
Cytoskeletal Regulation of Inflammation and Its Impact on Skin Blistering Disease Epidermolysis Bullosa Acquisita




Abstract
:
{kind=link}
{kind=link}
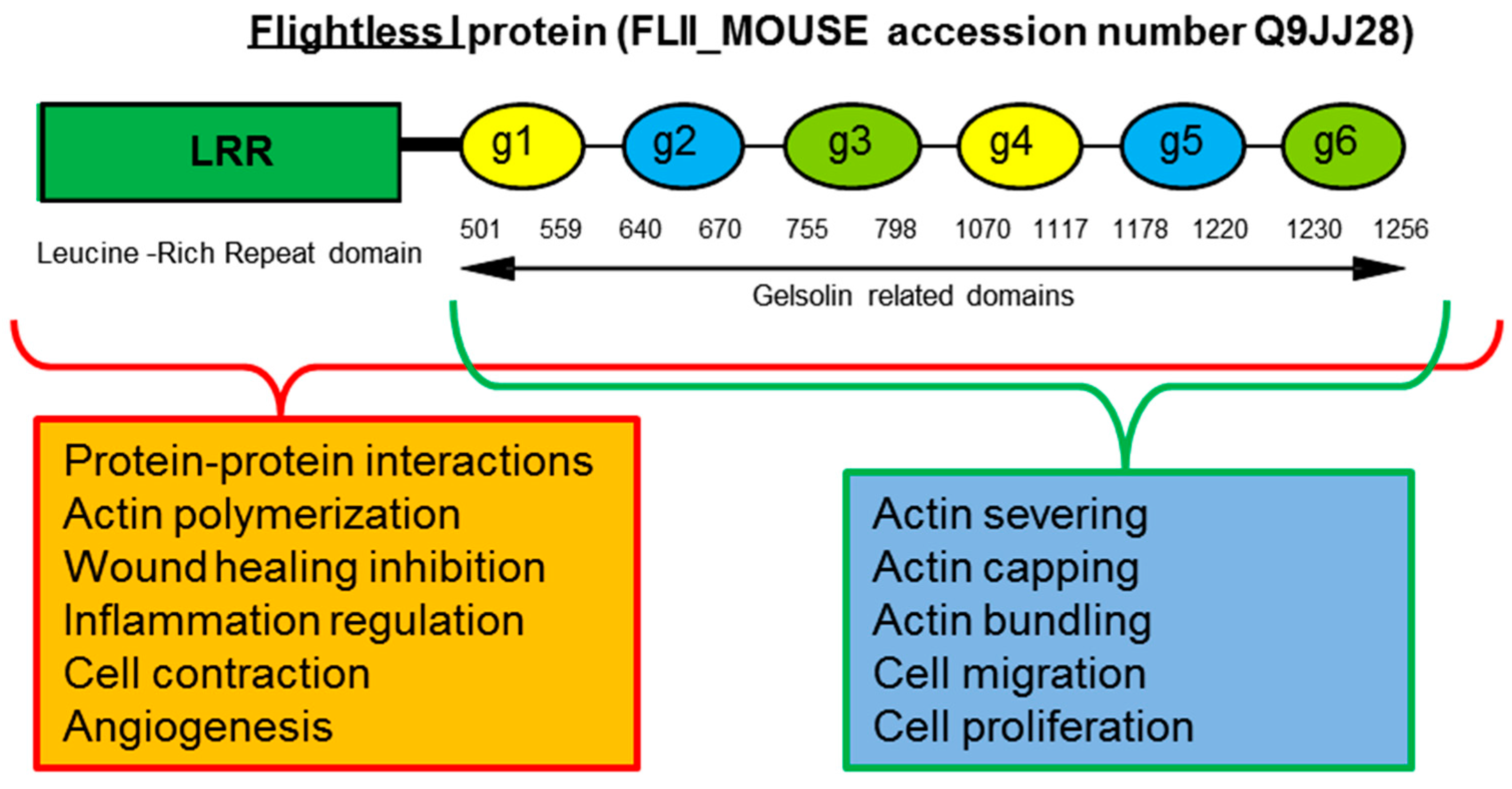{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Epidermolysis Bullosa Acquisita (EBA)
2. The Actin Cytoskeleton
3. The Role of the Actin Cytoskeleton in Inflammation and Autoimmune Inflammatory Conditions
4. The Gelsolin Family of Actin Remodelling Proteins
5. Gelsolin Family Member: Flightless I (Flii)
6. Flii Regulation of the Inflammatory Response
7. Role of Flightless I (Flii) in the Inflammatory Autoimmune Disease EBA
8. Therapeutic Antibodies for Treatments of Autoimmune Skin Conditions
9. Future Directions
Acknowledgments
Conflicts of Interest
References
- Baum, S.; Sakka, N.; Artsi, O.; Trau, H.; Barzilai, A. Diagnosis and classification of autoimmune blistering diseases. Autoimmun. Rev. 2014, 13, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Woodley, D.T.; Chen, M. Epidermolysis bullosa acquisita. Clin. Dermatol. 2012, 30, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Lehman, J.S.; Camilleri, M.J.; Gibson, L.E. Epidermolysis bullosa acquisita: Concise review and practical considerations. Int. J. Dermatol. 2009, 48, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, R.J. Clinical presentation, pathogenesis, diagnosis, and treatment of epidermolysis bullosa acquisita. ISRN Dermatol. 2013, 2013, 812029. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, R. Immune mechanism-targeted treatment of experimental epidermolysis bullosa acquisita. Expert Rev. Clin. Immunol. 2015, 11, 1365–1378. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, S.C. Epidermolysis bullosa acquisita. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 1204–1213. [Google Scholar] [CrossRef] [PubMed]
- Murrell, D.F. Autoimmune diseases of the skin. Immunol. Allergy Clin. N. Am. 2012, 32, xiii–xiv. [Google Scholar] [CrossRef] [PubMed]
- Damoiseaux, J. Bullous skin diseases: Classical types of autoimmune diseases. Scientifica 2013, 2013, 457982. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, R.J.; Kalies, K.; Kohl, J.; Zillikens, D.; Schmidt, E. Emerging treatments for pemphigoid diseases. Trends Mol. Med. 2013, 19, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Mihai, S.; Sitaru, C. Immunopathology and molecular diagnosis of autoimmune bullous diseases. J. Cell. Mol. Med. 2007, 11, 462–481. [Google Scholar] [CrossRef] [PubMed]
- Kasperkiewicz, M.; Sadik, C.D.; Bieber, K.; Ibrahim, S.M.; Manz, R.A.; Schmidt, E.; Zilikens, D.; Ludwig, R.J. Epidermolysis bullosa acquisita: From pathophysiology to novel therapeutic options. J. Investig. Dermatol. 2016, 136, 24–33. [Google Scholar] [PubMed]
