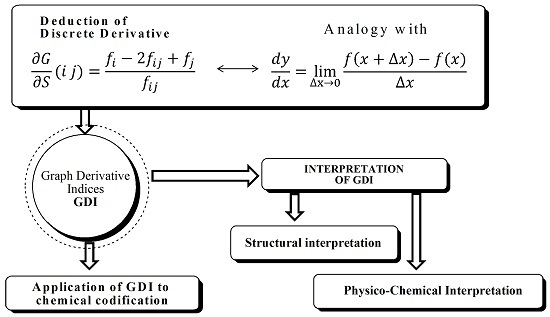
The GDIs have been previously used in several theoretical applications, providing relevant results fundamentally in QSAR/QSPR studies. [
10,
11,
12] However, little effort has been destined to the interpretation of these MDs in structural and/or physicochemical terms [
10,
11,
12,
16]. In this section, the results of different experiments designed to demonstrate the reliability of GDIs in structural and physicochemical hypotheses are presented. The GDIs interpretation will be developed using three different approaches, which will be detailed for every case study.
QSAR/QSPR models were developed by using Multiple Linear Regression (MLR) with the MobyDigs software [
26]. This program allows obtaining MLR equations using the genetic algorithm [
27] as MD selection method. Additionally, it allows for several model validation procedures such as: internal cross validation (Q
2loo), external validation (Q
2ext), bootstrapping (Q
2boot), and Y-randomization, as well as prediction analysis.
3.1. Structural Interpretation
Some of the 13 characteristics, proposed by M. Randic, that new TIs should ideally possess include: possible structural interpretation, isomers recognition, possibility for local definition, as well as present a correct dependence with size and gradual change with structural variations, among others [
28]. These properties are closely related and all of them indicate the direct correspondence that should exist among topological indices calculated for a molecule and its structure. If this condition is achieved the MDs can, at least in principle, describe any chemical information extracted from the connectivity of the molecular structure.
All calculations for the structural interpretation experiments performed in this section were made for derivatives over pairs of atoms with respect to the connected sub-graphs event (each event yields different LOVI values, but the interrelations among them are similar), using the generalized matrix and Pauling’s electronegativity over the incidence matrix as the weighting scheme The LOVIs for each atom in the molecular structures employed in the present section, as well as the corresponding Minkowsky norm for
p = 1(equivalent to the summation operator), the arithmetic mean, and the range (see
Table 1) are depicted in
Table 2,
Table 3,
Table 4 and
Table 5.
3.1.1. Differentiation among Homologs, Multiple Bond Codification and Positions
From
Table 2, it can be inferred that LOVIs for all the different atoms decrease as one get closer to the center of the carbon chain, belonging to the considered linear alkanes and their homologs (for a sake of simplicity, the hydrogen atoms are no taken into account in the representation of the G).
The value of every atom of the chain also increases as one moves from an inferior homolog to a superior one, maintaining the regularity among the LOVIs of atoms in the same molecule. Besides, the total invariants shown in this Table reflect a regular increase in their respective values when the number of methylene groups –CH2– in the structure increases from one homolog to another. The increase of the electronic density and its position in a specific region can be also quantified using this mathematical description. It is observed that atoms connected by multiple bonds also increase their LOVI values when the number of bonds among them increases. Consequently, the total (global) invariants from this molecule increase when the number of atoms increases. As illustrated these total invariants can be used for differentiating position isomers because their values decrease in the measure that the multiple bond is more embedded in the molecular structure.
3.1.2. Differentiation among Chain Isomers
In
Table 3, are the results of an experiment analogous to the previous one but now using chain isomers of saturated hydrocarbons with six carbon atoms.
Table 2 shows how the variations in the carbon chain from the molecule cause variations in the LOVIs values from each atom, according to the modifications experimented in the molecular structure, from one isomer to another.
It is interesting to note the peculiar variation of the different invariants when the quantities of ramifications increase, and consequently the length of the main chain decreases. Note that the invariants decrease their values, which is a logical result since LOVIs also experiment a decrease in their values in the measure that molecules are more ramified.
3.1.3. Codification of the Presence of Different Functions
Until this point, only molecules comprised exclusively of carbon and hydrogen have been described. Here, the capability of GDIs for describing structures containing different organic functions is evaluated.
The organic functions play a fundamental role in the properties of bio-active molecules; therefore their description cannot be ignored. In
Table 4 the results of the calculation of the atomic and total indices for a series of propane derivatives is summarized. As in the previous cases, the electronegativity according to Pauling’s scale is employed as label for differentiating the atoms in the molecule.
It is observed that when the electronegativity of atoms located in “X” position increases, the LOVIs values also increase and the total invariants increase in almost a regular way. It is interesting to detail that the increase observed in LOVI values, from a molecule to another, is proportional to the increase in the property used for characterizing the atoms. Besides, it should be pointed out that this increase in the LOVI values due to the presence of different X groups is identical to the summation of the increase of the rest of the chain.
3.1.4. Cycle, Conjugation and Aromaticity
Table 5 shows a set of cyclic structures and some few acyclic chains. The purpose of choosing these systems is to determine if GDIs are able to retrieve information about the conjugation, aromaticity and the presence of cycles, during the numerical description of the molecular structure.
From a detailed analysis of data in
Table 5, it can be observed that LOVIs from a cyclic structure are smaller in comparison to the values obtained for the corresponding acyclic structures with an isolated unsaturated bond. In the measure that the number of unsaturated bonds increase, the LOVIs values decrease, but these decrements are more significant when electronic conjugation is present in the structure, meaning that the aromatic molecules are characterized by the lowest values. The presence of heteroatoms in the systems increases the LOVIs values for carbon atoms compared to counterparts without heteroatoms. Moreover, atoms bound to heteroatoms present a smaller index value than the other ones, which is an indication that these atoms have a lower electronic density provoked by the direct interaction with a more electronegative atom. This result is consistent with the chemical reality of heteroatomic systems.
3.1.5. Molecular Symmetry Described in Terms of GDIs
A more complete structural description is obtained when the symmetry of the molecules is incorporated in the description achieved for the GDIs as previously shown.
In this sense, Shannon’s Entropy is used as a symmetry index (see Equation (9)) [
4]:
where
is the cardinality of the equivalent classes, which in our particular case is the number of equivalent LOVIs and N is the total number of atoms in the molecule. The symmetry index may be interpreted as a measure of the bidimensional symmetry of an organic molecule, and it takes values between 0 and 1 (
). A value of 0 corresponds to molecules with high 2D symmetry, whereas a value of 1 is associated with asymmetric molecules.
Table 6 shows the SI values computed for ten organic molecules, taking as base the cardinality of the LOVIs obtained with GDIs for derivatives over pairs of atoms with respect to the connected sub-graph event and by weighting the incidence matrix with Pauling’s electronegativity.
From the analysis of
Table 6, it can be inferred that the resulting SI are logical and acceptable if it is considered that this index contains information on the symmetry of the molecules based on the local vertex invariants calculated for these 2D structures.
