
Minimal Functional Sites in Metalloproteins and Their Usage in Structural Bioinformatics




Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
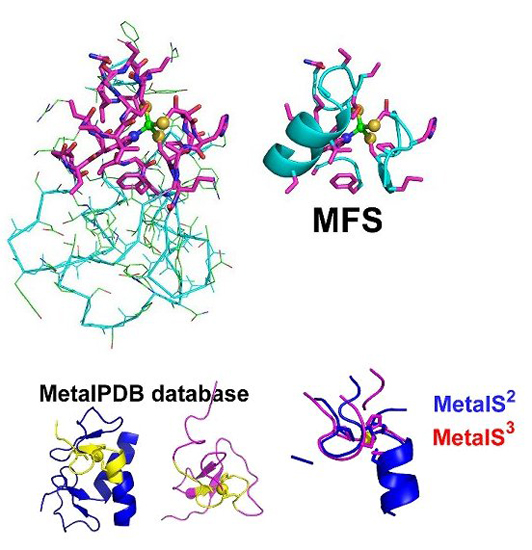
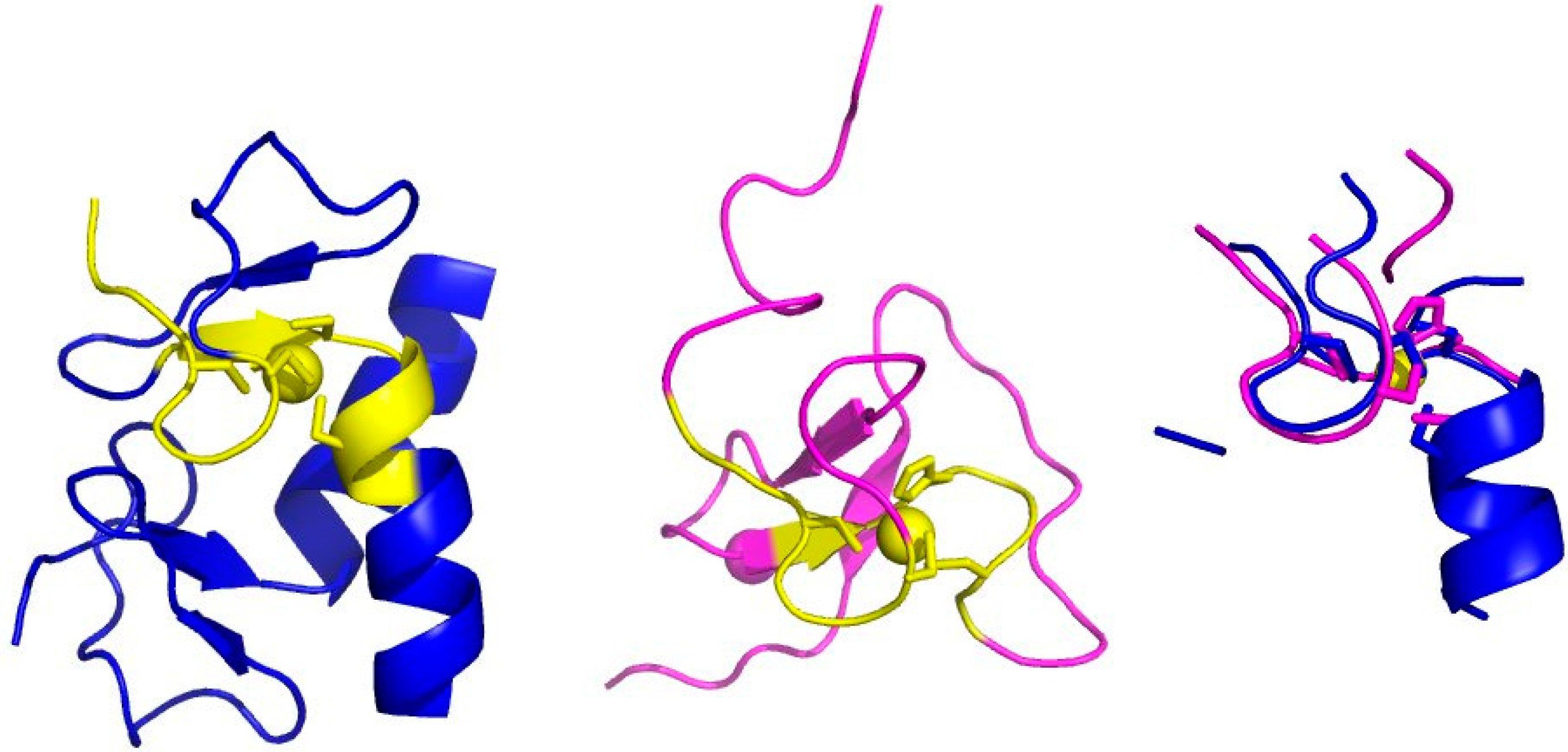
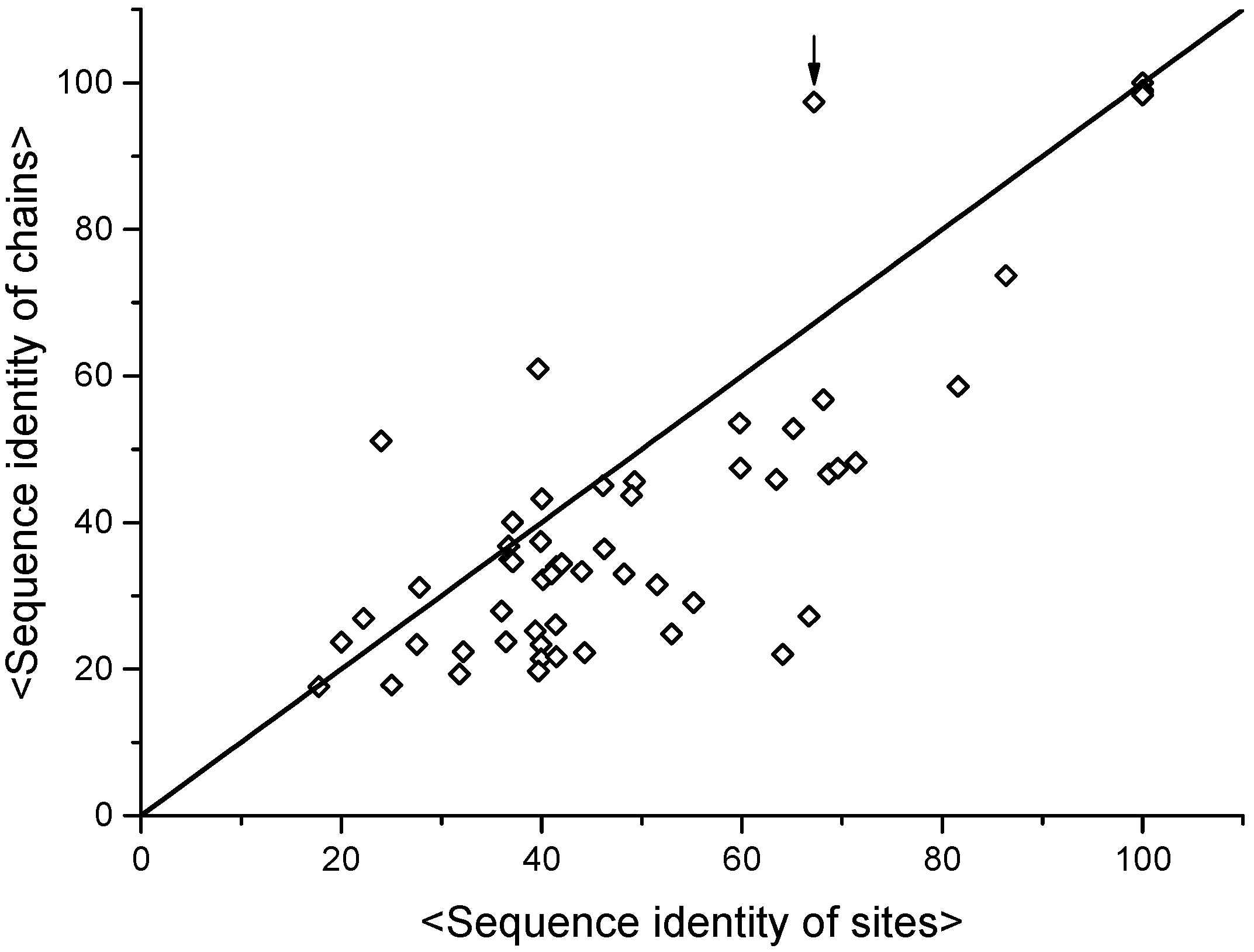