- Kopecki, Z.; Arkell, R.M.; Strudwick, X.L.; Hirose, M.; Ludwig, R.J.; Kern, J.S.; Bruckner-Tuderman, L.; Zilikens, D.; MurrelL, D.F.; Cowin, A.J. Overexpression of the Flii gene increases dermal-epidermal blistering in an autoimmune ColVII mouse model of epidermolysis bullosa acquisita. J. Pathol. 2011, 225, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Kopecki, Z.; Ruzehaji, N.; Turner, C.; Iwata, H.; Ludwig, R.J.; Zillikens, D.; Murrell, D.F.; Cowin, A.J. Topically applied flightless I neutralizing antibodies improve healing of blistered skin in a murine model of epidermolysis bullosa acquisita. J. Investig. Dermatol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.O.; Ryu, J.M.; Suh, H.N.; Park, S.H.; Oh, Y.M.; Lee, S.H.; Han, H.J. cAMP promotes cell migration through cell junctional complex dynamics and actin cytoskeleton remodeling: Implications in skin wound healing. Stem Cells Dev. 2015, 24, 2513–2524. [Google Scholar] [CrossRef] [PubMed]
- Kopecki, Z.; Yang, G.N.; Arkell, R.M.; Jackson, J.E.; Melville, E.; Iwata, H.; Ludwig, R.J.; Zilikens, D.; Murrell, D.F.; Cowin, A.J. Flightless I over-expression impairs skin barrier development, function and recovery following skin blistering. J. Pathol. 2014, 232, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Leduc, C.; Sobilo, L.; Toumi, H.; Mondon, P.; Lespessailles, E.; Ossant, F.; Kurfurst, R.; Pichon, C. TGF-β-induced early gene-1 overexpression promotes oxidative stress protection and actin cytoskeleton rearrangement in human skin fibroblasts. Biochim. Biophys. Acta 2016, 1860, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Cha, I.; Jeon, T.J. Dynamic localization of the actin-bundling protein cortexillin I during cell migration. Mol. Cells 2011, 32, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Dudnakova, T.; Spraggon, L.; Slight, J.; Hastie, N. Actin: A novel interaction partner of WT1 influencing its cell dynamic properties. Oncogene 2010, 29, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Gokhin, D.S.; Nowak, R.B.; Khoory, J.A.; Piedra Ade, L.; Ghiran, I.C.; Fowler, V.M. Dynamic actin filaments control the mechanical behavior of the human red blood cell membrane. Mol. Biol. Cell 2015, 26, 1699–1710. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, A.; van Troys, M.; Ampe, C. The actin cytoskeleton in normal and pathological cell motility. Int. J. Biochem. Cell Biol. 2004, 36, 1890–1909. [Google Scholar] [CrossRef] [PubMed]
- Le Clainche, C.; Carlier, M.F. Regulation of actin assembly associated with protrusion and adhesion in cell migration. Physiol. Rev. 2008, 88, 489–513. [Google Scholar] [CrossRef] [PubMed]
- Biro, M.; Munoz, M.A.; Weninger, W. Targeting Rho-GTPases in immune cell migration and inflammation. Br. J. Pharmacol. 2014, 171, 5491–5506. [Google Scholar] [CrossRef] [PubMed]
- Bach, C.T.; Schevzov, G.; Bryce, N.S.; Gunning, P.W.; O’Neill, G.M. Tropomyosin isoform modulation of focal adhesion structure and cell migration. Cell Adhes. Migr. 2010, 4, 226–234. [Google Scholar] [CrossRef]