3.2. Reactivity Based on Geometrical Effects
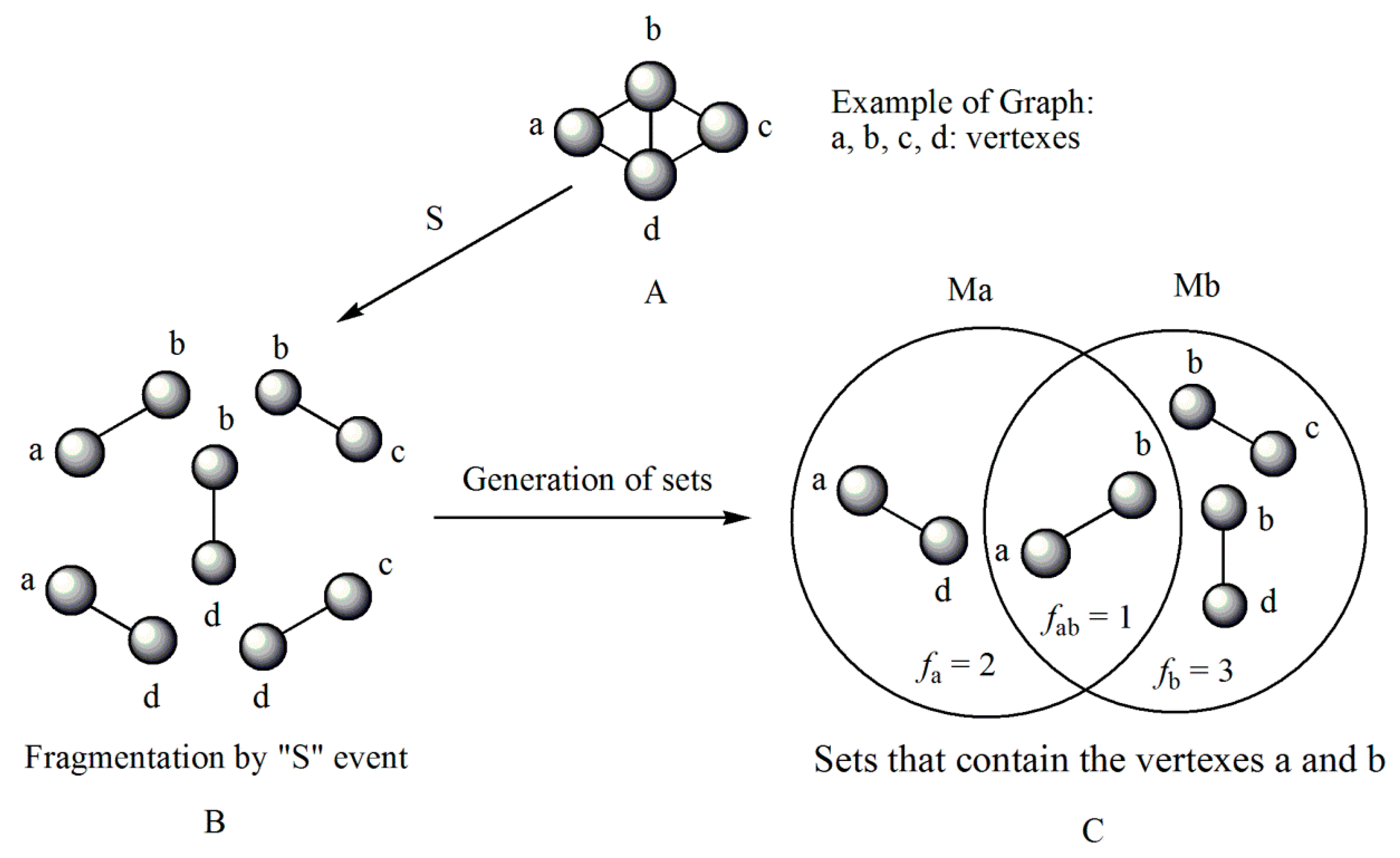
Consider a molecule fragmented with respect to the connected sub-graph (S) for order 1. Note that the individual frequencies are the number of total connections for each atom in the model generated by the considered event. For this particular case (S event for order 1), the individual frequencies correspond to the number of sigma bonds for each atom. In electronic terms this frequency may be written as:
where,
,
,
,
, and
are the total valence, the number of hydrogen atoms bond, the valence number with suppressed hydrogen atoms, the quantity of π bonds, the number of lone pairs associated to atom
i and the number of sigma bonds from the atom
i, respectively.
According to the latter expression, it can be observed that the individual frequency of order 1 can be simply defined as the number of sigma bonds of atom i. In the case where the generalized frequency matrix (including kth-order sub-graphs, k > 1) is employed, the interpretation of the individual frequency holds but should be identified as the number of connections between atoms separated by distance k, depending on the model generated by the chosen event and the maximum fragment order.
In this sense, the derivative evaluated for frequencies of order 1, over a pair of bonding atoms may be written as:
For carbon atoms in an organic molecule Equation (13) can be simplified to:
By analyzing Equations (12)–(14), it is evident that the derivative codifies information on the total number of sigma type connections associated to a pair of atoms (i j) connected at a specific distance. In this sense, the derivative can be defined as measure of the “accessibility” of a given region of a molecule.
The LOVI for each atom
i can be defined as the multiplicity of its sigma bonds affected or influenced by the respective surrounding multiplicities. This definition can be mathematically written as:
The term 2n appears from the expression of the derivative and it has the effect of cancelling out the elements repeated in both groups of bonds.
Based on these concepts, the LOVI value of an atom can be regarded as the degree of participation of this atom in the formation of the molecular structure; therefore, the accessibility
to an atom can be evaluated as the inverse of its LOVI because it is proportional to the total area in which an atom is likely to be attacked by any external entity. In this sense, the interaction capacity of two atoms located in different molecules can be evaluated as the product of their accessibilities
, defining the interaction capacity of an atom
i with any other atom
j as the addition of accessibility contributions of the atom
i with the rest
. Generalizing this idea, the interaction capacity of two molecules (
), understood as the total contribution of all the individual capacities of the constituent atoms determined from their accessibilities, should be proportional to the interaction probability of two molecules, which can be mathematically expressed as:
where,
is an operator, which involves the operation of the individual interaction capacity from each atom (
M would be the sum, product,
etc.),
P is the real probability of interaction between two molecules and
is the theoretical probability evaluated by taking as base the LOVIs from each atom. The real value of the intermolecular interaction probability
P depends on the structures (symmetry, form, size and distribution). These structural parameters are carefully quantified by GDIs as was showed in the previous epigraph.
In Collisions Kinetic Theory, the magnitude known as the Steric Factor (F), is considered as a consequence of the analysis of incongruences between theory and the experimentation results and it expresses the probability of the corresponding geometrical configuration during interactions [
29]. Posterior deductions showed that the Steric Factor from Collisions Kinetic Theory is analogous to the
term, which is related with the probability of existence of a viable configuration able to interact with another specie [
29] and
is the variation of entropy of the active complex and R a constant
.
The LOVIs, their inverses, and the parameters introduced in both kinetic theories, have a direct relationship with the probability of intermolecular interaction based on structural configurations during the interaction. In this sense, it is possible find analogies between these kinetic parameters and the local and total indices derived from the corresponding mathematical procedures implicit in GDIs calculation.
To evaluate the veracity of the aforementioned ideas, second order dimerization reactions, in gaseous states were studied as an example.
Table 7 summarizes the ΔS
# data for each studied reaction [
30].
In each case, the total interaction capacity was evaluated and it was related with the activation entropy for the process of dimerization of diverse unsaturated molecules, yielding a correlation of 97.58%. The operator taken to evaluate the totality of the atomic contributions was the standard deviation, which is a measure of the dispersion of the values.
The activation entropy is a parameter associated with the probability of an adequate interaction between two spaces, taking as reference the structural organization of the created transition state, which is product of a suitable interaction. In this sense, it is logical to find a strong relation between parameters obtained from GDIs calculation and this magnitude. This example can be taken as base for understanding the molecular graph derivative from a kinetic point of view, which is closely related with the possibility of interaction of the molecules based on their corresponding geometrical structures.
3.2.1. Accessibility as Measure of the Interaction’s Capacity
Accessibility is defined as the possibility of entrance or access capacity, in this case, to a specific region in a molecule. The Kier and Hall [
31,
32] molecular connectivity indices have been recently studied by Estrada in terms of the Relative Bond Accessibility Area (RBA),
Cij, which is expressed in square Randics (
R2) [
33,
34]. The reactivity of an atom is related with its accessibility, and the accessibility can by quantified from topological features of the molecular structure that contains this atom [
35,
36].
As was mentioned in the previous section, the inverse of a LOVI may be considered as a measure of the availability of a particular zone of a molecule (fully characterized by a pair of atoms) to interact with an external agent. From this definition, it can be stated that this MD is closely related to the steric effects controlling molecular reactivity or simply steric reactivity. The steric reactivity associated to a pair of bound atoms can be evaluated as
, where
is the LOVI of atom
i. In
Table 8, steric reactivity indices computed for a collection of molecules are summarized together with RBA in
R2 calculated by Estrada for these molecules [
33,
34].
