
Bacterial Expression and Kinetic Analysis of Carboxylesterase 001D from Helicoverpa armigera

 ,
,
Abstract
:1. Introduction
2. Results
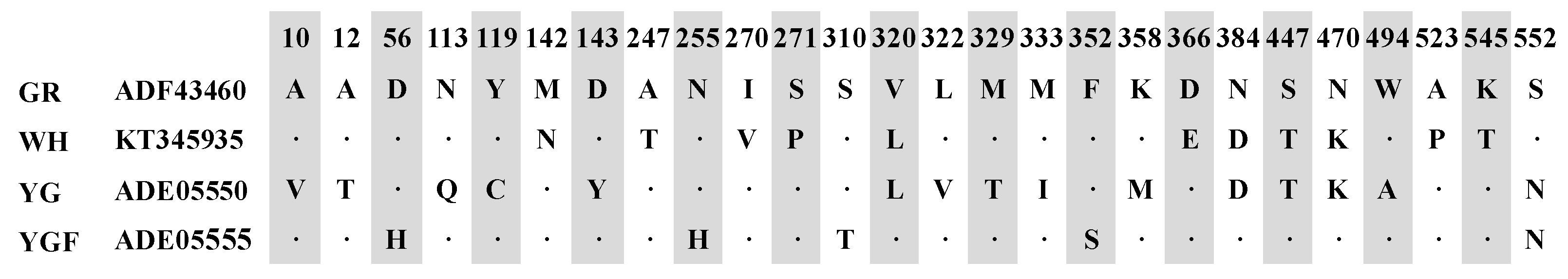
2.1. Cloning and Sequence Analysis
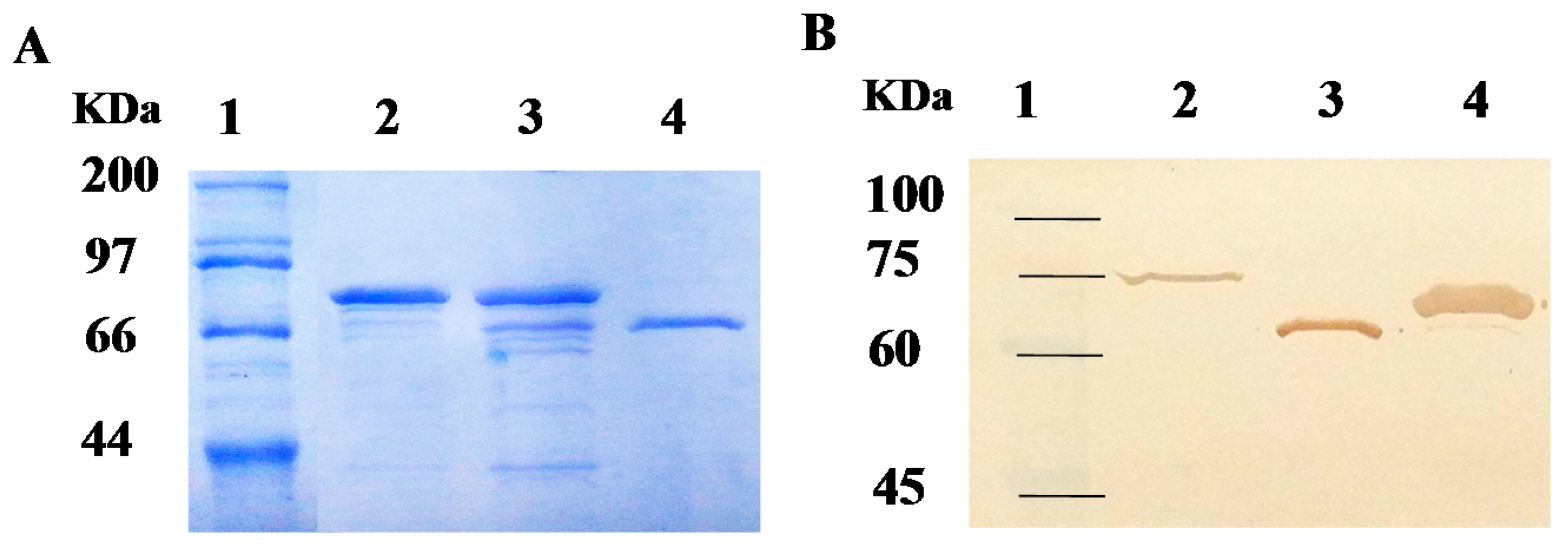
2.2. Purification of Recombinant Carboxylesterases and Western Blot Assay
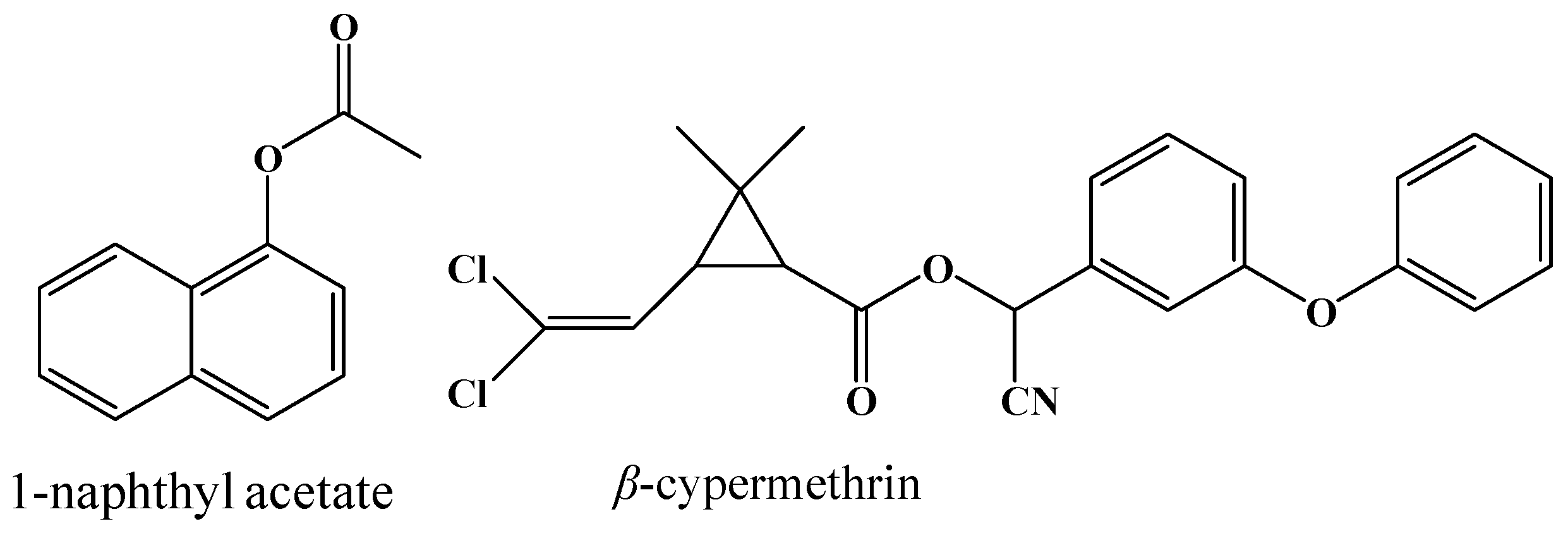
2.3. Enzymatic Activity of 001D towards an Artificial Carboxylester
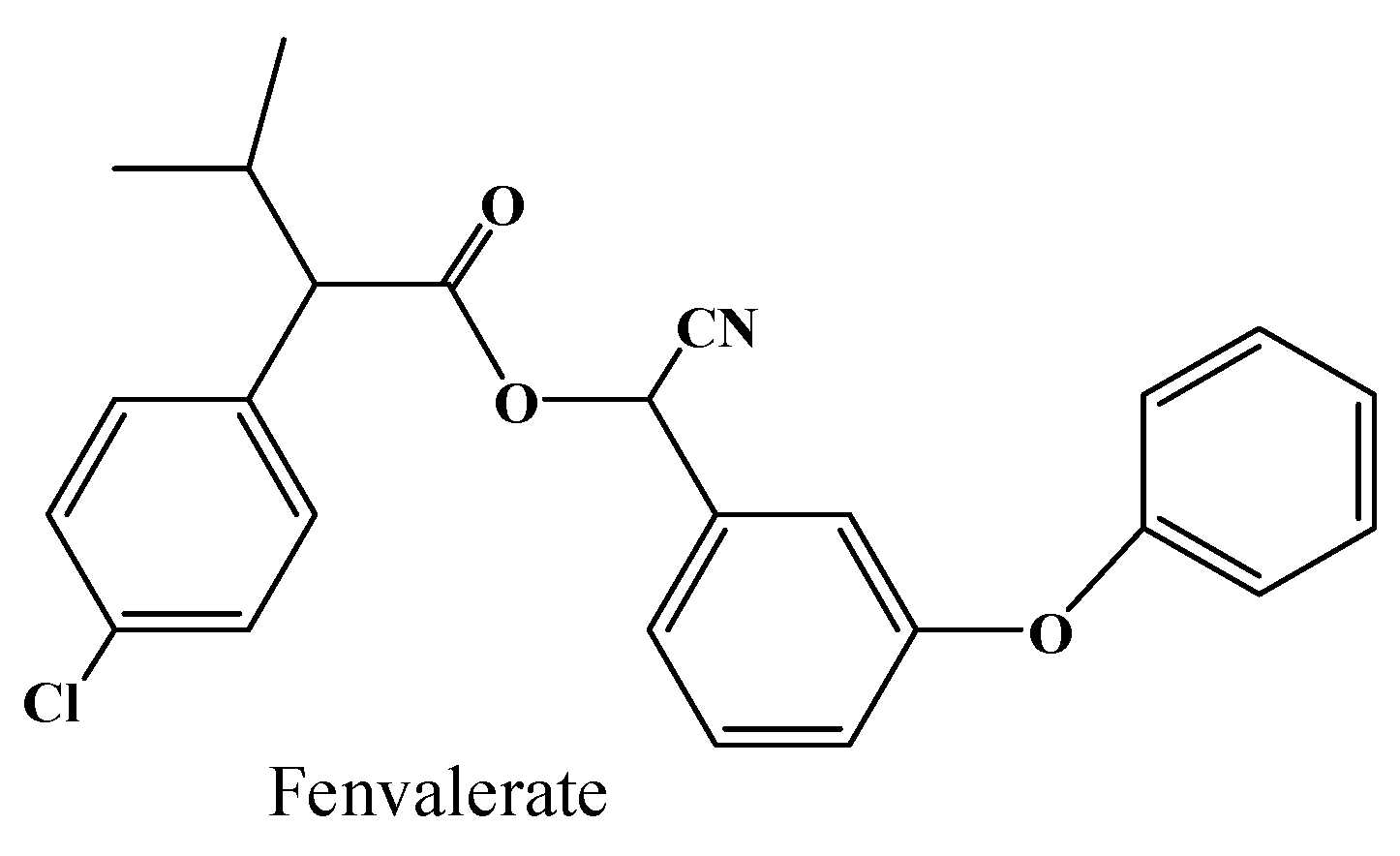
2.4. Hydrolysis Activity of 001D towards Pyrethroid Insecticides
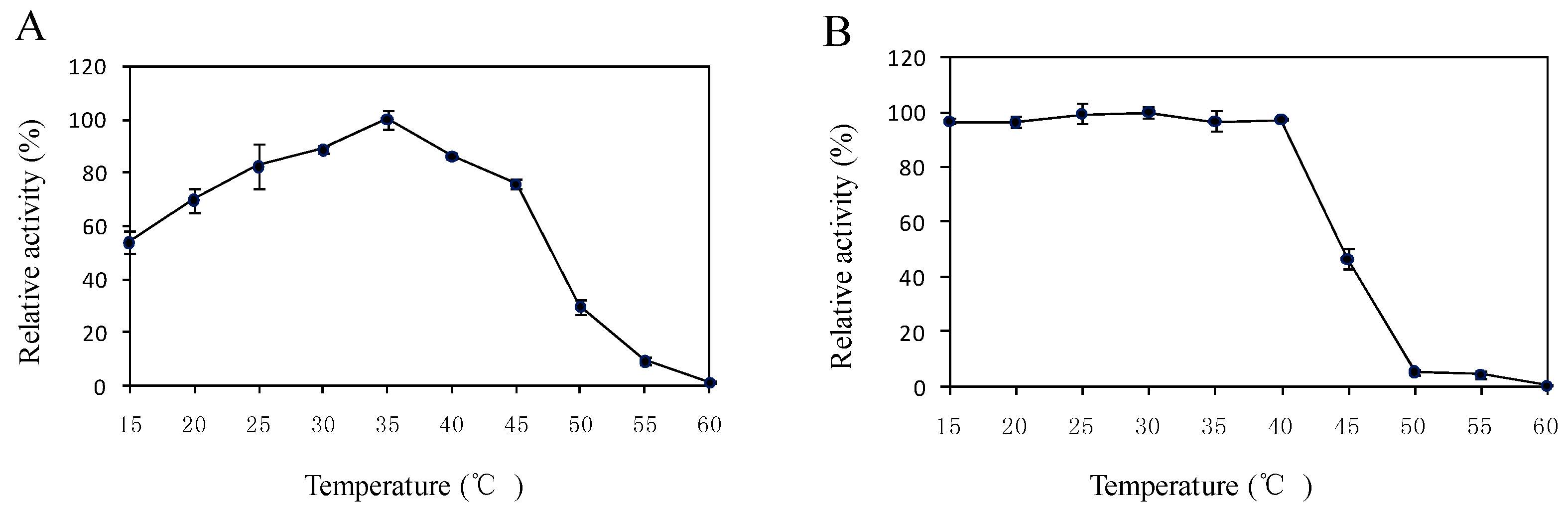
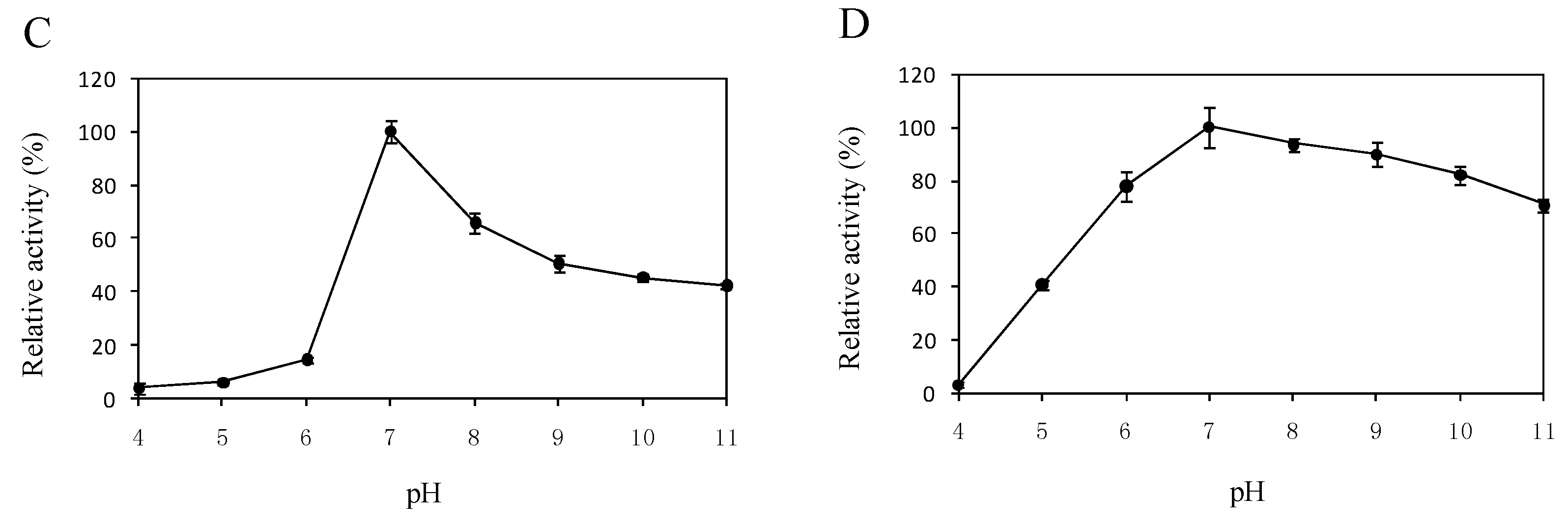
2.5. Effects of Temperature and pH on the Enzyme Activity
3. Discussion
4. Materials and Methods
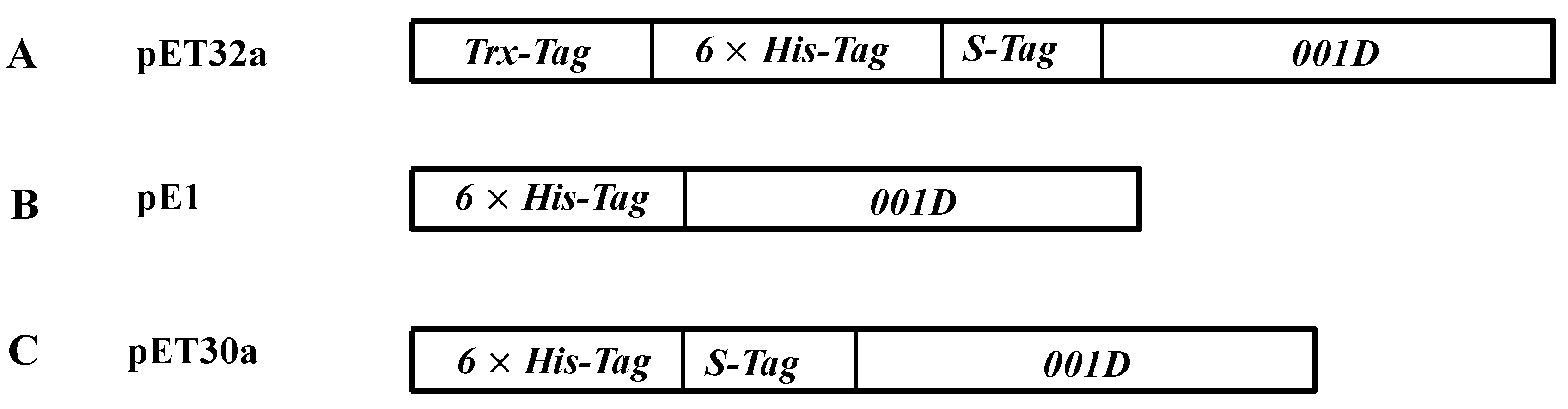
4.1. Chemicals and Plasmids
4.2. Insects
4.3. Sequencing of 001D from the H. armigera Wuhan (WH) Strain
4.4. Sequence Analysis
4.5. Cloning and Expression of Recombinant Proteins
4.6. Purification of Expressed Carboxylesterases and Western Blot Analysis
4.7. Assay of Enzymatic Activity
4.8. Thermostability and pH Stability
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 1-NA | 1-naphthyl acetate |
| OP | Organophosphate |
| CCE | Carboxy/cholinesterase(E.C.3.1.1) |
| PAGE | Polyacrylamide gel electrophoresis |
| pE1 | pEASY®-Blunt E1 vector |
References
- Forrester, N.W.; Cahill, M.; Bird, L.J.; Layland, J.K. Management of pyrethroid and endosulfan resistance in Helicoverpa armigera (Lepidoptera: Noctuidae) in Australia. In Bulletin of Entomological Research; International Institute of Entomology: Wallingford, Oxon, UK, 1993. [Google Scholar]
