
Recent Progress in Machine Learning-Based Methods for Protein Fold Recognition



Abstract
:1. Introduction
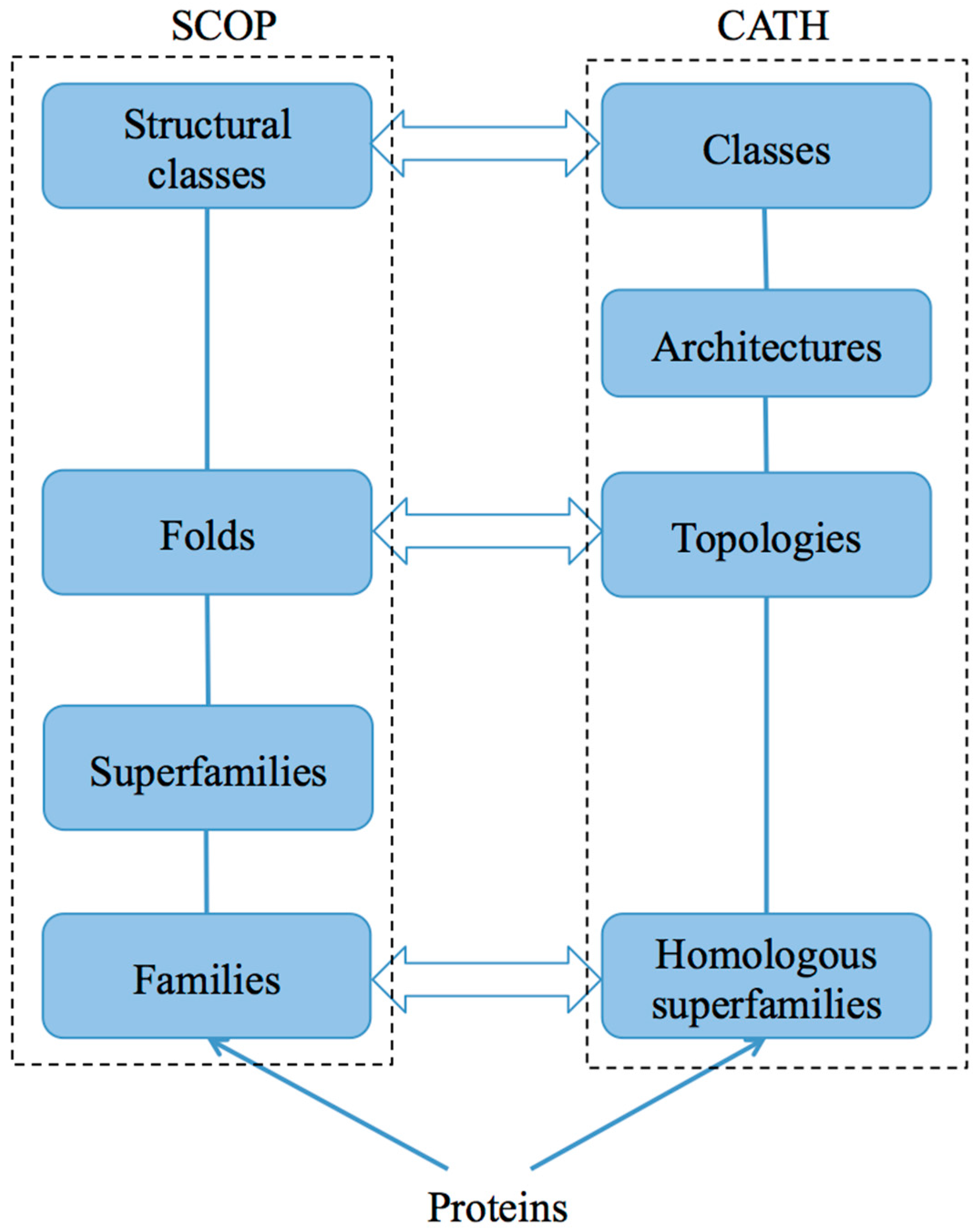
2. Databases
2.1. SCOP and SCOP2
2.2. CATH
3. Framework of Machine Learning-Based Methods
4. Recent Representative Methods for Protein Fold Recognition
4.1. Single Classifier-Based Methods
4.2. Ensemble Classifier-Based Methods
5. Comparisons with Different Methods on Benchmark Dataset
6. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jaroszewski, L.; Li, Z.; Cai, X.-H.; Weber, C.; Godzik, A. FFAS server: Novel features and applications. Nucleic Acids Res. 2011, 39, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Jaroszewski, L.; Li, Z.; Godzik, A. FFAS-3D: Improving fold recognition by including optimized structural features and template re-ranking. Bioinformatics 2013. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Blundell, T.L.; Mizuguchi, K. Fugue: Sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J. Mol. Biol. 2001, 310, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Källberg, M.; Margaryan, G.; Wang, S.; Ma, J.; Xu, J. RaptorX server: A resource for template-based protein structure modeling. Protein Struct. Predict. 2014, 17–27. [Google Scholar] [CrossRef]
- Peng, J.; Xu, J. RaptorX: Exploiting structure information for protein alignment by statistical inference. Proteins Struct. Funct. Bioinform. 2011, 79, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Ghouzam, Y.; Postic, G.; de Brevern, A.G.; Gelly, J.-C. Improving protein fold recognition with hybrid profiles combining sequence and structure evolution. Bioinformatics 2015. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; He, Z.; Zhang, C.; Zhang, L.; Xu, D. Transmembrane protein alignment and fold recognition based on predicted topology. PLoS ONE 2013, 8, e69744. [Google Scholar] [CrossRef] [PubMed]
- Moult, J.; Fidelis, K.; Kryshtafovych, A.; Rost, B.; Hubbard, T.; Tramontano, A. Critical assessment of methods of protein structure prediction—Round VII. Proteins Struct. Funct. Bioinform. 2007, 69, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.F.; Waterman, M.S. Identification of common molecular subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef]
- Wei, L.; Liao, M.; Gao, X.; Zou, Q. Enhanced protein fold prediction method through a novel feature extraction technique. IEEE Trans. Nanobiosci. 2015, 14, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.; Meyer, E.F.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The protein data bank. Eur. J. Biochem. 1977, 80, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Consortium, U. The universal protein resource (UniProt). Nucleic Acids Res. 2008, 36, D190–D195. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Murzin, A.G.; Brenner, S.E.; Hubbard, T.; Chothia, C. Scop: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995, 247, 536–540. [Google Scholar] [CrossRef]