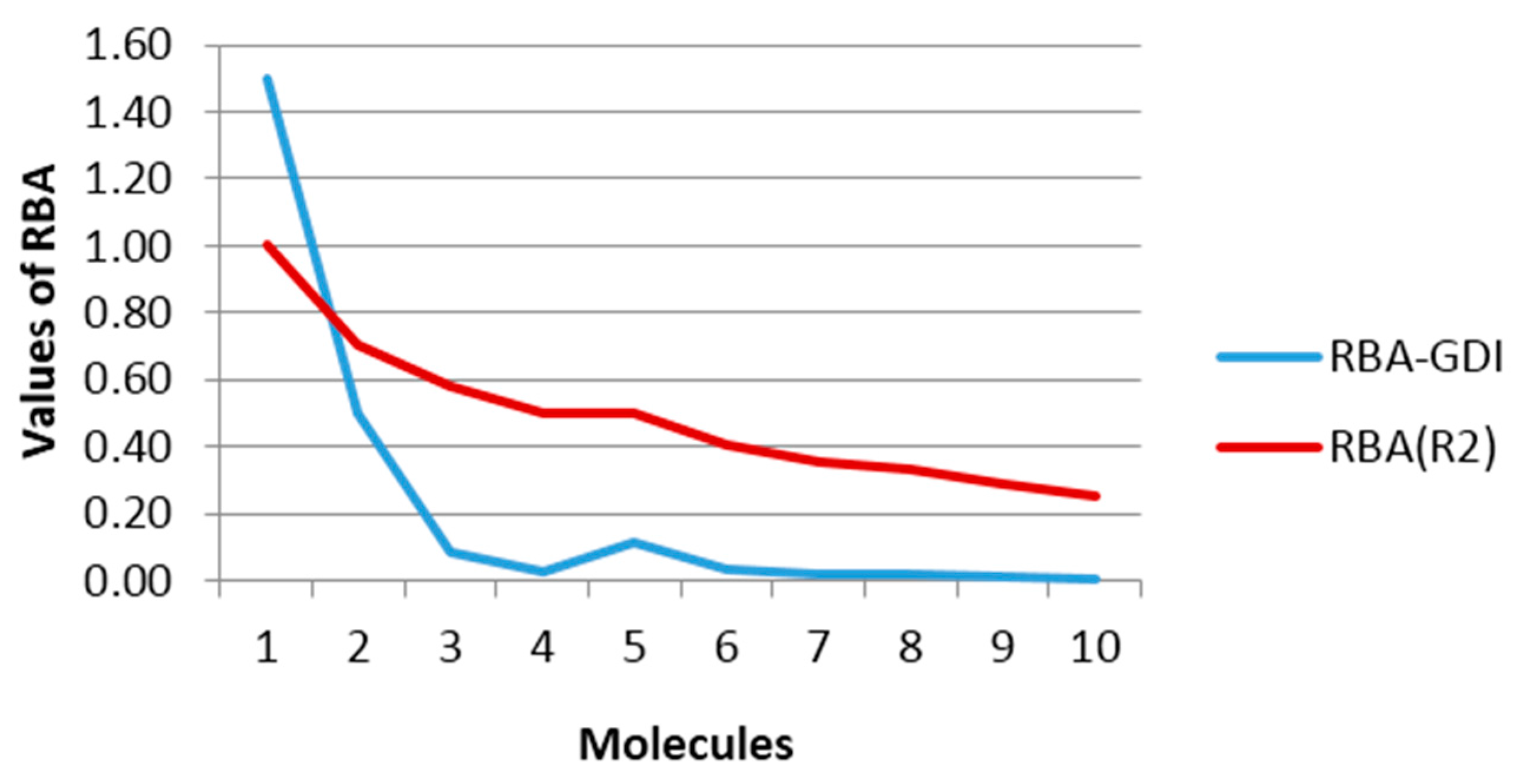
Both sets of data from
Table 8 are plotted in
Figure 4, where a qualitative agreement between the two curves may be observed. Note that for the pair of atoms (a,b) in the molecules
4 and
5 (2,2-dimethylpropane and butane, respectively) the same RBA values were obtained using the connectivity index, while the evaluation of the steric reactivity with GDIs calculations yielded better differentiation more consistent with the chemical reality because it is anticipated that the a–b bond in the 2,2-dimetylpropane molecule would be less accessible due to greater steric hindrance than the a–b bond in the butane molecule. This challenge of index degeneration in situations like these is not encountered with GDIs.
Another important aspect is the infinite value obtained for the trivial case of the ethane molecule. Although this value is useless for further statistic or algebraic developments, it is logical if we take into account that it is evaluating the accessibility of an entity external to a particular bond in each molecule. With this idea it plausible that the ethane molecule has infinite accessibility possibilities given that it is constituted by only one bond (G with suppressed H-atoms were considered in all cases). To visualize this effect in
Figure 4, an R-GDI value equal to 1.5 was arbitrarily assigned to this molecule to give the idea that it is superior and thus allowing for the visualization of the regularities of the rest without affecting the scale. Note, however, that this does not mean that GDIs would not have the capacity of codifying the structure of one molecule as simple as ethane, because this infinite value is only given for the order 1, which is the configuration used for simplifying the physical interpretation of the GDIs that may later be generalized to more complex systems (
i.e., using sub-graphs of superior orders in the event S, derivatives over
n-elements, using other events, and so on).
3.2.2. Specific Reaction Rate and Its Relation with GDI
The specific reaction rate constant is the speed of chemical transformation when the concentration of all the reagents is equal to 1 mol/L [
29,
30]. Thus, the rate constant is a magnitude proportional to the reactivity of the molecules that participate in a reaction, with the temperature of the system being constant [
29].
In the previous sections the derivative over a pair of bonded atoms was defined as the quantity of sigma connections of both atoms and was expressed as a difference of electronic contributions that express the part of the total electronic density destined to connections or sigma bonds. In this sense, the derivative over a pair of atoms i and j can also be understood as a measure of the interaction capacity of a bond with a neighboring molecule.
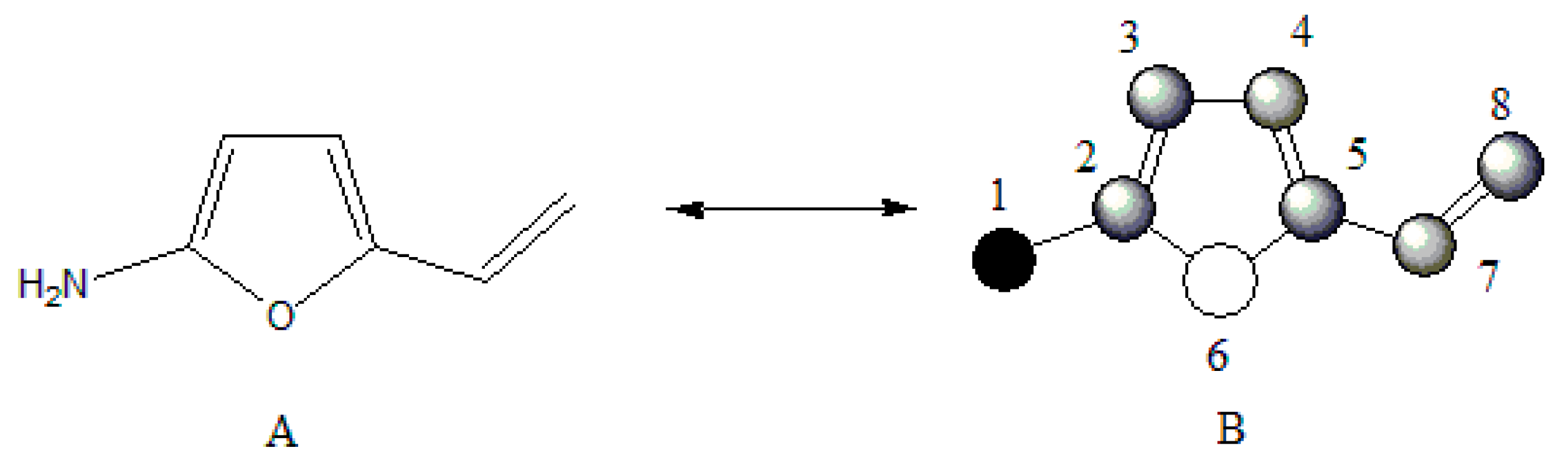
To evaluate the previous affirmation a data comprised of 34 derivatives of 2-vinylfuranes was employed, for which the specific rate constant for the nucleophylic addition to the double exocyclic bond from this molecules with mercaptoacetic acid has been reported [
2]. The best one variable regression shows that there exists a moderate relation between the property and the GDIs. However, taking into account that the best models reported by other authors for describing this property use seven variables [
2,
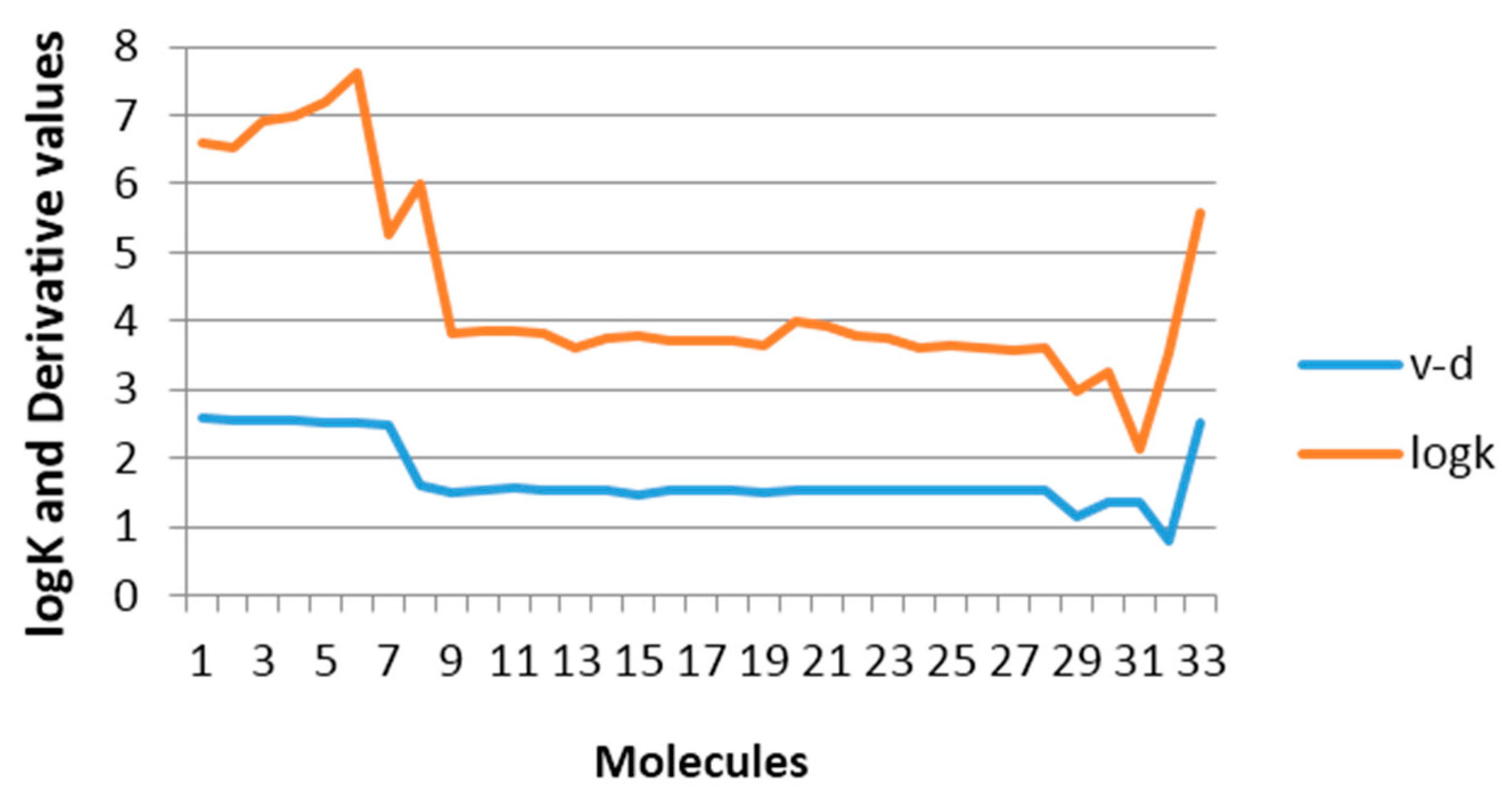
37], it is noteworthy that with only one descriptor (derivative over the double exocyclic bond) it is possible to explain approximately 80% of the variance of the experimental property.