2. The Minimal Functional Site (MFS)
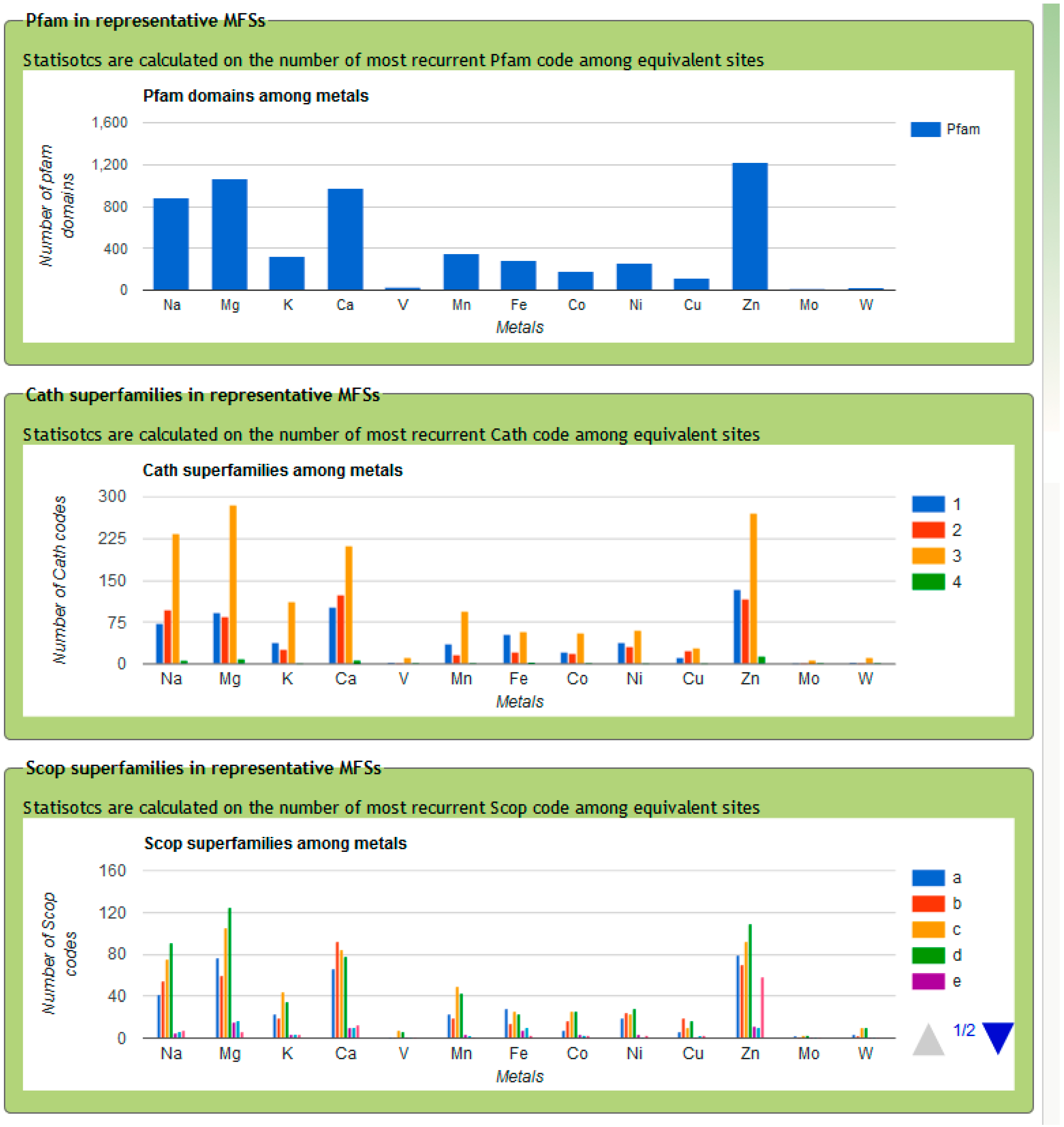
3. MetalPDB, a Database of Minimal Functional Sites in Metalloproteins
- (1)
- All metal-containing structures released after the last update are downloaded from the PDB.