- Wickramarachchi, D.C.; Theofilopoulos, A.N.; Kono, D.H. Immune pathology associated with altered actin cytoskeleton regulation. Autoimmunity 2010, 43, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Goyal, R.; Bulua, A.C.; Nikolov, N.P.; Schwartzberg, P.L.; Siegel, R.M. Rheumatologic and autoimmune manifestations of primary immunodeficiency disorders. Curr. Opin. Rheumatol. 2009, 21, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Lafouresse, F.; Vasconcelos, Z.; Cotta-de-Almeida, V.; Dupre, L. Actin cytoskeleton control of the comings and goings of T lymphocytes. Tissue Antigens 2013, 82, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Weninger, W.; Biro, M.; Jain, R. Leukocyte migration in the interstitial space of non-lymphoid organs. Nat. Rev. Immunol. 2014, 14, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Muller, W.A. Getting leukocytes to the site of inflammation. Vet. Pathol. 2013, 50, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Draber, P.; Sulimenko, V.; Draberova, E. Cytoskeleton in mast cell signaling. Front. Immunol. 2012, 3, 130. [Google Scholar] [CrossRef] [PubMed]
- Comrie, W.A.; Li, S.; Boyle, S.; Burkhardt, J.K. The dendritic cell cytoskeleton promotes T cell adhesion and activation by constraining ICAM-1 mobility. J. Cell Biol. 2015, 208, 457–473. [Google Scholar] [CrossRef] [PubMed]
- Stroka, K.M.; Hayenga, H.N.; Aranda-Espinoza, H. Human neutrophil cytoskeletal dynamics and contractility actively contribute to trans-endothelial migration. PLoS ONE 2013, 8, e61377. [Google Scholar] [CrossRef] [PubMed]
- McWhorter, F.Y.; Wang, T.; Nguyen, P.; Chung, T.; Liu, W.F. Modulation of macrophage phenotype by cell shape. Proc. Natl. Acad. Sci. USA 2013, 110, 17253–17258. [Google Scholar] [CrossRef] [PubMed]
- Claudianos, C.; Campbell, H.D. The novel flightless-I gene brings together two gene families, actin-binding proteins related to gelsolin and leucine-rich-repeat proteins involved in Ras signal transduction. Mol. Biol. Evol. 1995, 12, 405–414. [Google Scholar] [PubMed]
- Hu, Y.; Li, H.; Li, W.H.; Meng, H.X.; Fan, Y.Z.; Li, W.J.; Ji, Y.T.; Zhao, H.; Zhang, L.; Jin, X.M.; et al. The value of decreased plasma gelsolin levels in patients with systemic lupus erythematosus and rheumatoid arthritis in diagnosis and disease activity evaluation. Lupus 2013, 22, 1455–1461. [Google Scholar] [CrossRef] [PubMed]
- Osborn, T.M.; Verdrengh, M.; Stossel, T.P.; Tarkowski, A.; Bokarewa, M. Decreased levels of the gelsolin plasma isoform in patients with rheumatoid arthritis. Arthritis Res. Ther. 2008, 10, R117. [Google Scholar] [CrossRef] [PubMed]
- Gil, M.P.; Modol, T.; Espana, A.; Lopez-Zabalza, M.J. Inhibition of FAK prevents blister formation in the neonatal mouse model of pemphigus vulgaris. Exp. Dermatol. 2012, 21, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Yu, Q.; Han, C.; Hu, X.; Xu, S.; Wang, Q.; Wang, J.; Li, N.; Cao, X. LRRFIP2 negatively regulates NLRP3 inflammasome activation in macrophages by promoting Flightless-I-mediated caspase-1 inhibition. Nat. Commun. 2013, 4, 2075. [Google Scholar] [CrossRef] [PubMed]