Figure 5 shows the correspondence between the derivative values and the experimental values from log
K.
3.3. Interpretation of GDIs in Electronics Terms
The nature of the atoms and the molecules is determined by their electronic structure [
21], thus, by describing the dynamics, distribution and the electronic energy of these systems, it is feasible to establish a useful nexus for the better understanding of molecular structures and/or the methods used to codify them.
The detailed structural analysis in the previous epigraph showed that there is a relationship between GDIs and the influence of the electronic richness of atoms bound to a specific center in a molecule. Moreover, the obtained result demonstrated the possibility of a physicochemical interpretation in kinetic terms, on the basis of the quantification of the interaction potential of a molecule described by means of GDI and LOVIs obtained in geometrical framework. All these aspects are easily assimilable if we take into account the individual frequencies and the derivatives over atom pairs expressed in electronic terms as explained in the previous section.
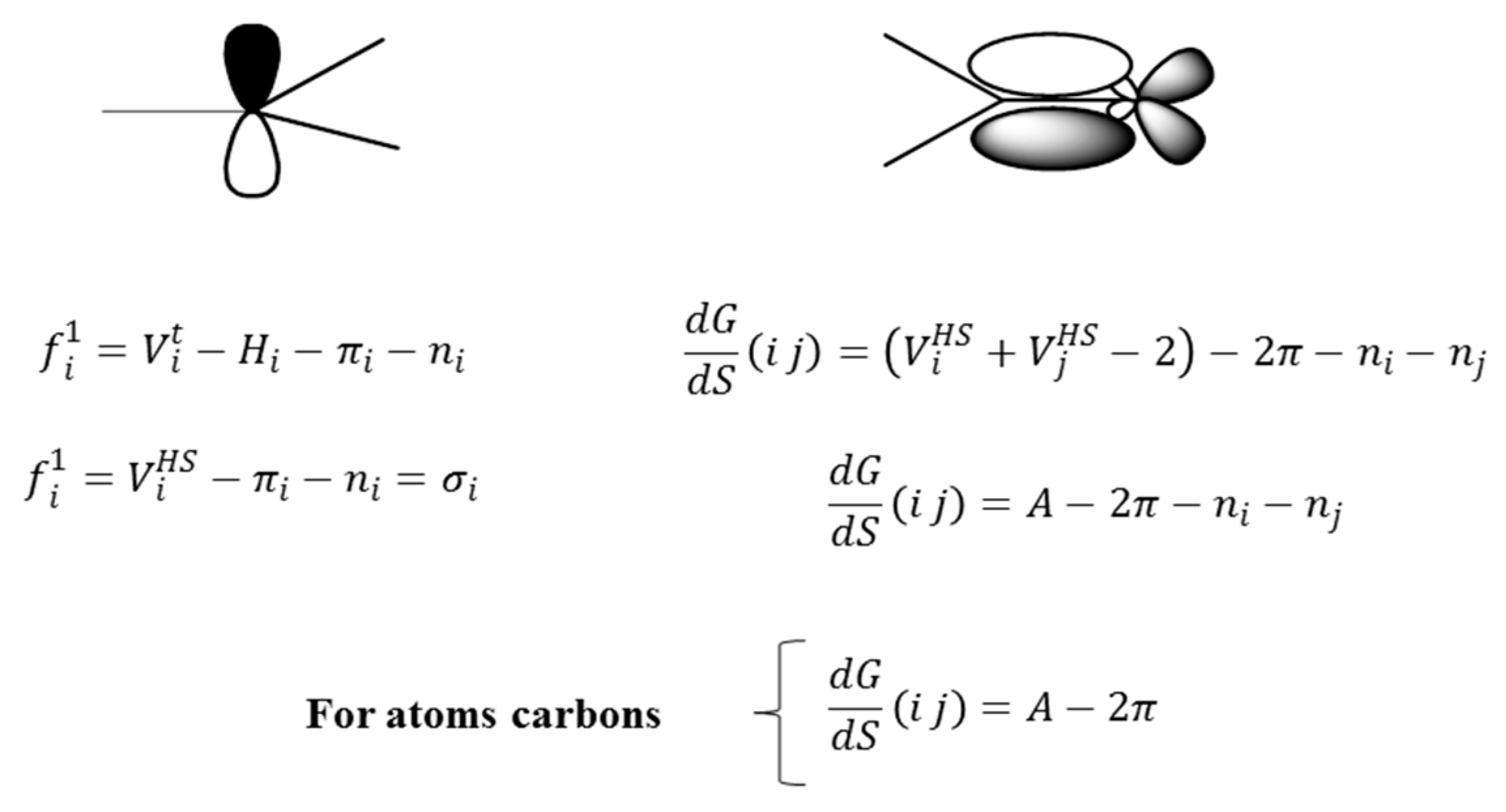
Figure 6 illustrates the electronic decomposition of the individual frequencies of order 1 and the derivatives of order 1 for a pair of bonded atoms.
Up to this point, only the quantity and distribution of bonds in the molecular structure has been taken into account for the GDIs interpretation. However, the knowledge about these bonds and their distribution around each vertex determines the electronic density in the environment of each atomic nucleus in the molecular structure, and thus motivating a deeper analysis of the GDIs from a Quantum Mechanics perspective.
From
Figure 6, the orbital description of the frequencies allows for two observations: (1) if the frequency is equal to the number of sigma bonds of an atom, it then also quantifies the hybridization of the atom and the symmetry of the sigma electronic distribution and (2) the parameters to the left suggest a separation of the electronic terms corresponding to the quantification of σ, π and non-shared electrons, which allows for the evaluation of the separated influence of these types of electrons, as it will be corroborated in the next part of the present study regarding the relation of the frequencies with Hückel’s free valence. It can be pointed out that only the frequency of order 1 is similar to the number of sigma bonds belonging to an atom. Nonetheless, it is anticipated that a similar concept holds in an abstract model based higher order frequencies obtained according to different fragmentation approaches.
For the case of the derivative, it can by described as a quantity related to the part of the total valence of the bonding atoms, which is destined to the establishment of connections in a molecular network. Equally, the separation of the π electrons offers a differentiated treatment to each type of electrons (it makes reference to the electrons that form part of the covalent bonds, π- or σ-type and non-shared electrons), considering the basic differences among their characteristics and expressing one in function of others, which shows the relation among them.
The fact that both the frequency and the derivative can be can be expressed in terms of the distribution of electrons around one atom or bond approximates to the idea of chemical reactivity, now understood from the perspective of the electronic interaction capacity. In next sections, carefully designed experiments will show the strength of these basic local descriptors from the discrete derivative algorithm in describing the electronic characteristics of atoms and molecules.