- Andreeva, A.; Howorth, D.; Chothia, C.; Kulesha, E.; Murzin, A.G. SCOP2 prototype: A new approach to protein structure mining. Nucleic Acids Res. 2014, 42, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Sillitoe, I.; Lewis, T.E.; Cuff, A.; Das, S.; Ashford, P.; Dawson, N.L.; Furnham, N.; Laskowski, R.A.; Lee, D.; Lees, J.G. Cath: Comprehensive structural and functional annotations for genome sequences. Nucleic Acids Res. 2015, 43, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Shamim, M.T.A.; Anwaruddin, M.; Nagarajaram, H.A. Support vector machine-based classification of protein folds using the structural properties of amino acid residues and amino acid residue pairs. Bioinformatics 2007, 23, 3320–3327. [Google Scholar] [CrossRef] [PubMed]
- Damoulas, T.; Girolami, M.A. Probabilistic multi-class multi-kernel learning: On protein fold recognition and remote homology detection. Bioinformatics 2008, 24, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Zhou, S.; Guan, J. A new taxonomy-based protein fold recognition approach based on autocross-covariance transformation. Bioinformatics 2009, 25, 2655–2662. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Chen, X. Improving taxonomy-based protein fold recognition by using global and local features. Proteins Struct. Funct. Bioinform. 2011, 79, 2053–2064. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Lyons, J.; Dehzangi, A.; Paliwal, K.K. A feature extraction technique using bi-gram probabilities of position specific scoring matrix for protein fold recognition. J. Theor. Biol. 2013, 320, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wu, J.; Chen, K. PFP-RFSM: Protein fold prediction by using random forests and sequence motifs. J. Biomed. Sci. Eng. 2013, 6, 1161–1170. [Google Scholar] [CrossRef]
- Lampros, C.; Simos, T.; Exarchos, T.P.; Exarchos, K.P.; Papaloukas, C.; Fotiadis, D.I. Assessment of optimized markov models in protein fold classification. J. Bioinform. Comput. Biol. 2014, 12, 1450016. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.B.; Chou, K.C. Predicting protein fold pattern with functional domain and sequential evolution information. J. Theor. Biol. 2009, 256, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Koonin, E.V. Iterated profile searches with PSI-BLAST—A tool for discovery in protein databases. Trends Biochem. Sci. 1998, 23, 444–447. [Google Scholar] [CrossRef]
- Jones, D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- John, G.H.; Langley, P. Estimating continuous distributions in bayesian classifiers. In Proceedings of the Eleventh Conference on Uncertainty in Artificial Intelligence, UAI’95, Montreal, QC, Canada, 18–20 August 1995; Morgan Kaufmann Publishers Inc.: San Francisco, CA, USA, 1995; pp. 338–345. [Google Scholar]
- Bouckaert, R.R. Bayesian Network Classifiers in Weka; Department of Computer Science, University of Waikato: Hamilton, New Zealand, 2004. [Google Scholar]
- Chang, C.-C.; Lin, C.-J. LIBSVM: A library for support vector machines. ACM Trans. Intell. Syst. Technol. 2011. [Google Scholar] [CrossRef]
- Platt, J. Fast training of support vector machines using sequential minimal optimization. In Advances in Kernel Methods—Support Vector Learning; MIT Press: Cambridge, MA, USA, 1999; Volume 3. [Google Scholar]
- Chen, D.; Tian, X.; Zhou, B.; Gao, J. Profold: Protein fold classification with additional structural features and a novel ensemble classifier. BioMed Res. Int. 2016, 2016, 6802832–6802842. [Google Scholar] [CrossRef] [PubMed]