- Martin, T.; Chandre, F.; Ochou, O.G.; Vaissayre, M.; Fournier, D. Pyrethroid resistance mechanisms in the cotton bollworm Helicoverpa armigera (Lepidoptera: Noctuidae) from West Africa. Pestic. Biochem. Phys. 2002, 74, 17–26. [Google Scholar] [CrossRef]
- Bues, R.; Bouvier, J.C.; Boudinhon, L. Insecticide resistance and mechanisms of resistance to selected strains of Helicoverpa armigera (Lepidoptera: Noctuidae) in the south of France. Crop. Prot. 2005, 24, 814–820. [Google Scholar] [CrossRef]
- Abd El-Latif, A.O.; Subrahmanyam, B. Pyrethroid resistance and esterase activity in three strains of the cotton bollworm, Helicoverpa armigera (Hubner). Pestic. Biochem. Phys. 2010, 96, 155–159. [Google Scholar] [CrossRef]
- Glenn, D.C.; Hoffmann, A.A.; Mcdonald, G. Resistance to pyrethroids in Helicoverpa armigera (Lepidoptera: Noctuidae) from corn: Adult resistance, larval resistance, and fitness effects. J. Econ. Entomol. 1994, 87, 1165–1171. [Google Scholar] [CrossRef]
- Armes, N.J.; Jadhav, D.R.; DeSouza, K.R. A survey of insecticide resistance in Helicoverpa armigera in the Indian subcontinent. Bull. Entomol. Res. 1996, 86, 499–514. [Google Scholar] [CrossRef]
- Djihinto, A.C.; Katary, A.; Prudent, P.; Vassal, J.M.; Vaissayre, M. Variation in resistance to pyrethroids in Helicoverpa armigera from Benin Republic, West Africa. J. Econ. Entomol. 2009, 102, 1928–1934. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.H.; Li, Y.P.; Wu, Y.D. Current status of insecticide resistance in Helicoverpa armigera after 15 years of Bt cotton planting in China. J. Econ. Entomol. 2013, 106, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Wheelock, C.E.; Shan, G.; Ottea, J. Overview of carboxylesterases and their role in the metabolism of insecticides. J. Pestic. Sci. 2005, 30, 75–83. [Google Scholar] [CrossRef]
- Li, X.C.; Schuler, M.A.; Berenbaum, M.R. Molecular mechanisms of metabolic resistance to synthetic and natural xenobiotics. Annu. Rev. Entomol. 2007, 52, 231–253. [Google Scholar] [CrossRef] [PubMed]
- Farnsworth, C.A.; Teese, M.G.; Yuan, G.R.; Li, Y.Q.; Scott, C.; Zhang, X.; Wu, Y.D.; Russell, R.J.; Oakeshott, J.G. Esterase-based metabolic resistance to insecticides in heliothine and spodopteran pests. J. Pestic. Sci. 2010, 35, 275–289. [Google Scholar] [CrossRef]
- Han, Y.C.; Wu, S.W.; Li, Y.P.; Liu, J.W.; Campbell, P.M.; Farnsworth, C.; Scott, C.; Russell, R.J.; Oakeshott, J.G.; Wu, Y.D. Proteomic and molecular analyses of esterases associated with monocrotophos resistance in Helicoverpa armigera. Pestic. Biochem. Phys. 2012, 104, 243–251. [Google Scholar] [CrossRef]
- Gong, Y.J.; Wang, Z.H.; Shi, B.C.; Kang, Z.J.; Zhu, L.; Jin, G.H.; Wei, S.J. Correlation between pesticide resistance and enzyme activity in the diamondback moth, Plutella xylostella. J. Insect Sci. 2013, 13. [Google Scholar] [CrossRef] [PubMed]