- (2)
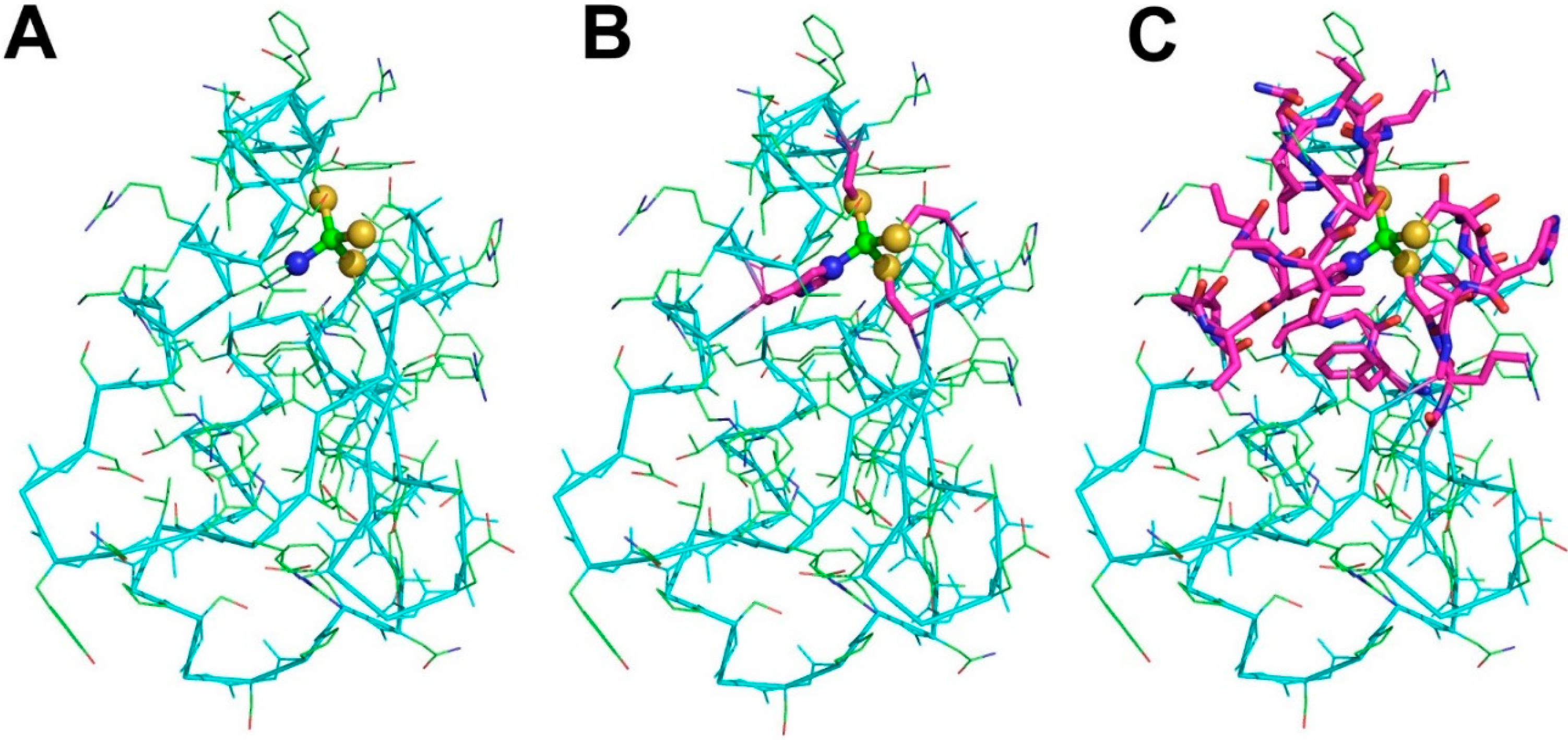
- For each metal ion in each structure from step (1) we identify the metal ligands, both within the polypeptide or polynucleotide chains (endogenous ligands) and different ions or molecules such as water, sulfide, acetate (exogenous ligands) (Figure 1B). Also organic cofactors such as heme are included in the exogenous ligands.
- (3)
- Each pair of metal ions having at least one common ligand or being at a distance lower than 5 Å is included into a single dinuclear site. This procedure is iterated such that if metal A and metal B form a single site and then metal B and metal C also form a single site, eventually a trinuclear site is defined that contains all three metal ions. In this way, e.g., each Fe4S4 cluster found in ferredoxins constitutes an individual four-nuclear site.
- (4)
- Identify the neighbors of all the metal ligands (both endogenous and exogenous) in each mono- or polynuclear site. Such neighbors are chemical species (residues in a polypeptide or a polynucleotide chain, or other molecules or ions) that contain at least one non-hydrogen atom at a distance smaller than 5 Å from the ligand itself. The ensemble of the neighbors, the ligands and the metal atom(s) constitute the MFS (Figure 1C).