- Landwehr, N.; Hall, M.; Frank, E. Logistic model trees. Mach. Learn. 2005, 59, 161–205. [Google Scholar] [CrossRef]
- Dehzangi, A.; Phon-Amnuaisuk, S.; Manafi, M.; Safa, S. Using rotation forest for protein fold prediction problem: An empirical study. In Evolutionary Computation, Machine Learning and Data Mining in Bioinformatics, Proceedings of the 8th European Conference, EvoBIO 2010, Istanbul, Turkey, 7–9 April 2010; Springer: Berlin/Heidelberg, Germany, 2010; pp. 217–227. [Google Scholar]
- Rodriguez, J.J.; Kuncheva, L.I.; Alonso, C.J. Rotation forest: A new classifier ensemble method. IEEE Trans. Pattern Anal. Mach. Intell. 2006, 28, 1619–1630. [Google Scholar] [CrossRef] [PubMed]
- Gama, J. Functional trees. Mach. Learn. 2004, 55, 219–250. [Google Scholar] [CrossRef]
- Liaw, A.; Wiener, M. Classification and regression by randomforest. R News 2002, 2, 18–22. [Google Scholar]
- Ding, C.H.; Dubchak, I. Multi-class protein fold recognition using support vector machines and neural networks. Bioinformatics 2001, 17, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Kurgan, L. Pfres: Protein fold classification by using evolutionary information and predicted secondary structure. Bioinformatics 2007, 23, 2843–2850. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Liu, X.; Huang, Y.; Jiang, Y.; Zou, Q.; Lin, C. Improved method for predicting protein fold patterns with ensemble classifiers. Genet. Mol. Res. 2012, 11, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, X.; Yang, M.Q.; Yang, J.Y. Ensemble of probabilistic neural networks for protein fold recognition. In Proceedings of the 7th IEEE International Conference on Bioinformatics and Bioengineering, 2007 (BIBE 2007), Boston, MA, USA, 14–17 Ocotober 2007; IEEE: Piscataway, NJ, USA, 2007; pp. 66–70. [Google Scholar]
- Chmielnicki, W. A hybrid discriminative/generative approach to protein fold recognition. Neurocomputing 2012, 75, 194–198. [Google Scholar] [CrossRef]
- Dehzangi, A.; Phon-Amnuaisuk, S.; Dehzangi, O. Using random forest for protein fold prediction problem: An empirical study. J. Inf. Sci. Eng. 2010, 26, 1941–1956. [Google Scholar]
- Ghanty, P.; Pal, N.R. Prediction of protein folds: Extraction of new features, dimensionality reduction, and fusion of heterogeneous classifiers. IEEE Trans. NanoBiosci. 2009, 8, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Zou, Y.; Qin, J.; Liu, X.; Jiang, Y.; Ke, C.; Zou, Q. Hierarchical classification of protein folds using a novel ensemble classifier. PLoS ONE 2013, 8, e56499. [Google Scholar] [CrossRef] [PubMed]
- Nanni, L. A novel ensemble of classifiers for protein fold recognition. Neurocomputing 2006, 69, 2434–2437. [Google Scholar] [CrossRef]
- Shen, H.-B.; Chou, K.-C. Ensemble classifier for protein fold pattern recognition. Bioinformatics 2006, 22, 1717–1722. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Kecman, V. Adaptive local hyperplane classification. Neurocomputing 2008, 71, 3001–3004. [Google Scholar] [CrossRef]