- Kwong, L.; Wozniak, M.A.; Collins, A.S.; Wilson, S.D.; Keely, P.J. R-Ras promotes focal adhesion formation through focal adhesion kinase and p130(Cas) by a novel mechanism that differs from integrins. Mol. Cell. Biol. 2003, 23, 933–949. [Google Scholar] [CrossRef] [PubMed]
- Rothenbach, P.A.; Dahl, B.; Schwartz, J.J.; O’Keefe, G.E.; Yamamoto, M.; Lee, W.M.; Horton, J.W.; Yin, H.L.; Turnage, R.H. Recombinant plasma gelsolin infusion attenuates burn-induced pulmonary microvascular dysfunction. J. Appl. Physiol. 2004, 96, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.Q.; Yamamoto, M.; Mejillano, M.; Yin, H.L. Gelsolin, a multifunctional actin regulatory protein. J. Biol. Chem. 1999, 274, 33179–33182. [Google Scholar] [CrossRef] [PubMed]
- Bucki, R.; Georges, P.C.; Espinassous, Q.; Funaki, M.; Pastore, J.J.; Chaby, R.; Janmey, P.A. Inactivation of endotoxin by human plasma gelsolin. Biochemistry 2005, 44, 9590–9597. [Google Scholar] [CrossRef] [PubMed]
- Li, G.H.; Arora, P.D.; Chen, Y.; McCulloch, C.A.; Liu, P. Multifunctional roles of gelsolin in health and diseases. Med. Res. Rev. 2012, 32, 999–1025. [Google Scholar] [CrossRef] [PubMed]
- Kobe, B.; Kajava, A.V. The leucine-rich repeat as a protein recognition motif. Curr. Opin. Struct. Biol. 2001, 11, 725–732. [Google Scholar] [CrossRef]
- Nag, S.; Larsson, M.; Robinson, R.C.; Burtnick, L.D. Gelsolin: The tail of a molecular gymnast. Cytoskeleton 2013, 70, 360–384. [Google Scholar] [CrossRef] [PubMed]
- Goshima, M.; Kariya, K.; Yamawaki-Kataoka, Y.; Okada, T.; Shibatohge, M.; Shima, F.; Fujimoto, E.; Kataoka, T. Characterization of a novel Ras-binding protein Ce-FLI-1 comprising leucine-rich repeats and gelsolin-like domains. Biochem. Biophys. Res. Commun. 1999, 257, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.T.; Yin, H.L. Identification of the binding partners for flightless, I.; A novel protein bridging the leucine-rich repeat and the gelsolin superfamilies. J. Biol. Chem. 1998, 273, 7920–7927. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, I.; Arora, P.D.; Naghibzadeh, Y.; Wang, Y.; Li, J.; Mascarenhas, W.; Janmey, P.A.; Dawson, J.F.; McCulloch, C.A. Flightless I is a focal adhesion-associated actin-capping protein that regulates cell migration. FASEB J. 2012, 26, 3260–3272. [Google Scholar] [CrossRef] [PubMed]
- Kopecki, Z.; O’Neill, G.M.; Arkell, R.M.; Cowin, A.J. Regulation of focal adhesions by flightless I involves inhibition of paxillin phosphorylation via a Rac1-dependent pathway. J. Investig. Dermatol. 2011, 131, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- McGough, A.M.; Staiger, C.J.; Min, J.K.; Simonetti, K.D. The gelsolin family of actin regulatory proteins: Modular structures, versatile functions. FEBS Lett. 2003, 552, 75–81. [Google Scholar] [CrossRef]