- Oakeshott, J.G.; Farnsworth, C.A.; East, P.D.; Scott, C.; Han, Y.C.; Wu, Y.D.; Russell, R.J. How many genetic options for evolving insecticide resistance in heliothine and spodopteran pests? Pest Manag. Sci. 2013, 69, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Newcomb, R.D.; Campbell, P.M.; Ollis, D.L.; Cheah, E.; Russell, R.J.; Oakeshott, J.G. A single amino acid substitution converts a carboxylesterase to an organophosphorus hydrolase and confers insecticide resistance on a blowfly. Proc. Natl. Acad. Sci. USA 1997, 94, 7464–7468. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.M.; Newcomb, R.D.; Russell, R.J.; Oakeshott, J.G. Two different amino acid substitutions in the ali-esterase, E3, confer alternative types of organophosphorus insecticide resistance in the sheep blowfly, Lucilia cuprina. Insect Biochem. Mol. 1998, 28, 139–150. [Google Scholar] [CrossRef]
- Claudianos, C.; Russell, R.J.; Oakeshott, J.G. The same amino acid substitution in orthologous esterases confers organophosphate resistance on the house fly and a blowfly. Insect Biochem. Mol. 1999, 29, 675–686. [Google Scholar] [CrossRef]
- Oakeshott, J.G.; Claudianos, C.; Newcomb, R.B.; Russell, R.J. Biochemical Genetics and Genomics of Insect Esterases; Elsevier: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Field, L.M.; Devonshire, A.L. Evidence that the E4 and FE4 esterase genes responsible for insecticide resistance in the aphid Myzus persicae (Sulzer) are part of a gene family. Biochem. J. 1998, 330, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Chevillon, C.; Raymond, M.; Guillemaud, T.; Lenormand, T.; Pasteur, N. Population genetics of insecticide resistance in the mosquito Culex pipiens. Biol. J. Linn. Soc. 1999, 68, 147–157. [Google Scholar] [CrossRef]
