
Sex-Based Selectivity of PPARγ Regulation in Th1, Th2, and Th17 Differentiation

Abstract
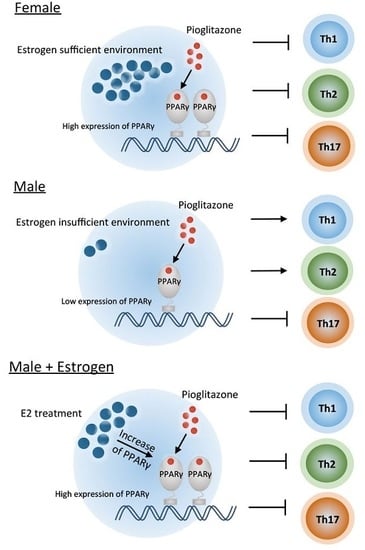
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
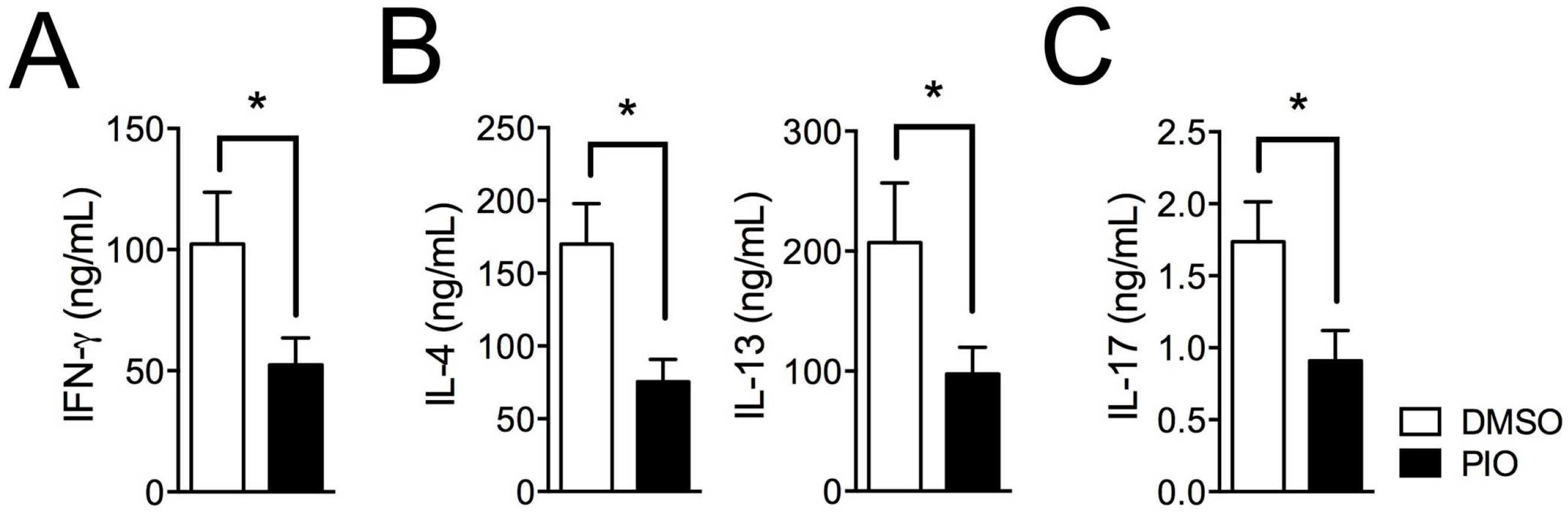
2.1. PPARγ Inhibits Th1, Th2, and Th17 Differentiation in Female Mouse Splenic T Cells
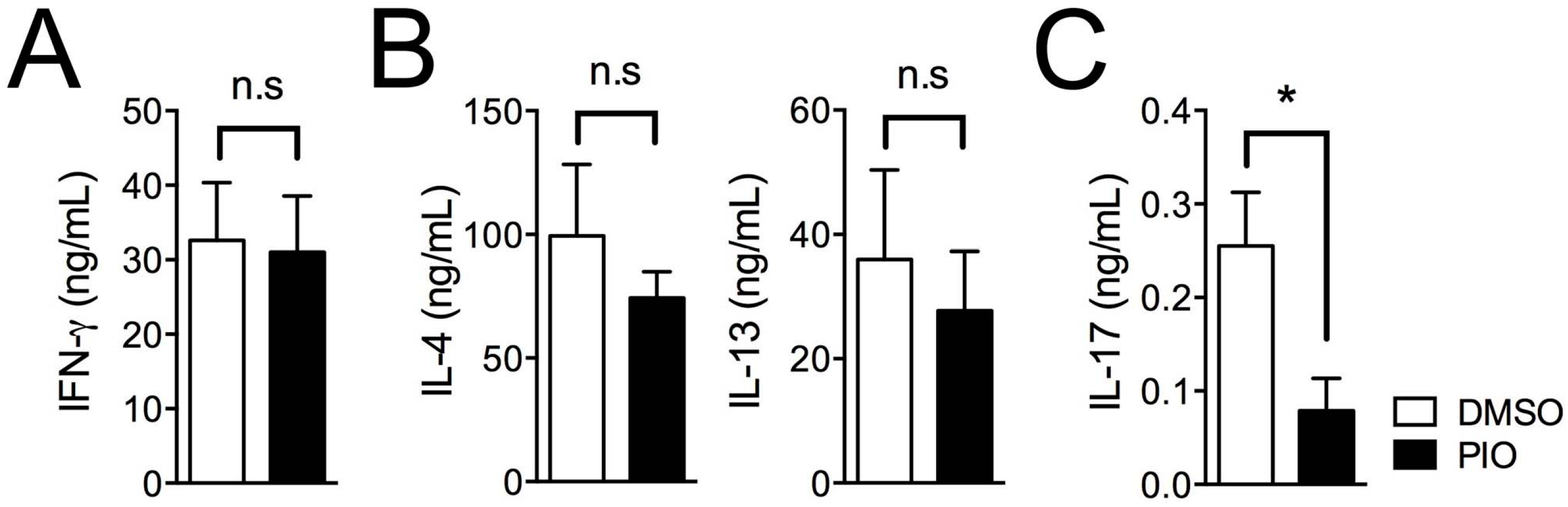
2.2. PPARγ Selectively Inhibits Th17 Differentiation in Male Mouse Splenic T Cells
2.3. Pioglitazone and Estradiol Co-Treatment Inhibits Th1, Th2, and Th17 Differentiation in Male Mouse Splenic T Cells
3. Discussion
4. Experimental Section
4.1. Mice
4.2. Naive T Cell Isolation
4.3. T Cell Differentiation
4.4. Flow Cytometry
4.5. ELISA
4.6. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Choi, J.M.; Bothwell, A.L. The nuclear receptor PPARs as important regulators of T-cell functions and autoimmune diseases. Mol. Cells 2012, 33, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Saijo, K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and t cells. Nat. Rev. Immunol. 2010, 10, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Hamm, J.K.; el Jack, A.K.; Pilch, P.F.; Farmer, S.R. Role of PPARγ in regulating adipocyte differentiation and insulin-responsive glucose uptake. Ann. N. Y. Acad. Sci. 1999, 892, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Farmer, S.R. Regulation of PPARγ activity during adipogenesis. Int. J. Obes. 2005, 29, S13–S16. [Google Scholar] [CrossRef] [PubMed]
- Siersbaek, R.; Nielsen, R.; Mandrup, S. PPARγ in adipocyte differentiation and metabolism—Novel insights from genome-wide studies. FEBS Lett. 2010, 584, 3242–3249. [Google Scholar] [CrossRef] [PubMed]
- Daynes, R.A.; Jones, D.C. Emerging roles of PPARs in inflammation and immunity. Nat. Rev. Immunol. 2002, 2, 748–759. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.B. The role of PPARs in inflammation and immunity. J. Leukoc. Biol. 2002, 71, 388–400. [Google Scholar] [PubMed]
- Straus, D.S.; Glass, C.K. Anti-inflammatory actions of PPAR ligands: New insights on cellular and molecular mechanisms. Trends Immunol. 2007, 28, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Wahli, W.; Michalik, L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol. Metab. 2012, 23, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Anderson, P.O.; Chen, S.; Paulsson, K.M.; Sjogren, H.O.; Li, S. Inhibition of the transcription factors AP-1 and NF-κB in CD4 T cells by peroxisome proliferator-activated receptor γ ligands. Int. Immunopharmacol. 2001, 1, 803–812. [Google Scholar] [CrossRef]