3.3.1. GDIs, Chemical Reactivity and Relation with Hückel’s Free Valence
A possible approximation for studying the chemical reactivity is to determine the degree in which the atoms in a molecule are united to the adjacent atoms, which is relative to their theoretical bonding capacity [
38], The degree in which one atom is united to its neighbors can be calculated summing all the values of the bond orders of that atom. If the sum of all the bond orders is subtracted from the value of the highest bonding capacity, we would obtain the free valence:
For conjugated hydrocarbons, the free valence index can be evaluated as:
where,
is the bond order for the atoms (
i j), determined by Hückel molecular orbitals method [
38,
39].
The individual frequency of each atom evaluates the number of connections of an atom in a specific model generated by an adequate event for describing the molecular structure. In the particular case of the event S, if only the order 1 matrix is taken into account, it is possible to relate this frequency with the quantity of sigma bonds of an atom.
Reorganizing the terms for the individual frequency shown in the
Figure 6, we obtain:
The term to the left of the Equation (19) describes the part of the total valence dedicated to forming π type bonds. The right term shows the number of π reactive electrons and the number of H-atoms bonded to each carbon. Both members from that equation show huge similarities with the Equations (17) and (18), although from different optics but explaining the same characteristic: chemical reactivity of each carbon atom in the molecular system, therefore, it should be hoped that:
Taking into account the equation 19, a study was performed on the behavior of the free valence calculated by the Hückel molecular orbitals method for specific atoms with different environments in a dataset composed of 19 conjugated hydrocarbons, with some of them being aromatic. This chemical dataset was primarily used by Kier and Hall [
39] with the goal of finding a relationship between the free valence and the Electropologic State index, which is a descriptor regularly employed in correlations with electronic properties of different molecular systems and known to yield good results [
39,
40].
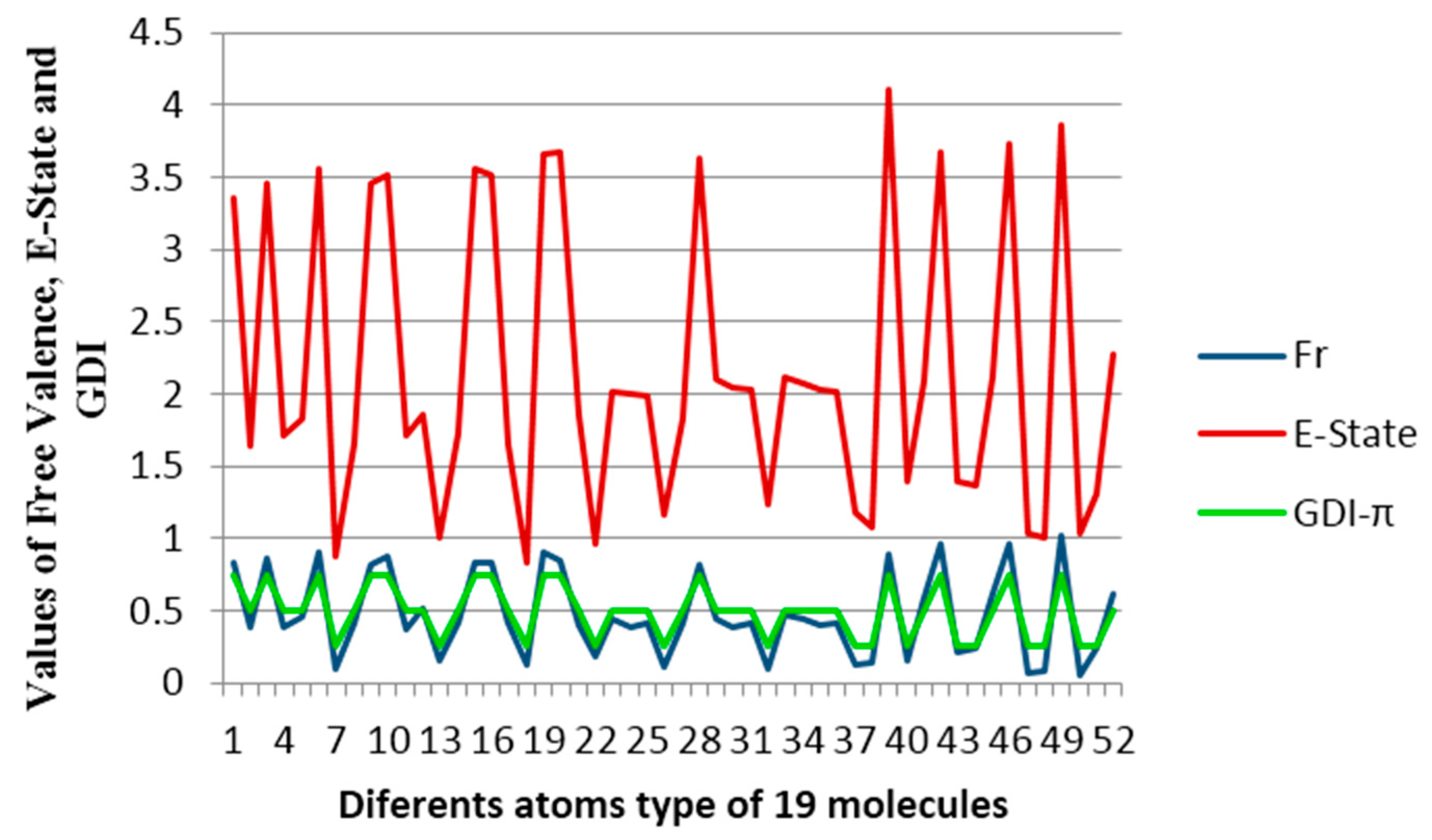
Figure 7 shows the free valence calculated by the Hückel’s molecular orbitals method (blue line), the Electropologic State index (red line) and the derivative indices taking as base the individual frequencies of the atoms. A uniform variation between the values obtained for Hückel’s free valence and the GDIs is observed and this evidently shows that the GDIs quantify electronic environments of atoms in the molecules and reinforcing the understanding of the GDIs in reactivity terms. In previous studies, Kier and Hall grouped the atom types in these molecules in three categories: carbon atoms with one hydrogen, carbon atoms with two hydrogens and carbon atoms without hydrogen [
39]. Likewise, a linear relationship is found in the present experiment:
where,
is the proportionality constant which adopts different values for each type of atom from the system and it allows finding reactivity values closer to the values evaluated by Huckel’s method.
Table 9 shows the six types of atoms that can appear in conjugated systems conformed by only one carbon and hydrogen.
The Equation (21) shows a simple relation to evaluate Hückel’s free valence taking as base the order 1 frequencies with the S event. It is obvious that in the Expression (21), the goal is not the substitution of the real calculation of Hückel’s free valence, but the use of this expression allows establishing in a faster and simple way an estimate of the chemical reactivity of any carbon atom in a conjugated molecule without evaluating the bond order, for which it is necessary to know the coefficients of each atomic wave function that participates in the linear combination to form the molecular wave function.
Another advantage of the Equations (20) and (21) is that they demonstrate a clear existence of a nexus between the electronic environment of the atoms and their corresponding atomic indices evaluated with derivative indices, reaffirming the notion that graph derivative indices describe electronic properties of atoms in a molecule and consequently their electro-chemical reactivity.
3.3.2. Electronic Interpretation
In the beginnings of 1950s, it was discovered that the resonance frequency of a nuclide depends not only of its magnetogyric ratio and the intensity of the magnetic field B
0, but that it also depends of the electronic environment where the nuclide is located [
41]. For one nuclide in a specific substance, there will be as many resonance frequencies as the electronic environments. This phenomenon known as chemical displacement (shift), is the base of the NMR (Nuclear Magnetic Resonance) [
22,
41] chemical applications. The chemical shift is a descriptor of the electronic characteristics of each atom from a molecule. In the following experiments we intend to find linear relationships between the chemical shift of some active nuclides in NMR, with the goal of discovering in what measure the electronic information of atoms and molecules is codified in the structural descriptions based on the GDI concept.
QSPRs of Chemical Shift of 17O-NMR for Aldehydes and Ketones
For this analysis a data of aldehydes and ketones was used, which have been previously studied by Kier and Hall [
40] with the Electropologic State index. All molecules are aliphatic and their chemical shifts of the
17O have been reported in the literature (see
Table 10).
The variables used for linear regression were obtained by calculating the duplex derivative with respect to 10 different events (using chemical, physical and graph-theoretical atom-labels) and several norms, means, statistical and classic invariants as total and local MDs. For this experiment the MobyDigs software was used [
26].