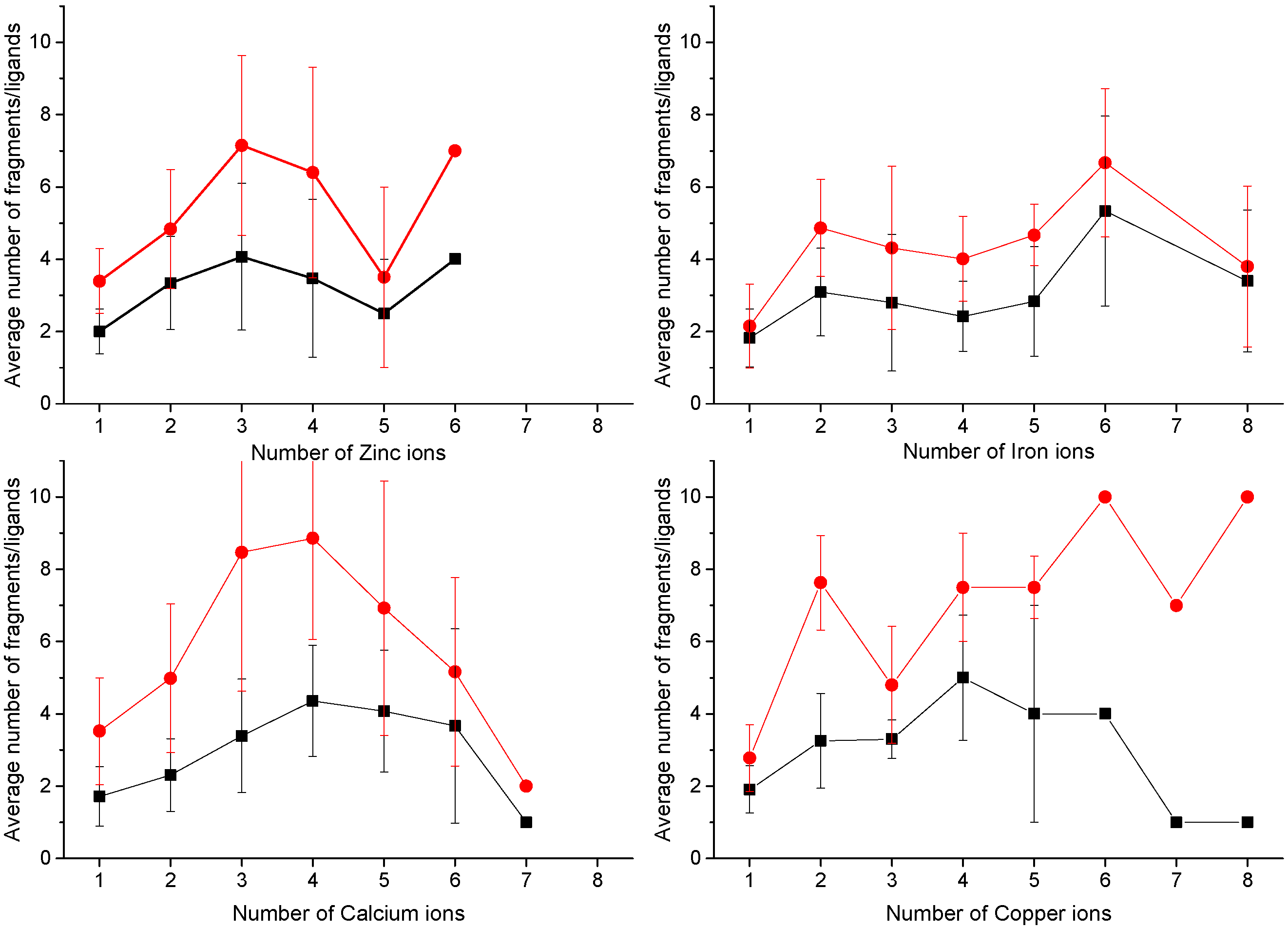
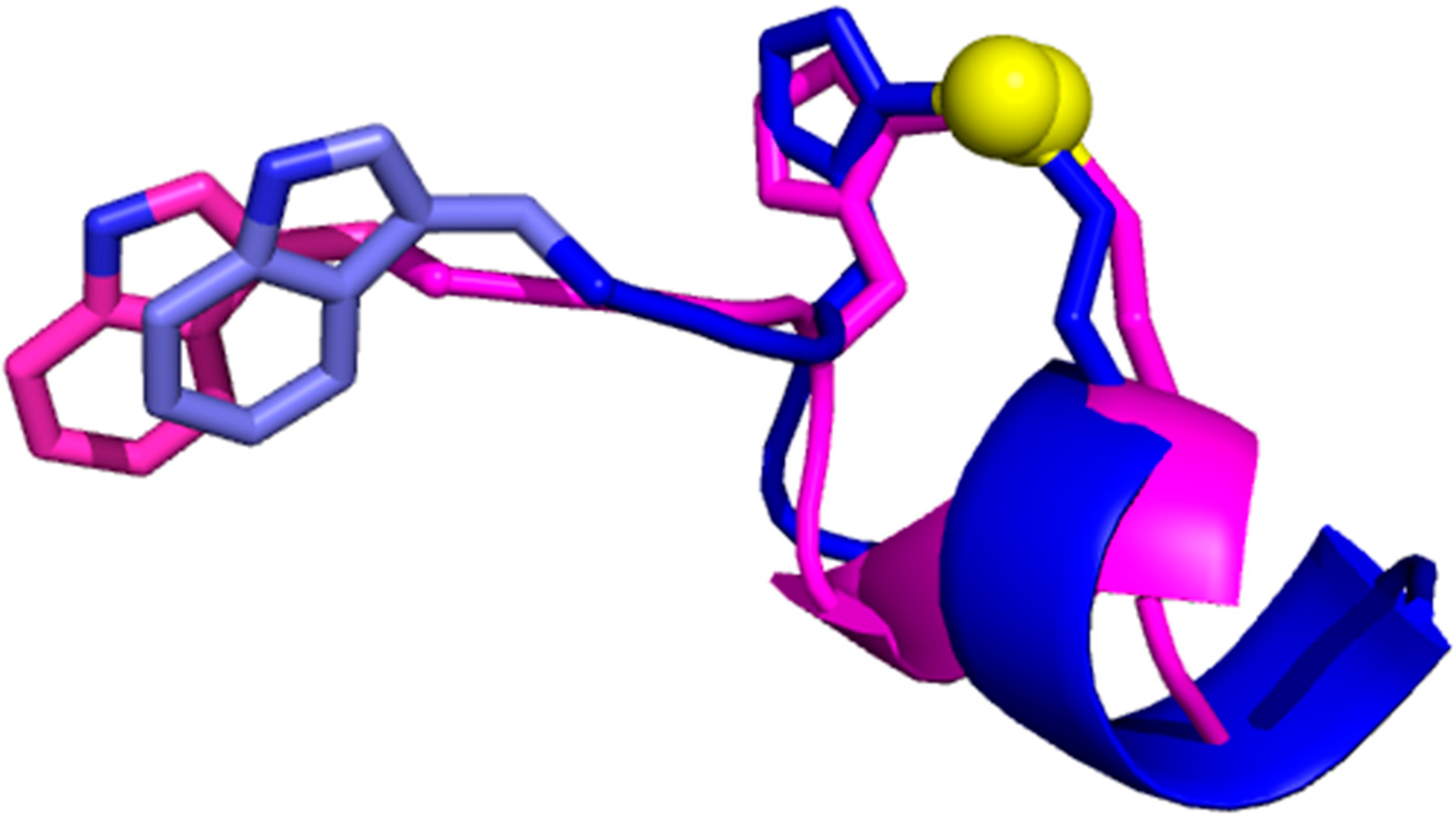
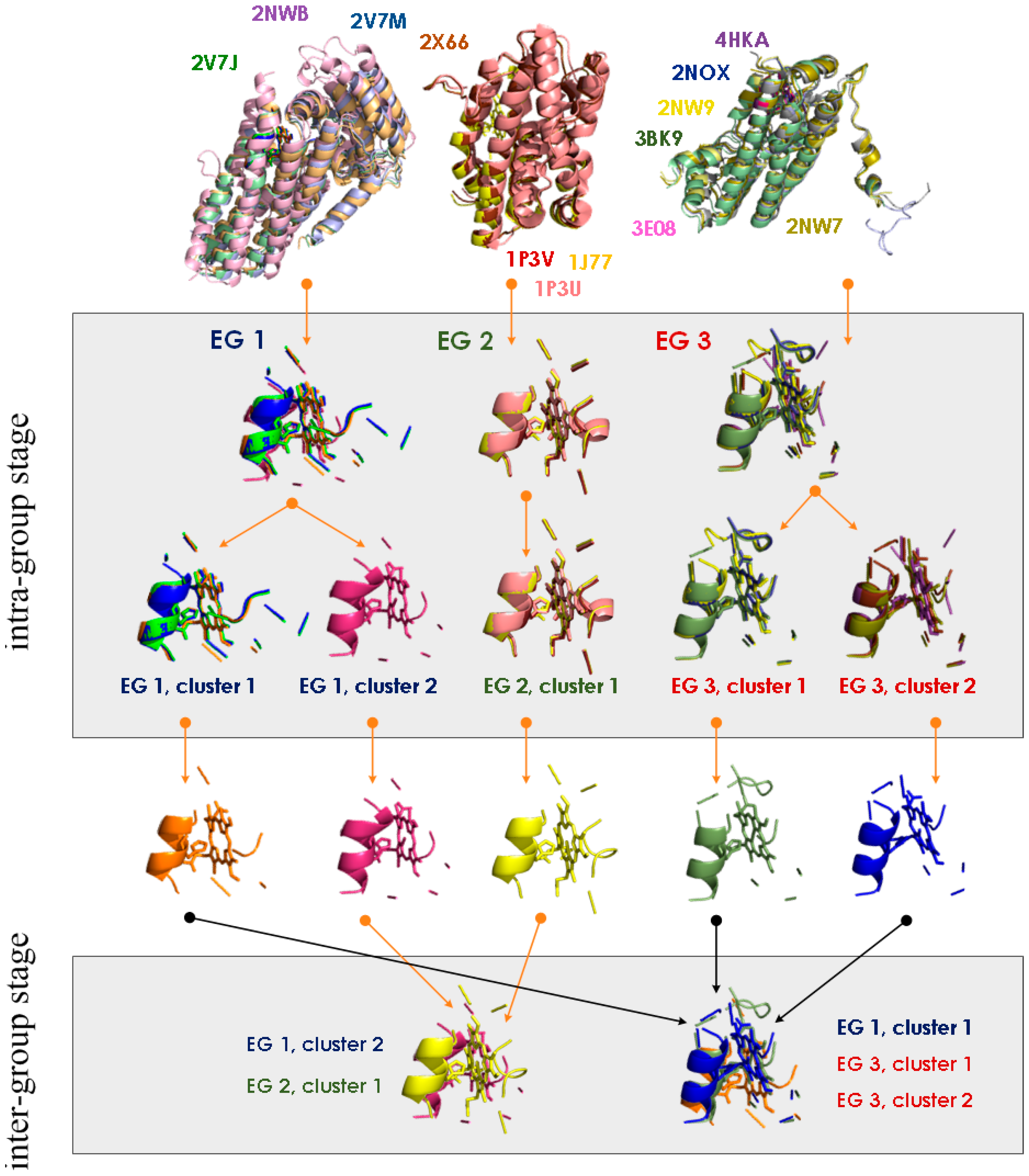
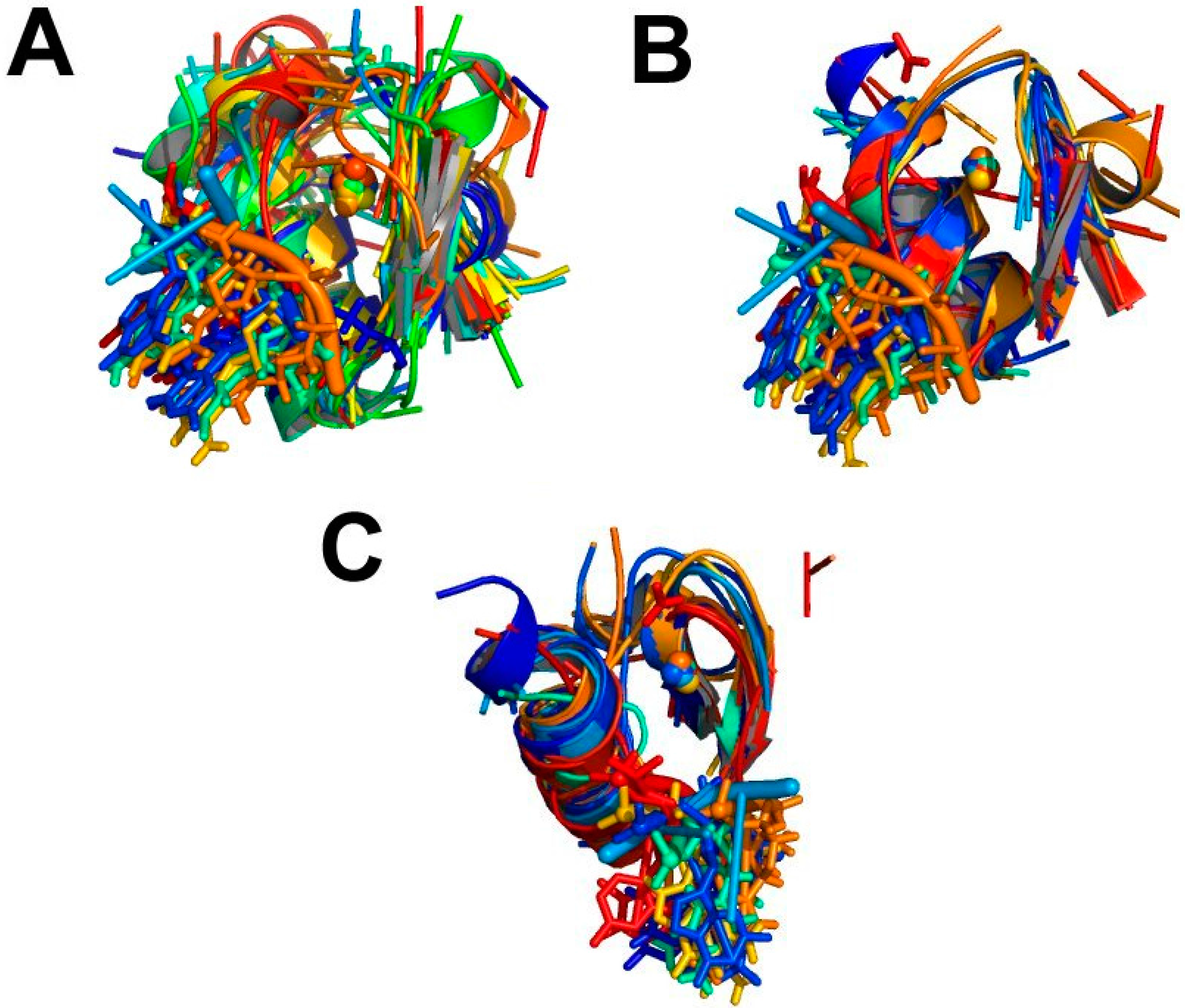
4. MetalS2: A Tool for the 3D Structural Comparison of MFSs
- the fragmentation of the alignment, by measuring how many fragments the alignment is broken into (F) and how long each fragment is (nf), N being the total alignment length
- the relative coverage of the two sites, by comparing the total number of Cα and Cβ atoms in the shortest site (Cmax) to the number of atoms effectively put in correspondence (c)
- the biochemical similarity of the residues put in correspondence, by comparing the BLOSUM62 similarity score (S) to the maximum possible score (Smax).
5. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| MBP | Metal-binding pattern |
| MFS | Minimal Functional Site |
| PDB | Protein Data Bank |
References
- Frausto da Silva, J.J.R.; Williams, R.J.P. The Biological Chemistry of the Elements: The Inorganic Chemistry of Life; Oxford University Press: New York, NY, USA, 2001. [Google Scholar]
- Bertini, I.; Gray, H.B.; Stiefel, E.I.; Valentine, J.S. Biological Inorganic Chemistry; University Science Books: Sausalito, CA, USA, 2006. [Google Scholar]
- Andreini, C.; Bertini, I.; Cavallaro, G.; Holliday, G.L.; Thornton, J.M. Metal ions in biological catalysis: From enzyme databases to general principles. J. Biol. Inorg. Chem. 2008, 13, 1205–1218. [Google Scholar] [CrossRef] [PubMed]
- Bertini, I.; Rosato, A. Bioinorganic chemistry in the post-genomic era. Proc. Natl. Acad. Sci. USA 2003, 100, 3601–3604. [Google Scholar] [CrossRef] [PubMed]
- Arnesano, F.L.; Banci, L.; Bertini, I.; Capozzi, F.; Ciurli, S.; Luchinat, C.; Mangani, S.; Ciofi-Baffoni, S.; Rosato, A.; Turano, P.; et al. An Italian contribution to structural genomics: Understanding metalloproteins. Coord. Chem. Rev. 2006, 250, 1419–1450. [Google Scholar] [CrossRef]
- Bowman, S.E.; Bridwell-Rabb, J.; Drennan, C.L. Metalloprotein crystallography: More than a structure. Acc. Chem. Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Chance, M.R. Metalloproteomics: Forward and reverse approaches in metalloprotein structural and functional characterization. Curr. Opin. Chem. Biol. 2011, 15, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Zhan, C.; Ignatov, A.; Manjasetty, B.A.; Marinkovic, N.; Sullivan, M.; Huang, R.; Chance, M.R. Metalloproteomics: High-throughput structural and functional annotation of proteins in structural genomics. Structure 2005, 13, 1473–1486. [Google Scholar] [CrossRef] [PubMed]
- Barnett, J.P.; Scanlan, D.J.; Blindauer, C.A. Protein fractionation and detection for metalloproteomics: Challenges and approaches. Anal. Bioanal. Chem. 2012, 402, 3311–3322. [Google Scholar] [CrossRef] [PubMed]