- Clark, R.B.; Bishop-Bailey, D.; Estrada-Hernandez, T.; Hla, T.; Puddington, L.; Padula, S.J. The nuclear receptor PPAR and immunoregulation: PPAR mediates inhibition of helper T cell responses. J. Immunol. 2000, 164, 1364–1371. [Google Scholar] [CrossRef] [PubMed]
- Tobiasova, Z.; Zhang, L.; Yi, T.; Qin, L.; Manes, T.D.; Kulkarni, S.; Lorber, M.I.; Rodriguez, F.C.; Choi, J.M.; Tellides, G.; et al. Peroxisome proliferator-activated receptor-γ agonists prevent in vivo remodeling of human artery induced by alloreactive T cells. Circulation 2011, 124, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.; Burgdorf, S.; Dani, I.; Saijo, K.; Flossdorf, J.; Hucke, S.; Alferink, J.; Nowak, N.; Beyer, M.; Mayer, G.; et al. The nuclear receptor PPARγ selectively inhibits Th17 differentiation in a T cell-intrinsic fashion and suppresses CNS autoimmunity. J. Exp. Med. 2009, 206, 2079–2089. [Google Scholar] [CrossRef] [PubMed]
- Hontecillas, R.; Bassaganya-Riera, J. Peroxisome proliferator-activated receptor γ is required for regulatory CD4+ T cell-mediated protection against colitis. J. Immunol. 2007, 178, 2940–2949. [Google Scholar] [CrossRef] [PubMed]
- Ricote, M.; Glass, C.K. PPARs and molecular mechanisms of transrepression. Biochim. Biophys. Acta 2007, 1771, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Diab, A.; Deng, C.; Smith, J.D.; Hussain, R.Z.; Phanavanh, B.; Lovett-Racke, A.E.; Drew, P.D.; Racke, M.K. Peroxisome proliferator-activated receptor-γ agonist 15-deoxy-Δ12,1412,14-prostaglandin J2 ameliorates experimental autoimmune encephalomyelitis. J. Immunol. 2002, 168, 2508–2515. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, D.L.; Galea, E.; Gavrilyuk, V.; Brosnan, C.F.; Whitacre, C.C.; Dumitrescu-Ozimek, L.; Landreth, G.E.; Pershadsingh, H.A.; Weinberg, G.; Heneka, M.T. Peroxisome proliferator-activated receptor-γ agonists prevent experimental autoimmune encephalomyelitis. Ann. Neurol. 2002, 51, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Niino, M.; Iwabuchi, K.; Kikuchi, S.; Ato, M.; Morohashi, T.; Ogata, A.; Tashiro, K.; Onoe, K. Amelioration of experimental autoimmune encephalomyelitis in C57BL/6 mice by an agonist of peroxisome proliferator-activated receptor-γ. J. Neuroimmunol. 2001, 116, 40–48. [Google Scholar] [CrossRef]