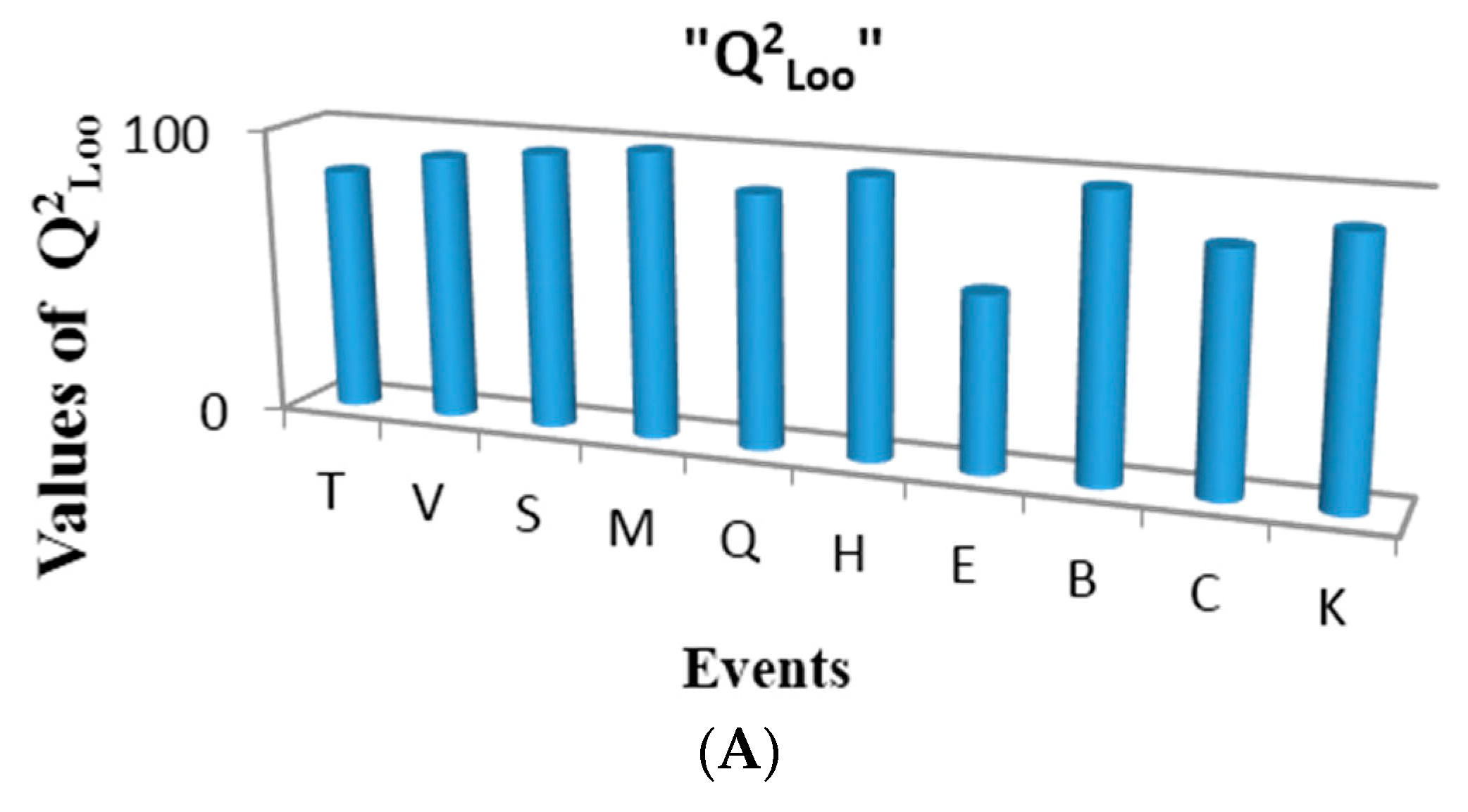
Figure 8A shows the performance of one variable regressions built for each of the events in the present study, according to the cross validation parameter Q
2Loo. As can be observed, the events with the best correlations for the studied property are Sub-Structure (B) and Multiplicity (M), respectively. This observation is logical considering the fact that first one is a fingerprint-based event [
3,
16], and it conforms the incidence matrix only with substructures with functional groups and/or atom types of chemical interest, while the second one is a topological description of the connections at one step topological distances and their multiplicities (simple, double and triple bonds between pairs of atoms in a molecule) [
3,
16]. These two events yield matrices that reflect the electronic richness of the molecule, fragmented in individual bonds and their multiplicities. The best regression (based on the sub-structure event) obtained in the present study is shown below (see Equation (22)):
R2 = 98.65%, Q2Loo = 98.1%, Q2Boot = 98.21%, s = 2.304, scv = 2.41, ysc = −0.051, F = 512.25.
As it can be observed from the statistics of this equation, a good correlation (
R2 = 98.65% and Q
2Loo = 98.10%) is obtained between the calculated MDs and the experimental chemical shift for
17O. An analysis of the descriptor contained in this model shows that it expresses arithmetic mean of the LOVIs of the carbon and oxygen from carbonyl group (IS: unsaturated bond), weighted by the valence degree, which is a topological expression of each atom [
4]. This is a logical result taking into consideration that the value of the chemical shift of oxygen mainly depends on the electronic environment of this atom as well as of the influence of the electronic density of its unique adjacent atom (carbonyl carbon); therefore, the average is a direct quantitative measure of the electronic richness in the model, which is the main factor that influences the chemical shift of the nuclide of
17O.
QSPRs of Chemical Shift of 17O-NMR for Eithers
A similar study was performed using a dataset composed of 10 aliphatic ethers (
Table 11), for which the chemical shift of
17O-NMR was reported [
40]. The best one-variable model obtained in this study is shown below (Equation (23)):
R2 = 99.28%, Q2Loo = 98.94%, Q2Boot = 99.05%, s = 3.588, scv = 3.891, ysc = −0.046, F = 1102.54.
As can be seen, this model is statistically robust (R2 = 99.28% and Q2 = 98.94%). It is especially interesting that the variable that best correlates with the modeled property is the one that uses the topological polar surface area (T) to weight the oxygen LOVI. This property is an expression of the electronic and steric environment of the oxygen nucleus and the LOVI explains the influence of the groups adjacent to oxygen. Therefore, it is logical that this local atomic index has greater influence in modeling the studied property.
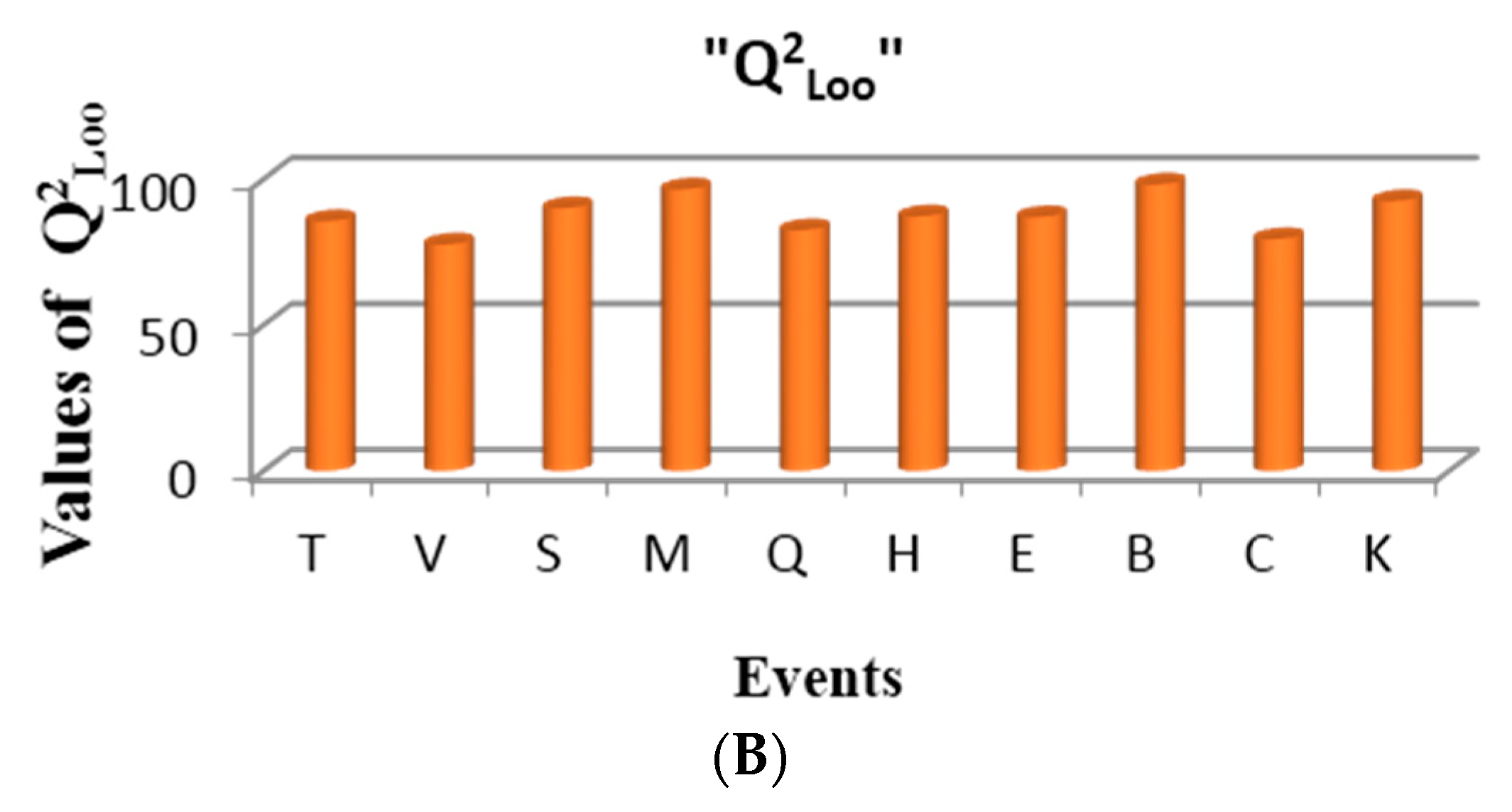
Figure 8B shows the performance of one variable models built for the events in the present study, according to the cross validation parameter Q
2Loo. As can be observed, the best events for describing the electronic properties of atoms in these molecules are: multiplicity (M) and sub-structure events (B), respectively. Good performance is also achieved with connected sub-graphs (S) and Sach’s sub-graphs (H).