- Lin, I.J.; Gebel, E.B.; Machonkin, T.E.; Westler, W.M.; Markley, J.L. Changes in hydrogen-bond strenght explain reduction potentials in 10 ruvredoxin variants. Proc. Natl. Acad. Sci. USA 2005, 102, 14581–14586. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Jenney, F.E., Jr.; Adams, M.W.; Babini, E.; Takahashi, Y.; Fukuyama, K.; Hodgson, K.O.; Hedman, B.; Solomon, E.I. Solvent tuning of electrochemical potentials in the active sites of HiPIP versus ferredoxin. Science 2007, 318, 1464–1468. [Google Scholar] [CrossRef] [PubMed]
- Andreini, C.; Bertini, I.; Cavallaro, G.; Najmanovich, R.J.; Thornton, J.M. Structural analysis of metal sites in proteins: Non-heme iron sites as a case study. J. Mol. Biol. 2009, 388, 356–380. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.M.; Lim, C. Factors controlling the reactivity of zinc finger cores. J. Am. Chem. Soc. 2011, 133, 8691–8703. [Google Scholar] [CrossRef] [PubMed]
- Dudev, T.; Lim, C. Competition among metal ions for protein binding sites: Determinants of metal ion selectivity in proteins. Chem. Rev. 2014, 114, 538–556. [Google Scholar] [CrossRef] [PubMed]
- Valasatava, Y.; Rosato, A.; Cavallaro, G.; Andreini, C. MetalS3, a database-mining tool for the identification of structurally similar metal sites. J. Biol. Inorg. Chem. 2014, 19, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Valasatava, Y.; Andreini, C.; Rosato, A. Hidden relationship between metalloproteins unveiled by structural comparison of their metal sites. Sci. Rep. 2015, 5, 9486. [Google Scholar] [CrossRef] [PubMed]
- Andreini, C.; Bertini, I.; Cavallaro, G. Minimal functional sites allow a classification of zinc sites in proteins. PLoS ONE 2011, 10, e26325. [Google Scholar] [CrossRef] [PubMed]
- Hasnain, S.S.; Hodgson, K.O. Structure of metal centres in proteins at subatomic resolution. J. Synch. Rad. 1999, 6, 852–864. [Google Scholar] [CrossRef]
- Cotelesage, J.J.H.; Pushie, M.J.; Grochulski, P.; Pickering, I.J.; George, G.N. Metalloprotein active site structure determination: Synergy between X-ray absorption spectroscopy and X-ray crystallography. J. Inorg. Biochem. 2012, 115, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Punta, M.; Bohon, J.; Sauder, J.M.; D’Mello, R.; Sullivan, M.; Toomey, J.; Abel, D.; Lippi, M.; Passerini, A.; et al. Characterization of metalloproteins by high-throughput X-ray absorption spectroscopy. Genome Res. 2011, 21, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Hsin, K.; Sheng, Y.; Harding, M.M.; Taylor, P.; Walkinshaw, M.D. MESPEUS: A database of the geometry of metal sites in proteins. J. Appl. Cryst. 2008, 41, 963–968. [Google Scholar] [CrossRef]
- Tus, A.; Rakipovic, A.; Peretin, G.; Tomic, S.; Sikic, M. BioMe: Biologically relevant metals. Nucleic Acids Res. 2012, 40, W352–W357. [Google Scholar] [CrossRef] [PubMed]
- Schnabl, J.; Suter, P.; Sigel, R.K.O. MINAS—A database of Metal Ions in Nucleic Acids. Nucleic Acids Res. 2012, 40, D434–D438. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.W.; Beran, B.; Bi, C.; Bluhm, W.F.; Dimitropoulos, D.; Goodsell, D.S.; Prlic, A.; Quesada, M.; Quinn, G.B.; Westbrook, J.D.; et al. The RCSB Protein Data Bank: Redesigned web site and web services. Nucleic Acids Res. 2011, 39, D392–D401. [Google Scholar] [CrossRef] [PubMed]