- Davy, D.A.; Campbell, H.D.; Fountain, S.; de Jong, D.; Crouch, M.F. The flightless I protein colocalizes with actin- and microtubule-based structures in motile Swiss 3T3 fibroblasts: Evidence for the involvement of PI 3-kinase and Ras-related small GTPases. J. Cell Sci. 2001, 114(Pt. 3), 549–562. [Google Scholar] [PubMed]
- Bella, J.; Hindle, K.L.; McEwan, P.A.; Lovell, S.C. The leucine-rich repeat structure. Cell. Mol. Life Sci. 2008, 65, 2307–2333. [Google Scholar] [CrossRef] [PubMed]
- Cowin, A.J.; Lei, N.; Franken, L.; Ruzehaji, N.; Offenhauser, C.; Kopecki, Z.; Murray, R.Z. Lysosomal secretion of Flightless I upon injury has the potential to alter inflammation. Commun. Integr. Biol. 2012, 5, 546–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruzehaji, N.; Grose, R.; Krumbiegel, D.; Zola, H.; Dasari, P.; Wallace, H.; Stacey, M.; Fitridge, R.; Cowin, A.J. Cytoskeletal protein Flightless (Flii) is elevated in chronic and acute human wounds and wound fluid: Neutralizing its activity in chronic but not acute wound fluid improves cellular proliferation. Eur. J. Dermatol. 2012, 22, 740–750. [Google Scholar] [PubMed]
- Lei, N.; Franken, L.; Ruzehaji, N.; Offenhauser, C.; Cowin, A.J.; Murray, R.Z. Flightless, secreted through a late endosome/lysosome pathway, binds LPS and dampens cytokine secretion. J. Cell Sci. 2012, 125(Pt. 18), 4288–4296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seward, M.E.; Easley, C.A., IV; McLeod, J.J.; Myers, A.L.; Tombes, R.M. Flightless-I, a gelsolin family member and transcriptional regulator, preferentially binds directly to activated cytosolic CaMK-II. FEBS Lett. 2008, 582, 2489–2495. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.K.; Chai, J.Y.; Kong, Y.; Waikagul, J.; Insisiengmay, B.; Rim, H.J.; Eom, K.S. Differential diagnosis of Taenia asiatica using multiplex PCR. Exp. Parasitol. 2009, 121, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Campbell, H.D.; Stallcup, M.R. Developmentally essential protein flightless I is a nuclear receptor coactivator with actin binding activity. Mol. Cell. Biol. 2004, 24, 2103–2117. [Google Scholar] [CrossRef] [PubMed]
- Jeong, K.W. Flightless I (Drosophila) homolog facilitates chromatin accessibility of the estrogen receptor alpha target genes in MCF-7 breast cancer cells. Biochem. Biophys. Res. Commun. 2014, 446, 608–613. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Chen, H.; Zhu, Y.; Meng, J.; Li, Y.; Li, M.; Yang, D.; Zhang, P.; Feng, M.; Tonh, X. Flightless I homolog negatively regulates ChREBP activity in cancer cells. Int. J. Biochem. Cell Biol. 2013, 45, 2688–2697. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Stallcup, M.R. Interplay of Fli-I and FLAP1 for regulation of β-catenin dependent transcription. Nucleic Acids Res. 2006, 34, 5052–5059. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Chuang, T.H.; Ronni, T.; Gu, S.; Du, Y.C.; Cai, H.; Sun, H.Q.; Yin, H.L.; Chen, X. Flightless I homolog negatively modulates the TLR pathway. J. Immunol. 