- Small, G.J.; Hemingway, J. Molecular characterization of the amplified carboxylesterase gene associated with organophosphorus insecticide resistance in the brown planthopper, Nilaparvata lugens. Insect Mol. Biol. 2000, 9, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Cui, F.; Qu, H.; Cong, J.; Liu, X.L.; Qiao, C.L. Do mosquitoes acquire organophosphate resistance by functional changes in carboxylesterases? FASEB J. 2007, 21, 3584–3591. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.W.; Yang, Y.H.; Yuan, G.R.; Campbell, P.M.; Teese, M.G.; Russell, R.J.; Oakeshott, J.G.; Wu, Y.D. Overexpressed esterases in a fenvalerate resistant strain of the cotton bollworm, Helicoverpa armigera. Insect Biochem. Mol. 2011, 41, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Gunning, R.V.; Moores, G.D.; Devonshire, A.L. Esterases and esfenvalerate resistance in Australian Helicoverpa armigera (Hubner) Lepidoptera: Noctuidae. Pestic. Biochem. Phys. 1996, 54, 12–23. [Google Scholar] [CrossRef]
- Gunning, R.V.; Moores, G.D.; Devonshire, A.L. Esterases and fenvalerate resistance in a field population of Helicoverpa punctigera (Lepidoptera: Noctuidae) in Australia. Pestic. Biochem. Phys. 1997, 58, 155–162. [Google Scholar] [CrossRef]
- Campbell, B.E. The Role of Esterases in Pyrethroid Resistance in Australian Polulations of the Cotton Bollworm, Helicoverpa armigera (Hübner) (Lepidoptera: Noctuidae); Australian National University: Canberra, Australia, 2001. [Google Scholar]