- Klotz, L.; Schmidt, M.; Giese, T.; Sastre, M.; Knolle, P.; Klockgether, T.; Heneka, M.T. Proinflammatory stimulation and pioglitazone treatment regulate peroxisome proliferator-activated receptor gamma levels in peripheral blood mononuclear cells from healthy controls and multiple sclerosis patients. J. Immunol. 2005, 175, 4948–4955. [Google Scholar] [CrossRef] [PubMed]
- Su, C.G.; Wen, X.; Bailey, S.T.; Jiang, W.; Rangwala, S.M.; Keilbaugh, S.A.; Flanigan, A.; Murthy, S.; Lazar, M.A.; Wu, G.D. A novel therapy for colitis utilizing PPAR-γ ligands to inhibit the epithelial inflammatory response. J. Clin. Investig. 1999, 104, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Marquillies, P.; Capron, M.; Dombrowicz, D. Peroxisome proliferator-activated receptor gamma is expressed in airways and inhibits features of airway remodeling in a mouse asthma model. J. Allergy Clin. Immunol. 2004, 113, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Park, S.J.; Hwang, P.H.; Yi, H.K.; Song, C.H.; Chai, O.H.; Kim, J.S.; Lee, M.K.; Lee, Y.C. PPAR-γ modulates allergic inflammation through up-regulation of PTEN. FASEB J. 2005, 19, 1033–1035. [Google Scholar] [CrossRef] [PubMed]
- Hammad, H.; de Heer, H.J.; Soullie, T.; Angeli, V.; Trottein, F.; Hoogsteden, H.C.; Lambrecht, B.N. Activation of peroxisome proliferator-activated receptor-γ in dendritic cells inhibits the development of eosinophilic airway inflammation in a mouse model of asthma. Am. J. Pathol. 2004, 164, 263–271. [Google Scholar] [CrossRef]
- Woerly, G.; Honda, K.; Loyens, M.; Papin, J.P.; Auwerx, J.; Staels, B.; Capron, M.; Dombrowicz, D. Peroxisome proliferator-activated receptors α and γ down-regulate allergic inflammation and eosinophil activation. J. Exp. Med. 2003, 198, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Huang, Y.; He, J.; Li, C.; Deng, W.; Ran, X.; Wang, D. Rosiglitazone, a peroxisome proliferator-activated receptor-γ agonist, attenuates airway inflammation by inhibiting the proliferation of effector T cells in a murine model of neutrophilic asthma. Immunol. Lett. 2014, 157, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.Y.; Wang, L.H.; Chen, T.; Hodge, D.R.; Resau, J.H.; DaSilva, L.; Farrar, W.L. Activation of human t lymphocytes is inhibited by peroxisome proliferator-activated receptor γ (PPARγ) agonists. PPARγ co-association with transcription factor NFAT. J. Biol. Chem. 2000, 275, 4541–4544. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.W.; Bayliffe, A.I.; Warren, A.P.; Lamb, J.R. Interleukin-10 is upregulated by nanomolar rosiglitazone treatment of mature dendritic cells and human CD4+ T cells. Cytokine 2007, 39, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Berthier, C.C.; Lewis, E.E.; McCune, W.J.; Kretzler, M.; Kaplan, M.J. The peroxisome-proliferator activated receptor-γ agonist pioglitazone modulates aberrant T cell responses in systemic lupus erythematosus. Clin. Immunol. 2013, 149, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Wohlfert, E.A.; Nichols, F.C.; Nevius, E.; Clark, R.B. Peroxisome proliferator-activated receptor γ (PPARγ) and immunoregulation: Enhancement of regulatory T cells through PPARγ-dependent and -independent mechanisms. J. Immunol. 2007, 178, 4129–4135. [Google Scholar] [CrossRef] [PubMed]
- Cipolletta, D.; Feuerer, M.; Li, A.; Kamei, N.; Lee, J.; Shoelson, S.E.; Benoist, C.; Mathis, D. PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue treg cells. Nature 2012, 486, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Kim, D.H.; Choi, J.Y.; Kim, W.J.; Kim, J.Y.; Senejani, A.G.; Hwang, S.S.; Kim, L.K.; Tobiasova, Z.; Lee, G.R.; et al. PPARγ negatively regulates T cell activation to prevent follicular helper T cells and germinal center formation. PLoS ONE 2014, 9, e99127. [Google Scholar] [CrossRef] [PubMed]
- Housley, W.J.; Adams, C.O.; Vang, A.G.; Brocke, S.; Nichols, F.C.; LaCombe, M.; Rajan, T.V.; Clark, R.B. Peroxisome proliferator-activated receptor γ is required for CD4+ T cell-mediated lymphopenia-associated autoimmunity. J. Immunol. 2011, 187, 4161–4169. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Park, H.S.; Lee, J.U.; Bothwell, A.L.; Choi, J.M. Gender-specific differences in PPARγ regulation of follicular helper T cell responses with estrogen. Sci. Rep. 2016, 6, 28495. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Kim, D.H.; Lim, S.H.; Kim, W.J.; Youn, J.; Choi, Y.S.; Choi, J.M. Insights into the role of follicular helper t cells in autoimmunity. Immune Netw. 2014, 14, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Vlckova, V.; Cornelius, V.; Kasliwal, R.; Wilton, L.; Shakir, S. Hypoglycaemia with pioglitazone: Analysis of data from the prescription-event monitoring study. J. Eval. Clin. Pract. 