Table 11 shows the values of the variable that best correlates with the mentioned property. In the measure that chemical shift values increase, the LOVIs values decrease in a regular way, having a negative contribution in the model.
This linear relationship between the LOVIs from the oxygen atom and its chemical shift in NMR implicates that the LOVIs from the oxygen atom codify electronic information varying in an almost uniform way with the chemical shift values.
Comparison between Real Spectrums and Atomic GDIs for Alkanes
To comprehend better the relationship of the GDIs with the electronic properties of the atoms of a molecule, some simple alkane molecules have been selected and their protonic Nuclear Magnetic Resonance spectrum (
1H-NMR) predicted. Posteriorly, these are superimposed with the LOVIs values computed for the atoms in each molecule. The spectra were predicted using the ChemDraw program [
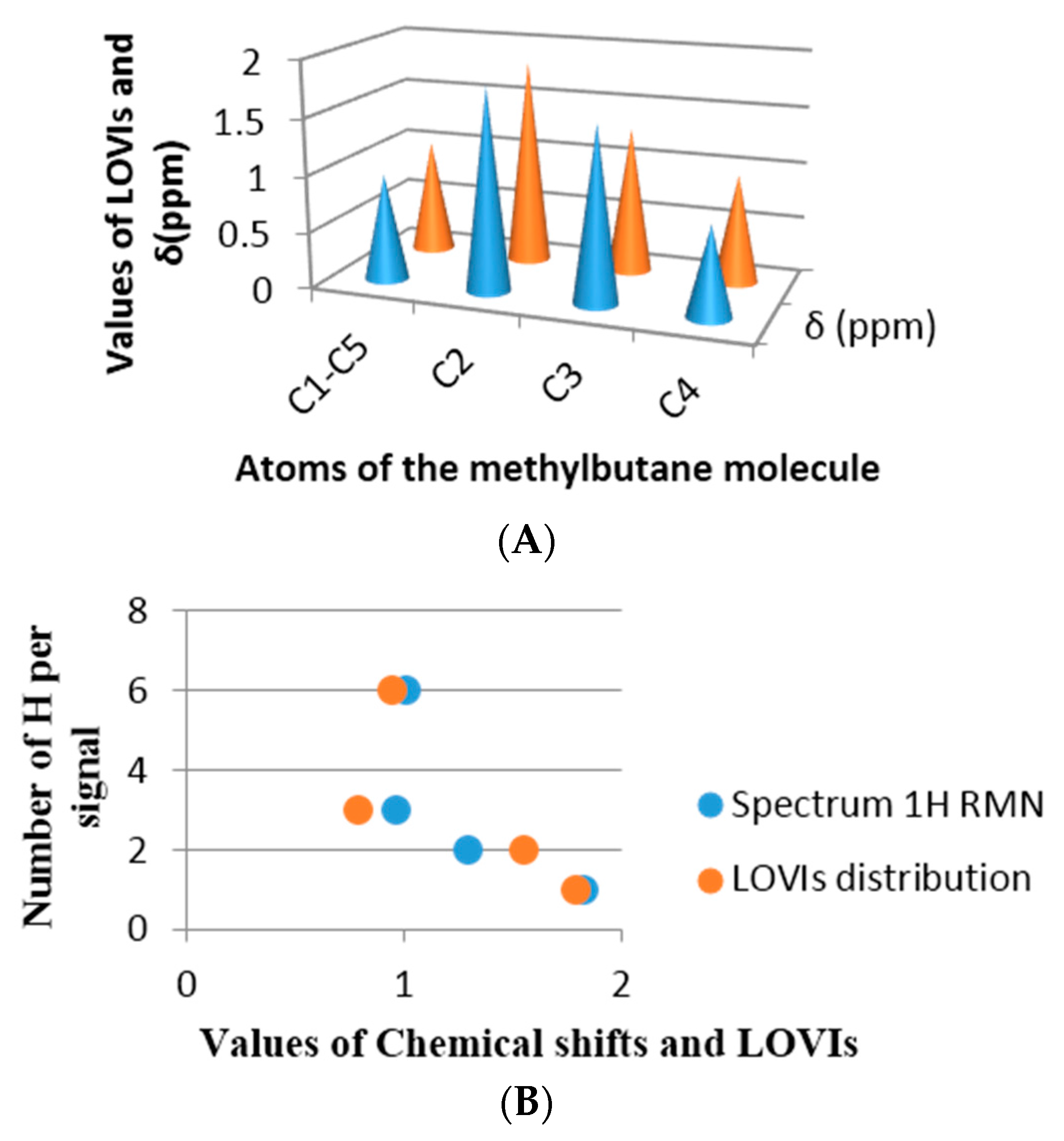
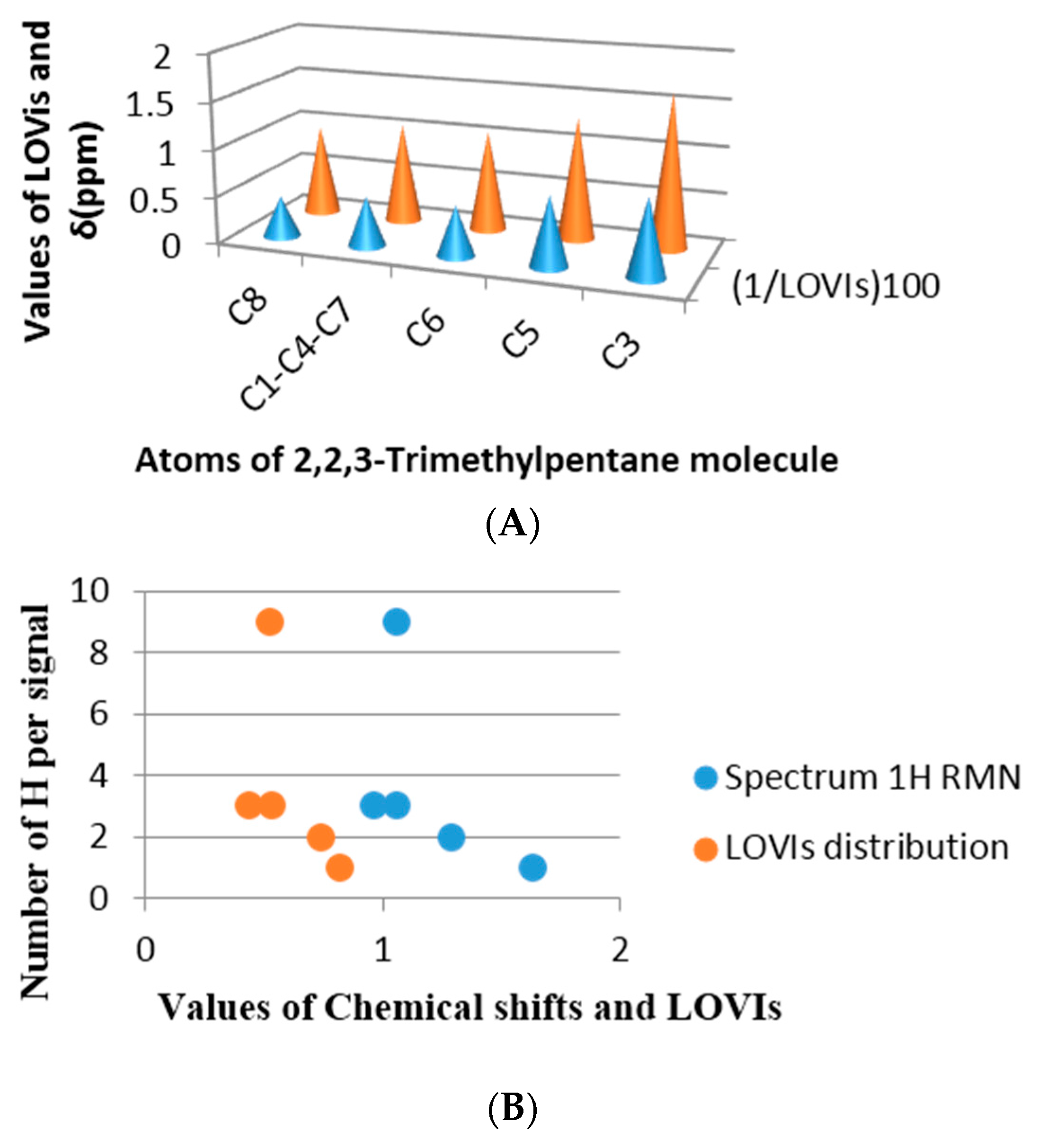
42] and in both examples a good estimation was achieved. The selected molecules were methylbutane and 2,2,3-trimethylpentane, both molecules have all their sp3 carbon with different environments.