- Guo, X.; Gao, X. A novel hierarchical ensemble classifier for protein fold recognition. Protein Eng. Des. Sel. 2008, 21, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Kecman, V.; Cao, L.; Zhang, C.; Huang, J.Z. Margin-based ensemble classifier for protein fold recognition. Expert Syst. Appl. 2011, 38, 12348–12355. [Google Scholar] [CrossRef]
- Kavousi, K.; Sadeghi, M.; Moshiri, B.; Araabi, B.N.; Moosavi-Movahedi, A.A. Evidence theoretic protein fold classification based on the concept of hyperfold. Math. Biosci. 2012, 240, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hu, X. Recognition of 27-class protein folds by adding the interaction of segments and motif information. BioMed. Res. Int. 2014, 2014, 262850–262859. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hu, X.; Jiang, Z.; Song, H.; Ashraf, M.A. The recognition of multi-class protein folds by adding average chemical shifts of secondary structure elements. Saudi J. Biol. Sci. 2016, 23, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Jo, T.; Hou, J.; Eickholt, J.; Cheng, J. Improving protein fold recognition by deep learning networks. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. Mutationtaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.-C.; Zhang, Z. Snpdryad: Predicting deleterious non-synonymous human snps using only orthologous protein sequences. Bioinformatics 2014, 30, 1112–1119. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Li, S.C.; Wang, L. Protein–protein binding sites prediction by 3D structural similarities. J. Chem. Inf. Model. 2011, 51, 3287–3294. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Li, S.C.; Du, P.; Wang, L. Probabilistic models for capturing more physicochemical properties on protein–protein interface. J. Chem. Inf. Model. 2014, 54, 1798–1809. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Li, S.C.; Ma, W.; Wang, L. Detecting protein conformational changes in interactions via scaling known structures. J. Comput. Biol. 2013, 20, 765–779. [Google Scholar] [CrossRef] [PubMed]
- Alipanahi, B.; Delong, A.; Weirauch, M.T.; Frey, B.J. Predicting the sequence specificities of DNA-and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015, 33, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.-C.; Li, Y.; Peng, C.; Moses, A.M.; Zhang, Z. Computational learning on specificity-determining residue-nucleotide interactions. Nucleic Acids Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Tang, J.; Zou, Q. Local-DPP: An improved DNA-binding protein prediction method by exploring local evolutionary information. Inf. Sci. 2016, in press. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Database Sources | Websites | References |
|---|---|---|
| PDB | http://www.rcsb.org/pdb/ | [14] |
| UniProt | http://www.uniprot.org/ | [15] |
| DSSP | http://swift.cmbi.ru.nl/gv/dssp/ | [16] |
| SCOP | http://scop.mrc-lmb.cam.ac.uk/ | [17] |
| SCOP2 | http://scop2.mrc-lmb.cam.ac.uk/ | [18] |
| CATH | http://www.cathdb.info/ | [19] |
| Index | Fold Identifier | Fold Name | STrain | STest | Total |
|---|---|---|---|---|---|
| 1 | a.1 | Globin-like | 13 | 6 | 19 |
| 2 | a.3 | Cytochrome c | 7 | 9 | 16 |
| 3 | a.4 | DNA/RNA-binding 3-helical bundle | 12 | 30 | 32 |
| 4 | a.24 | 4-Helical up-and-down bundle | 7 | 8 | 15 |