- Achaleke, J.; Martin, T.; Ghogomu, R.T.; Vaissayre, M.; Brevault, T. Esterase-mediated resistance to pyrethroids in field populations of Helicoverpa armigera (Lepidoptera: Noctuidae) from Central Africa. Pest Manag. Sci. 2009, 65, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Teese, M.G.; Campbell, P.M.; Scott, C.; Gordon, K.H.J.; Southon, A.; Hovan, D.; Robin, C.; Russell, R.J.; Oakeshott, J.G. Gene identification and proteomic analysis of the esterases of the cotton bollworm, Helicoverpa armigera. Insect Biochem. Mol. 2010, 40, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Teese, M.G.; Farnsworth, C.A.; Li, Y.Q.; Coppin, C.W.; Devonshire, A.L.; Scott, C.; East, P.; Russell, R.J.; Oakeshott, J.G. Heterologous expression and biochemical characterisation of fourteen esterases from Helicoverpa armigera. PLoS ONE 2013, 8, e65951. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Q.; Farnsworth, C.A.; Coppin, C.W.; Teese, M.G.; Liu, J.W.; Scott, C.; Zhang, X.; Russell, R.J.; Oakeshott, J.G. Organophosphate and pyrethroid hydrolase activities of mutant esterases from the cotton bollworm Helicoverpa armigera. PLoS ONE 2013, 8, e77685. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.; Li, Y.; Farnsworth, C.A.; Coppin, C.W.; Devonshire, A.L.; Scott, C.; Russell, R.J.; Wu, Y.; Oakeshott, J.G. Isomer-specific comparisons of the hydrolysis of synthetic pyrethroids and their fluorogenic