- Laitaoja, M.; Valjakka, J.; Janis, J. Zinc coordination spheres in protein structures. Inorg. Chem. 2013, 52, 10983–10991. [Google Scholar] [CrossRef] [PubMed]
- Raczynska, J.; Wlodawer, A.; Jaskolski, M. Prior knowledge or freedom of interpretation? A critical look at a recently published classification of “novel” Zn binding sites. Proteins 2016. [Google Scholar] [CrossRef] [PubMed]
- Degtyarenko, K.; Contrino, S. COMe: The ontology of bioinorganic proteins. BMC Struct. Biol. 2004, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degtyarenko, K.N. Bioinorganic motifs: Towards functional classification of metalloproteins. Bioinformatics 2000, 16, 851–864. [Google Scholar] [CrossRef] [PubMed]
- Andreini, C.; Bertini, I.; Rosato, A. A hint to search for metalloproteins in gene banks. Bioinformatics 2004, 20, 1373–1380. [Google Scholar] [CrossRef] [PubMed]
- Passerini, A.; Lippi, M.; Frasconi, P. MetalDetector v2.0: Predicting the geometry of metal binding sites from protein sequence. Nucleic Acids Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Passerini, A.; Andreini, C.; Menchetti, S.; Rosato, A.; Frasconi, P. Predicting zinc binding at the proteome level. BMC Bioinform. 2007, 5, 8–39. [Google Scholar]
- Andreini, C.; Bertini, I.; Rosato, A. Metalloproteomes: A bioinformatic approach. Acc. Chem. Res. 2009, 42, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- Shu, N.; Zhou, T.; Hovmoller, S. Prediction of zinc-binding sites in proteins from sequence. Bioinformatics 2008, 24, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Andreini, C.; Bertini, I.; Cavallaro, G.; Decaria, L.; Rosato, A. A simple protocol for the comparative analysis of the structure and occurence of biochemical pathways across superkingdoms. J. Chem. Inf. Model. 2011, 51, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Karlin, S.; Zhu, Z.Y.; Karlin, K.D. The extended environment of mononuclear metal centers in protein structures. Proc. Natl. Acad. Sci. USA 1997, 94, 14225–14230. [Google Scholar] [CrossRef] [PubMed]
- Dudev, T.; Lin, Y.L.; Dudev, M.; Lim, C. First-second shell interactions in metal binding sites in proteins: A PDB survey and DFT/CDM calculations. J. Am. Chem. Soc. 2003, 125, 3168–3180. [Google Scholar] [CrossRef] [PubMed]
- Dudev, T.; Lim, C. Metal binding affinity and selectivity in metalloproteins: Insights from computational studies. Annu. Rev. Biophys. 2008, 37, 97–116. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Bertini, I.; del Conte, R.; Mangani, S.; Meyer-Klaucke, W. X-ray absorption spectroscopy study of CopZ, a copper chaperone in Bacillus subtilis. The coordination properties of the copper ion. Biochemistry 2003, 8, 2467–2474. [Google Scholar] [CrossRef] [PubMed]