2006, 176, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Seta, V.; Aucouturier, F.; Bonnefoy, J.; Le Roux-Villet, C.; Pendaries, V.; Alexandre, M.; Grootenboer-Mignot, H.M.; Lievre, N.; Laroche, L.; Caux, F.; et al. Comparison of 3 type VII collagen (C7) assays for serologic diagnosis of epidermolysis bullosa acquisita (EBA). J. Am. Acad. Dermatol. 2016, 74, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Monack, D.M. Newly described pattern recognition receptors team up against intracellular pathogens. Nat. Rev. Immunol. 2013, 13, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Dai, P.; Jeong, S.Y.; Yu, Y.; Leng, T.; Wu, W.; Xie, L.; Chen, X. Modulation of TLR signaling by multiple MyD88-interacting partners including leucine-rich repeat Fli-I-interacting proteins. J. Immunol. 2009, 182, 3450–3460. [Google Scholar] [CrossRef] [PubMed]
- Hamerman, J.A.; Pottle, J.; Ni, M.; He, Y.; Zhang, Z.Y.; Buckner, J.H. Negative regulation of TLR signaling in myeloid cells—Implications for autoimmune diseases. Immunol. Rev. 2016, 269, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Piccinini, A.M.; Midwood, K.S. Endogenous control of immunity against infection: Tenascin-C regulates TLR4-mediated inflammation via microRNA-155. Cell Rep. 2012, 2, 914–926. [Google Scholar] [CrossRef] [PubMed]
- Ishibe, S.; Joly, D.; Zhu, X.; Cantley, L.G. Phosphorylation-dependent paxillin-ERK association mediates hepatocyte growth factor-stimulated epithelial morphogenesis. Mol. Cell 2003, 12, 1275–1285. [Google Scholar] [CrossRef]
- Li, J.; Yin, H.L.; Yuan, J. Flightless-I regulates proinflammatory caspases by selectively modulating intracellular localization and caspase activity. J. Cell Biol. 2008, 181, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Kopecki, Z.; Arkell, R.; Powell, B.C.; Cowin, A.J. Flightless I regulates hemidesmosome formation and integrin-mediated cellular adhesion and migration during wound repair. J. Investig. Dermatol. 2009, 129, 2031–2045. [Google Scholar] [CrossRef] [PubMed]
- Kopecki, Z.; Murrell, D.F.; Cowin, A. Raising the roof on epidermolysis bullosa (EB): A focus on new therapies. Wound Pract. Res. 2009, 17, 76–82. [Google Scholar]
- Morizane, S.; Takiguchi, T.; Ikeda, K.; Suzuki, D.; Hirai, Y.; Aoyama, Y. Widespread chronic skin ulcers in epidermolysis bullosa patients trigger Castleman’s disease-like condition through Toll-like receptors. J. Investig. Dermatol. 2012, 132, S108–S114. [Google Scholar]
- Cowin, A.; Adams, D.; Strudwick, X.; Chan, H.; Hooper, J.; Sander, G.; Ryner, T.E.; Matthaei, K.I.; Powell, B.C.; Campbell, H.D. Flightless I deficiency enhances wound repair by increasing cell migration and proliferation. J. Pathol. 2007, 211, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.H.; Ruzehaji, N.; Strudwick, X.L.; Greenwood, J.E.; Campbell, H.D.; Arkell, R.; Cowin, A.J. Attenuation of Flightless, I.; an actin-remodelling protein, improves burn injury repair via modulation of transforming growth factor (TGF)-β1 and TGF-β3. Br. J. Dermatol. 