analogues by esterases from the cotton bollworm Helicoverpa armigera. Pestic. Biochem. Phys. 2015, 121, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.H.; Yangb, B.J.; Go, J.H.; Yao, X.M.; Zhang, Y.X.; Song, F.; Liu, Z.W. Molecular cloning and characterization of a juvenile hormone esterase gene from brown planthopper, Nilaparvata lugens. J. Insect Physiol. 2008, 54, 1495–1502. [Google Scholar] [CrossRef] [PubMed]
- Rai, M.; Padh, H. Expression systems for production of heterologous proteins. Curr. Sci. India 2001, 80, 1121–1128. [Google Scholar]
- Cheesman, M.J.; Traylor, M.J.; Hilton, M.E.; Richards, K.E.; Taylor, M.C.; Daborn, P.J.; Russell, R.J.; Gillam, E.M.J.; Oakeshott, J.G. Soluble and membrane-bound Drosophila melanogaster CYP6G1 expressed in Escherichia coli: Purification, activity, and binding properties toward multiple pesticides. Insect Biochem. Mol. 2013, 43, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.N.; Zhang, L.; Zhang, X.T.; Xiwu, G. Molecular cloning and recombinant expression of cytochrome P450 CYP6B6 from Helicoverpa armigera in Escherichia coli. Mol. Biol. Rep. 2013, 40, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.Q.; Zhang, Y.L. Effect of temperature and sorbitol in improving the solubility of carboxylesterases protein CpCE-1 from Cydia pomonella and biochemical characterization. Appl. Microbiol. Biot. 2013, 97, 10423–10433. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.R.; (Southwest University, Chongqing, China). Personal communiation, 23 May 2015.