- Solomon, E.I.; Heppner, D.E.; Johnston, E.M.; Ginsbach, J.W.; Cirera, J.; Qayyum, M.; Kieber-Emmons, M.T.; Kjaergaard, C.H.; Hadt, R.G.; Tian, L. Copper active sites in biology. Chem. Rev. 2014, 114, 3659–3853. [Google Scholar] [CrossRef] [PubMed]
- Abriata, L.A. Analysis of copper-ligand bond lengths in X-ray structures of different types of copper sites in proteins. Acta Crystallogr. Sect. D 2012, 68, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chakraborty, S.; Hosseinzadeh, P.; Yu, Y.; Tian, S.L.; Petrik, I.; Bhagi, A.; Lu, Y. Metalloproteins containing cytochrome, iron-sulfur, or copper Redox centers. Chem. Rev. 2014, 114, 4366–4469. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.F.; Lopes, A.B.; Fernandes, P.A.; Ramos, M.J. The Zinc proteome: A tale of stability and functionality. Dalton Trans. 2009, 14, 7946–7956. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Chruszcz, M.; Lasota, P.; Lebioda, L.; Minor, W. Data mining of metal ion environments present in protein structures. J. Inorg. Biochem. 2008, 102, 1765–1776. [Google Scholar] [CrossRef] [PubMed]
- Andreini, C.; Cavallaro, G.; Lorenzini, S.; Rosato, A. MetalPDB: A database of metal sites in biological macromolecular structures. Nucleic Acids Res. 2013, 41, D312–D319. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Smith, R.D.; Clark, J.J.; Dunbar, J.B., Jr.; Carlson, H.A. Recent improvements to Binding MOAD: A resource for protein-ligand binding affinities and structures. Nucleic Acids Res. 2015, 43, D465–D469. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Kang, H.; Park, H. MetLigDB: A web-based database for identification of chemical groups to design metalloprotein inhibitors. J. Appl. Cryst. 2011, 44, 878–881. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. PFAM: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Sillitoe, I.; Lewis, T.E.; Cuff, A.; Das, S.; Ashford, P.; Dawson, N.L.; Furnham, N.; Laskowski, R.A.; Lee, D.; Lees, J.G.; et al. CATH: Comprehensive structural and functional annotations for genome sequences. Nucleic Acids Res. 2015, 43, D376–D381. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, A.; Howorth, D.; Chothia, C.; Kulesha, E.; Murzin, A.G. SCOP2 prototype: A new approach to protein structure mining. Nucleic Acids Res. 2014, 42, D310–D314. [Google Scholar] [CrossRef] [PubMed]
- Andreini, C.; Cavallaro, G.; Rosato, A.; Valasatava, Y. MetalS2: A tool for the structural alignment of minimal functional sites in metal-binding proteins and nucleic acids. J. Chem. Inf. Model. 2013, 53, 3064–3075. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Chordia, M.D.; Cooper, D.R.; Chruszcz, M.; Muller, P.; Sheldrick, G.M.; Minor, W. Validation of metal-binding sites in macromolecular structures with the CheckMyMetal web server. Nat. Protoc. 2014, 9, 156–170. [Google Scholar] [CrossRef] [PubMed]