Figure 9 shows the correspondence obtained between the chemical shift in ppm from each proton and the inverse of the LOVI value for each carbon corresponding to that proton (or group of protons). It can be observed that there is an unequivocal numerical proximity between both groups of values.
If the chemical shift is a numerical expression for electronic density of a nuclei as well as the surrounding electronic environment, influenced by the electronic richness of the adjacent atoms, then a linear relationship between the chemical shift and the GDIs may be found, which means that our MDs codify steric and electronic information of the atoms in the molecules.
Figure 9B and
Figure 10B in each previous case are an approximation of the real spectrum (without taking into account the signal’s multiplicities), where the similarity of both spectra may be observed: the spectrum obtained from the chemical shift and the one obtained from the calculated GDIs. However, when a more realistic spectrum is needed, additional considerations are required: Calculation of the integrated intensity and multiplicity.
The Integrated Intensity (quantities of hydrogen bonded to the atom (
i), which provokes the signal) is computed according to the Equation (24):
On the other hand, the signal’s Multiplicity is determined by Equation (25):
where δ is the vertex degree of an atom and the atoms in the sum are those that possess derivative values different from zero with the atom
i, for the order 1, according to the Connected Sub-graph (CS) or Multiplicity (M) event.
Description of Global Electronic Properties. Energy of Resonance
The resonance energy or stabilization energy by resonance is the difference between the energy corresponding to the structure with double (or triple) rigid bonds established in positions located in molecules with alternate unsaturated bonds (the most probable Kekule’s structure) and the real energy of the substance. The latter (
i.e., real energy) is less than the former and this decrement is associated with electronic delocalization [
43,
44]. In this study the correlation between the resonance energy from a dataset composed of 17 aromatic molecules and values calculated by GDIs was determined using several atomic labels (chemical, physical and graph-theoretical atomic properties) and 10 events. The existence of a linear correlation between the previous mentioned property and GDIs implies that they are able to characterize electronic densities and their delocalization capacity (which would corroborate with the study in Epigraph 3.1.4).
The best one and two variable models together with their corresponding statistical parameters are shown in Equations (26) and (27), respectively:
R2 = 98.25,
s = 5.286,
Q2Loo = 97.41,
sCV = 6.049,
Q2Boot = 97.60,
ysc = −0.038,
F = 844.03.
R2 = 99.24, s = 3.613, Q2Loo = 98.99, sCV = 3.764, Q2Boot = 99.01, ysc = 0.017, F = 910.02.
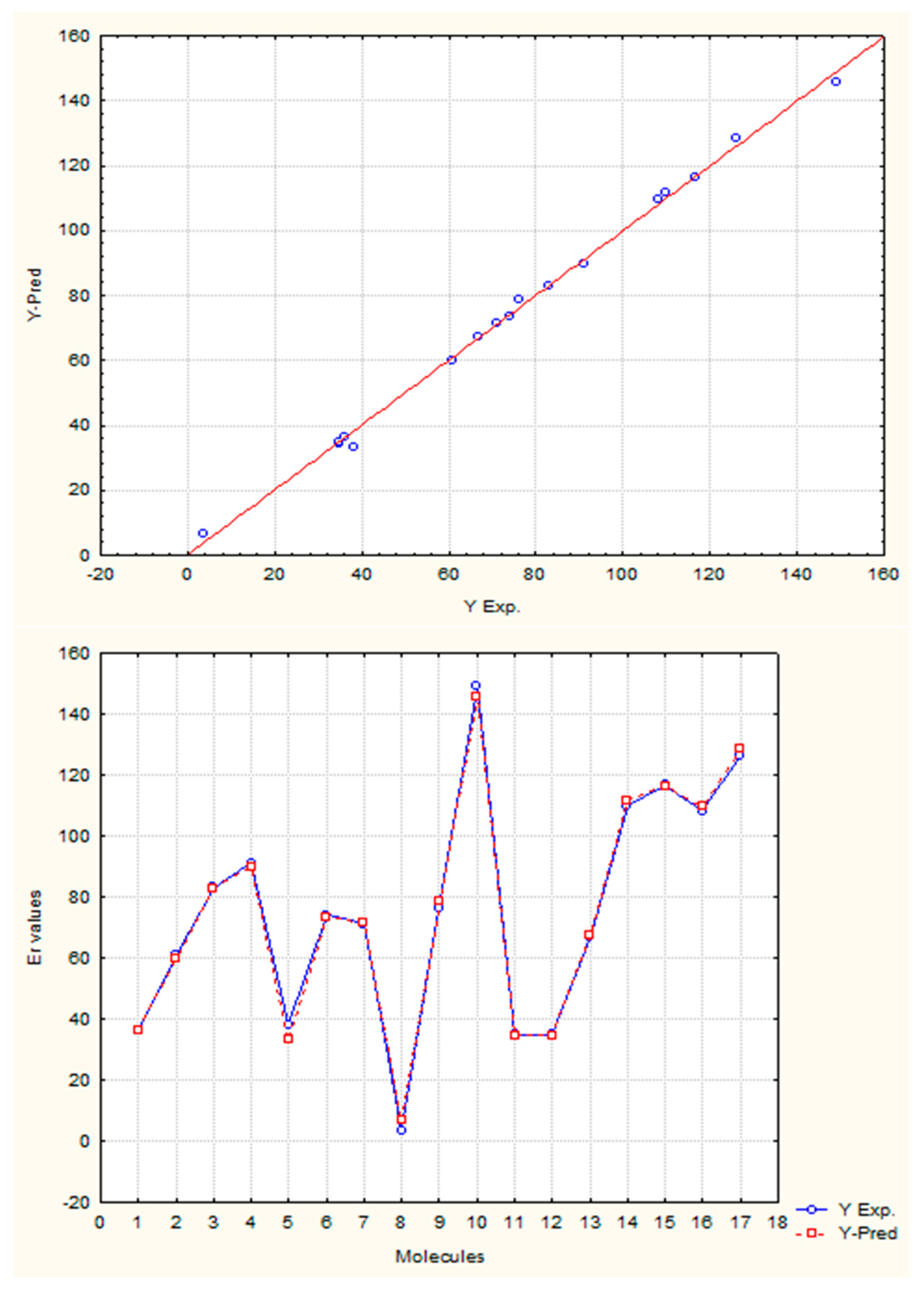
The satisfactory statistical parameters from these models demonstrate the close linear relationship between GDIs and the resonance energy for this group of molecules. The Equation (26) with only one variable is able to explain more than the 98% of the variance of the property.
Table 12 shows the experimental and calculated values with the Equations (26) and (27), and their corresponding residuals. It is interesting to point out that the invariant that best correlates with this electronic property is the norm 1, which is the linear combination of the individual LOVI values.
In the previous experiment it was demonstrated that the atomic index values have a close relationship with the electronic properties of atoms in their molecular environment; therefore, as expected one total descriptor such as the norm 1 (N
1, see
Table 1 for more details) adequately codifies all the contributions and it expresses the electronic characteristics of the molecule, showing also a good correlation with the resonance energy, which is a property that express the electronic behavior product of conjugation. The two variable model contains a local descriptor for atoms forming part of unsaturated bonds. This variable quantifies the electronic effects of all the atoms with these characteristics in the molecule. Both descriptors from Equation (27) were derived with respect to the Sach’s sub-graphs (H) event, because its fragments only take into account the sub-graphs of order 1 and ring sub-graphs [
3,
16,
45,
46,
47].
The present study adds value to the previous experiments, which show the capacity of quantifying the electronic environment in atoms and molecules with this mathematical approach. The
Figure 11 shows the regression graphs and the comparative behavior between experimental and theoretical values from the Equation (27).



 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



















