- Baneyx, F.; Mujacic, M. Recombinant protein folding and misfolding in Escherichia coli. Nat. Biotechnol. 2004, 22, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.W.; Boucher, Y.; Stokes, H.W.; Ollis, D.L. Improving protein solubility: The use of the Escherichia coli dihydrofolate reductase gene as a fusion reporter. Protein Expr. Purif. 2006, 47, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.W.; Hadler, K.S.; Schenk, G.; Ollis, D. Using directed evolution to improve the solubility of the C-terminal domain of Escherichia coli aminopeptidase P—Implications for metal binding and protein stability. FEBS J. 2007, 274, 4742–4751. [Google Scholar] [CrossRef] [PubMed]
- Harel, M.; Kryger, G.; Rosenberry, T.L.; Mallender, W.D.; Lewis, T.; Fletcher, R.J.; Guss, J.M.; Silman, I.; Sussman, J.L. Three-dimensional structures of Drosophila melanogaster acetylcholinesterase and of its complexes with two potent inhibitors. Protein Sci. 2000, 9, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Pezzementi, L.; Johnson, K.; Tsigelny, I.; Cotney, J.; Manning, E.; Barker, A.; Merritt, S. Amino acids defining the acyl pocket of an invertebrate cholinesterase. Comp. Biochem. Phys. B 2003, 136, 813–832. [Google Scholar] [CrossRef]
- Devonshire, A.; Heidari, R.; Bell, K.; Campbell, P.; Campbell, B.; Odgers, W.; Oakeshott, J.; Russell, R. Kinetic efficiency of mutant carboxylesterases implicated in organophosphate insecticide resistance. Pestic. Biochem. Phys. 2003, 76, 1–13. [Google Scholar] [CrossRef]
- Ruan, Z.Y.; Zhai, Y.; Song, J.L.; Shi, Y.H.; Li, K.; Zhao, B.; Yan, Y.C. Molecular cloning and characterization of a newly isolated pyrethroid-degrading esterase gene from a genomic library of Ochrobactrum anthropi YZ-1. PLoS ONE 2013, 8, e77329. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [PubMed]
- Coppin, C.W.; Jackson, C.J.; Sutherland, T.; Hart, P.J.; Devonshire, A.L.; Russell, R.J.; Oakeshott, J.G. Testing the evolvability of an insect carboxylesterase for the detoxification of synthetic pyrethroid insecticides. Insect Biochem. Mol. 2012, 42, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Xie, J.; Zhao, Y.; Zhang, H.; Liu, Z.; Lu, Z. Improving methyl parathion hydrolase to enhance its chlorpyrifos-hydrolysing efficiency. Lett. Appl. Microbiol. 2014, 58, 53–59. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Specific Activity | Km | kcat |
|---|---|---|---|
| pET32a-001D | 2.24(0.14) | 7.61(0.63) | 2.29(0.08) |
| pE1-001D | 0.34(0.14) | 19.72(6.69) | 0.35(0.05) |
| pET30a-001D | 0.57(0.07) | 12.51(2.14) | 0.60(0.06) |
| Enzyme | β-Cypermethrin | Fenvalerate |
|---|---|---|
| pET32a-001D | 0.15(0.02) | 0.32(0.02) |
| pE1-001D | 0.19(0.02) | 0.13(0.06) |
| pET30a-001D | 0.41(0.04) | 0.67(0.17) |
| Control | 0.10(0.01) | 0.03(0.02) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Liu, J.; Lu, M.; Ma, Z.; Cai, C.; Wang, Y.; Zhang, X. Bacterial Expression and Kinetic Analysis of Carboxylesterase 001D from Helicoverpa armigera. Int. J. Mol. Sci. 2016, 17, 493. https://doi.org/10.3390/ijms17040493
Li Y, Liu J, Lu M, Ma Z, Cai C, Wang Y, Zhang X. Bacterial Expression and Kinetic Analysis of Carboxylesterase 001D from Helicoverpa armigera. International Journal of Molecular Sciences. 2016; 17(4):493. https://doi.org/10.3390/ijms17040493
Chicago/Turabian StyleLi, Yongqiang, Jianwei Liu, Mei Lu, Zhiqing Ma, Chongling Cai, Yonghong Wang, and Xing Zhang. 2016. "Bacterial Expression and Kinetic Analysis of Carboxylesterase 001D from Helicoverpa armigera" International Journal of Molecular Sciences 17, no. 4: 493. https://doi.org/10.3390/ijms17040493
APA StyleLi, Y., Liu, J., Lu, M., Ma, Z., Cai, C., Wang, Y., & Zhang, X. (2016). Bacterial Expression and Kinetic Analysis of Carboxylesterase 001D from Helicoverpa armigera. International Journal of Molecular Sciences, 17(4), 493. https://doi.org/10.3390/ijms17040493