| 5 | a.26 | 4-Helical cytokines | 9 | 9 | 18 |
| 6 | a.39 | EF hand-like | 6 | 9 | 15 |
| 7 | b.1 | Immunoglobulin-like β-sandwich | 30 | 44 | 74 |
| 8 | b.6 | Cupredoxin-like | 9 | 12 | 21 |
| 9 | b.121 | Nucleoplasmin-like/VP | 16 | 13 | 29 |
| 10 | b.29 | ConA-like lectins/glucanases | 7 | 6 | 13 |
| 11 | b.34 | SH3-like barrel | 8 | 8 | 16 |
| 12 | b.40 | OB-Fold | 13 | 19 | 32 |
| 13 | b.42 | β-Trefoil | 8 | 4 | 12 |
| 14 | b.47 | Trypsin-like serine proteases | 9 | 4 | 13 |
| 15 | b.60 | Lipocalins | 9 | 7 | 16 |
| 16 | c.1 | TIM β/α-barrel | 29 | 48 | 77 |
| 17 | c.2 | FAD/NAD(P)-binding domain | 11 | 12 | 23 |
| 18 | c.3 | Flavodoxin-like | 11 | 13 | 24 |
| 19 | c.23 | NAD(P)-binding Rossmann | 13 | 27 | 40 |
| 20 | c.37 | P-loop containing NTH | 10 | 12 | 22 |
| 21 | c.47 | Thioredoxin-fold | 9 | 8 | 17 |
| 22 | c.55 | Ribonuclease H-like motif | 10 | 12 | 22 |
| 23 | c.69 | α/β-Hydrolases | 11 | 7 | 18 |
| 24 | c.93 | Periplasmic binding protein-like | 11 | 4 | 15 |
| 25 | d.15 | β-Grasp (ubiquitin-like) | 7 | 8 | 15 |
| 26 | d.58 | Ferredoxin-like | 13 | 27 | 40 |
| 27 | g.3 | Knottins (small inhibitors, toxins, lectins) | 13 | 27 | 40 |
| Total | 311 | 383 | 694 | ||
| Index | Methods | Classifier Type | References | Overall Accuracy (%) |
|---|---|---|---|---|
| 1 | Nanni et al. (2006) | Ensemble | [49] | 61.1 |
| 2 | PFP-Pred (2006) | Ensemble | [50] | 62.1 |
| 3 | Shamim et al. (2007) | Single (SVM) | [20] | 60.5 |
| 4 | PFRES (2007) | Ensemble | [42] | 68.4 |
| 5 | Damoulas et al. (2008) | Single (SVM) | [21] | 68.1 |
| 6 | ALHK (2008) | Ensemble | [51] | 61.8 |
| 7 | GAOEC (2008) | Ensemble | [52] | 64.7 |
| 8 | PFP-FunDSeqE (2009) | Ensemble | [27] | 70.5 |
| 9 | ACCFold_AC (2009) | Single (SVM) | [22] | 65.3 |
| 10 | ACCFold_ACC (2009) | Single (SVM) | [22] | 66.6 |
| 11 | Ghanty et al. (2009) | Ensemble | [47] | 68.6 |
| 12 | TAXFOLD (2011) | Single (SVM) | [23] | 71.5 |
| 13 | Alok Sharma et al. (2012) | Single (SVM) | [24] | 69.5 |
| 14 | Marfold (2012) | Ensemble | [53] | 71.7 |
| 15 | Kavousi et al. (2012) | Ensemble | [54] | 73.1 |
| 16 | PFP-RFSM (2013) | Single (RF) | [25] | 73.7 |
| 17 | Feng and Hu (2014) | Ensemble | [55] | 70.2 |
| 18 | PFPA (2015) | Ensemble | [13] | 73.6 |
| 19 | Feng et al. (2016) | Ensemble | [56] | 70.8 |
| 20 | ProFold (2016) | Ensemble | [35] | 76.2 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, L.; Zou, Q. Recent Progress in Machine Learning-Based Methods for Protein Fold Recognition. Int. J. Mol. Sci. 2016, 17, 2118. https://doi.org/10.3390/ijms17122118
Wei L, Zou Q. Recent Progress in Machine Learning-Based Methods for Protein Fold Recognition. International Journal of Molecular Sciences. 2016; 17(12):2118. https://doi.org/10.3390/ijms17122118
Chicago/Turabian StyleWei, Leyi, and Quan Zou. 2016. "Recent Progress in Machine Learning-Based Methods for Protein Fold Recognition" International Journal of Molecular Sciences 17, no. 12: 2118. https://doi.org/10.3390/ijms17122118
APA StyleWei, L., & Zou, Q. (2016). Recent Progress in Machine Learning-Based Methods for Protein Fold Recognition. International Journal of Molecular Sciences, 17(12), 2118. https://doi.org/10.3390/ijms17122118