2010, 16, 1124–1128. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.; Anderson, R.J.; Rappaport, E.B. Rosiglitazone monotherapy improves glycaemic control in patients with type 2 diabetes: A twelve-week, randomized, placebo-controlled study. Diabetes Obes. Metab. 1999, 1, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Ormseth, M.J.; Oeser, A.M.; Cunningham, A.; Bian, A.; Shintani, A.; Solus, J.; Tanner, S.; Stein, C.M. Peroxisome proliferator-activated receptor γ agonist effect on rheumatoid arthritis: A randomized controlled trial. Arthritis Res. Ther. 2013, 15, R110. [Google Scholar] [CrossRef] [PubMed]
- Cuzzocrea, S.; Mazzon, E.; Dugo, L.; Patel, N.S.; Serraino, I.; di Paola, R.; Genovese, T.; Britti, D.; de Maio, M.; Caputi, A.P.; et al. Reduction in the evolution of murine type II collagen-induced arthritis by treatment with rosiglitazone, a ligand of the peroxisome proliferator-activated receptor γ. Arthritis Rheumatol. 2003, 48, 3544–3556. [Google Scholar] [CrossRef] [PubMed]
- Aprahamian, T.; Bonegio, R.G.; Richez, C.; Yasuda, K.; Chiang, L.K.; Sato, K.; Walsh, K.; Rifkin, I.R. The peroxisome proliferator-activated receptor γ agonist rosiglitazone ameliorates murine lupus by induction of adiponectin. J. Immunol. 2009, 182, 340–346. [Google Scholar] [CrossRef] [PubMed]






© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.-J.; Park, H.-S.; Lee, J.-U.; Bothwell, A.L.M.; Choi, J.-M. Sex-Based Selectivity of PPARγ Regulation in Th1, Th2, and Th17 Differentiation. Int. J. Mol. Sci. 2016, 17, 1347. https://doi.org/10.3390/ijms17081347
Park H-J, Park H-S, Lee J-U, Bothwell ALM, Choi J-M. Sex-Based Selectivity of PPARγ Regulation in Th1, Th2, and Th17 Differentiation. International Journal of Molecular Sciences. 2016; 17(8):1347. https://doi.org/10.3390/ijms17081347
Chicago/Turabian StylePark, Hong-Jai, Hyeon-Soo Park, Jae-Ung Lee, Alfred L. M. Bothwell, and Je-Min Choi. 2016. "Sex-Based Selectivity of PPARγ Regulation in Th1, Th2, and Th17 Differentiation" International Journal of Molecular Sciences 17, no. 8: 1347. https://doi.org/10.3390/ijms17081347
APA StylePark, H. -J., Park, H. -S., Lee, J. -U., Bothwell, A. L. M., & Choi, J. -M. (2016). Sex-Based Selectivity of PPARγ Regulation in Th1, Th2, and Th17 Differentiation. International Journal of Molecular Sciences, 17(8), 1347. https://doi.org/10.3390/ijms17081347