, H.; Di Cosimo, S.; De Azambuja, E.; Aura, C.; Gómez, H.; Dinh, P.; Fauria, K.; Van Dooren, V.; et al. Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): A randomised, open-label, multicentre, phase 3 trial. Lancet 2012, 379, 633–640. [Google Scholar] [CrossRef]
- Zabransky, D.J.; Yankaskas, C.L.; Cochran, R.L.; Wong, H.Y.; Croessmann, S.; Chu, D.; Kavuri, S.M.; Red Brewer, M.; Rosen, D.M.; Dalton, W.B.; et al. HER2 missense mutations have distinct effects on oncogenic signaling and migration. Proc. Natl. Acad. Sci. USA 2015, 112, E6205–E6214. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Reis-Filho, J.S. Activating mutations in HER2: Neu opportunities and Neu challenges. Cancer Discov. 2013, 3, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Chen, W.S.; Xiao, N.; Bender, R.; Ghazalpour, A.; Tan, Z.; Swensen, J.; Millis, S.Z.; Basu, G.; Gatalica, Z.; et al. Mutations in the kinase domain of the HER2/ErbB2 gene identified in a wide variety of human cancers. J. Mol. Diagn. 2015, 17, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Acharyya, S. Activating mutations and senescence secretome: New insights into HER2 activation, drug sensitivity and metastatic progression. Breast Cancer Res. 2013, 15, 309. [Google Scholar] [CrossRef] [PubMed]
- Bose, R.; Kavuri, S.M.; Searleman, A.C.; Shen, W.; Shen, D.; Koboldt, D.C.; Monsey, J.; Goel, N.; Aronson, A.B.; Li, S.; et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013, 3, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Kourie, H.R.; Chaix, M.; Gombos, A.; Aftimos, P.; Awada, A. Pharmacodynamics, pharmacokinetics and clinical efficacy of neratinib in HER2-positive breast cancer and breast cancer with HER2 mutations. Expert Opin. Drug Metab. Toxicol. 2016, 12, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Boulbes, D.R.; Arold, S.T.; Chauhan, G.B.; Blachno, K.V.; Deng, N.; Chang, W.C.; Jin, Q.; Huang, T.H.; Hsu, J.M.; Brady, S.W.; et al. HER family kinase domain mutations promote tumor progression and can predict response to treatment in human breast cancer. Mol. Oncol. 2015, 9, 586–600. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IHC Score | Represent | Numbers of HER2 Receptors | Staining Pattern | Percentage of Cells Stained |
|---|---|---|---|---|
| 0 | Negative | <20,000 | No staining | 0 |
| 1+ | Negative | about 100,000 | Faint incomplete staining | <10% |
| 2+ | Weak positive | about 500,000 | Light to moderate complete staining | >10% |
| 3+ | Strong positive | about 2,300,000 | Strong complete staining | >30% |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lv, Q.; Meng, Z.; Yu, Y.; Jiang, F.; Guan, D.; Liang, C.; Zhou, J.; Lu, A.; Zhang, G. Molecular Mechanisms and Translational Therapies for Human Epidermal Receptor 2 Positive Breast Cancer. Int. J. Mol. Sci. 2016, 17, 2095. https://doi.org/10.3390/ijms17122095
Lv Q, Meng Z, Yu Y, Jiang F, Guan D, Liang C, Zhou J, Lu A, Zhang G. Molecular Mechanisms and Translational Therapies for Human Epidermal Receptor 2 Positive Breast Cancer. International Journal of Molecular Sciences. 2016; 17(12):2095. https://doi.org/10.3390/ijms17122095
Chicago/Turabian StyleLv, Quanxia, Ziyuan Meng, Yuanyuan Yu, Feng Jiang, Daogang Guan, Chao Liang, Junwei Zhou, Aiping Lu, and Ge Zhang. 2016. "Molecular Mechanisms and Translational Therapies for Human Epidermal Receptor 2 Positive Breast Cancer" International Journal of Molecular Sciences 17, no. 12: 2095. https://doi.org/10.3390/ijms17122095
APA StyleLv, Q., Meng, Z., Yu, Y., Jiang, F., Guan, D., Liang, C., Zhou, J., Lu, A., & Zhang, G. (2016). Molecular Mechanisms and Translational Therapies for Human Epidermal Receptor 2 Positive Breast Cancer. International Journal of Molecular Sciences, 17(12), 2095. https://doi.org/10.3390/ijms17122095