- Morshed, N.; Echols, N.; Adams, P.D. Using support vector machines to improve elemental ion identification in macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Konc, J.; Janezic, D. ProBiS-ligands: A web server for prediction of ligands by examination of protein binding sites. Nucleic Acids Res. 2014, 42, W215–W220. [Google Scholar] [CrossRef] [PubMed]
- Roche, D.B.; Brackenridge, D.A.; McGuffin, L.J. Proteins and their interacting partners: An introduction to protein-ligand binding site prediction methods. Int. J. Mol. Sci. 2015, 16, 29829–29842. [Google Scholar] [CrossRef] [PubMed]
- Babor, M.; Gerzon, S.; Raveh, B.; Sobolev, V.; Edelman, M. Prediction of transition metal-binding sites from apo protein structures. Proteins Struct. Funct. Bioinf. 2008, 70, 208–217. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Liang, Z.; Teng, M.; Niu, L. mFASD: A structure-based algorithm for discriminating different types of metal-binding sites. Bioinformatics 2015, 31, 1938–1944. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Tang, G.W.; Altman, R.B. High resolution prediction of calcium-binding sites in 3D protein structures using FEATURE. J. Chem. Inf. Model. 2015, 55, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Valasatava, Y.; Rosato, A.; Andreini, C. Systematic classification of metalloproteins based on three-dimensional structural similarity of their metal sites. Protocol. Exch. 2015. [Google Scholar] [CrossRef]
- Sharma, S.; Cavallaro, G.; Rosato, A. A systematic investigation of multi-heme c-type cytochromes in prokaryotes. J. Biol. Inorg. Chem. 2010, 15, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Tebo, A.G.; Pecoraro, V.L. Artificial metalloenzymes derived from three-helix bundles. Curr. Opin. Chem. Biol. 2015, 25, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.T.; Cangelosi, V.M.; Zastrow, M.L.; Tegoni, M.; Plegaria, J.S.; Tebo, A.G.; Mocny, C.S.; Ruckthong, L.; Qayyum, H.; Pecoraro, V.L. Protein design: Toward functional metalloenzymes. Chem. Rev. 2014, 114, 3495–3578. [Google Scholar] [CrossRef] [PubMed]








© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosato, A.; Valasatava, Y.; Andreini, C. Minimal Functional Sites in Metalloproteins and Their Usage in Structural Bioinformatics. Int. J. Mol. Sci. 2016, 17, 671. https://doi.org/10.3390/ijms17050671
Rosato A, Valasatava Y, Andreini C. Minimal Functional Sites in Metalloproteins and Their Usage in Structural Bioinformatics. International Journal of Molecular Sciences. 2016; 17(5):671. https://doi.org/10.3390/ijms17050671
Chicago/Turabian StyleRosato, Antonio, Yana Valasatava, and Claudia Andreini. 2016. "Minimal Functional Sites in Metalloproteins and Their Usage in Structural Bioinformatics" International Journal of Molecular Sciences 17, no. 5: 671. https://doi.org/10.3390/ijms17050671
APA StyleRosato, A., Valasatava, Y., & Andreini, C. (2016). Minimal Functional Sites in Metalloproteins and Their Usage in Structural Bioinformatics. International Journal of Molecular Sciences, 17(5), 671. https://doi.org/10.3390/ijms17050671