2009, 161, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Ruzehaji, N.; Kopecki, Z.; Melville, E.; Appleby, S.L.; Bonder, C.S.; Arkell, R.M.; Cowin, A.J. Attenuation of flightless I improves wound healing and enhances angiogenesis in a murine model of type 1 diabetes. Diabetologia 2014, 57, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Gurcan, H.M.; Ahmed, A.R. Current concepts in the treatment of epidermolysis bullosa acquisita. Expert Opin. Pharmacother. 2011, 12, 1259–1268. [Google Scholar] [CrossRef] [PubMed]
- Samavedam, U.K.; Iwata, H.; Muller, S.; Schulze, F.S.; Recke, A.; Schmidt, E.; Zilikens, D.; Ludwig, R.J. GM-CSF modulates autoantibody production and skin blistering in experimental epidermolysis bullosa acquisita. J. Immunol. 2014, 192, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Tukaj, S.; Hellberg, L.; Ueck, C.; Hansel, M.; Samavedam, U.; Zillikens, D.; Ludwig, R.J.; Laskay, T.; Kasperkiewicz, M. Heat shock protein 90 is required for ex vivo neutrophil-driven autoantibody-induced tissue damage in experimental epidermolysis bullosa acquisita. Exp. Dermatol. 2015, 24, 471–473. [Google Scholar] [CrossRef] [PubMed]
- Tukaj, S.; Zillikens, D.; Kasperkiewicz, M. Heat shock protein 90: A pathophysiological factor and novel treatment target in autoimmune bullous skin diseases. Exp. Dermatol. 2015, 24, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.E.; Kopecki, Z.; Adams, D.H.; Cowin, A.J. Flii neutralizing antibodies improve wound healing in porcine preclinical studies. Wound Repair Regen. 2012, 20, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Casacó, A.; Fuente, D.; Ledón, N.; Fernández, A.; Crombet, T. Anti-epidermal growth factor/epidermal growth factor receptortherapeutic anti-cancer drugs and the wound healing process. J. Cancer Sci. Ther. 2012, 4, 324–329. [Google Scholar] [CrossRef]





© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kopecki, Z.; Ludwig, R.J.; Cowin, A.J. Cytoskeletal Regulation of Inflammation and Its Impact on Skin Blistering Disease Epidermolysis Bullosa Acquisita. Int. J. Mol. Sci. 2016, 17, 1116. https://doi.org/10.3390/ijms17071116
Kopecki Z, Ludwig RJ, Cowin AJ. Cytoskeletal Regulation of Inflammation and Its Impact on Skin Blistering Disease Epidermolysis Bullosa Acquisita. International Journal of Molecular Sciences. 2016; 17(7):1116. https://doi.org/10.3390/ijms17071116
Chicago/Turabian StyleKopecki, Zlatko, Ralf J. Ludwig, and Allison J. Cowin. 2016. "Cytoskeletal Regulation of Inflammation and Its Impact on Skin Blistering Disease Epidermolysis Bullosa Acquisita" International Journal of Molecular Sciences 17, no. 7: 1116. https://doi.org/10.3390/ijms17071116
APA StyleKopecki, Z., Ludwig, R. J., & Cowin, A. J. (2016). Cytoskeletal Regulation of Inflammation and Its Impact on Skin Blistering Disease Epidermolysis Bullosa Acquisita. International Journal of Molecular Sciences, 17(7), 1116. https://doi.org/10.3390/ijms17071116