
Growth Hormone Releasing Peptide-2 Attenuation of Protein Kinase C-Induced Inflammation in Human Ovarian Granulosa Cells

Abstract
:1. Introduction
2. Results
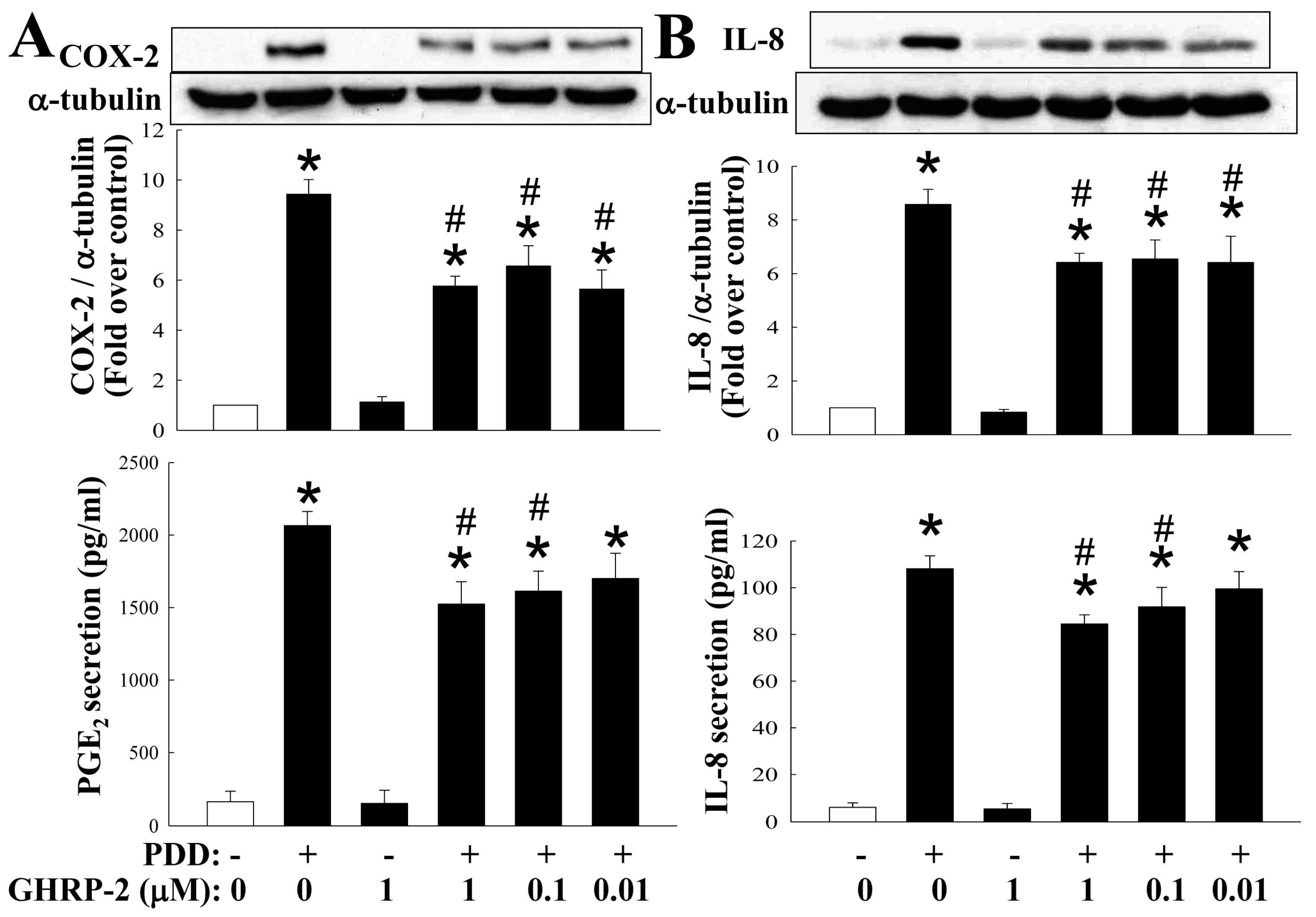
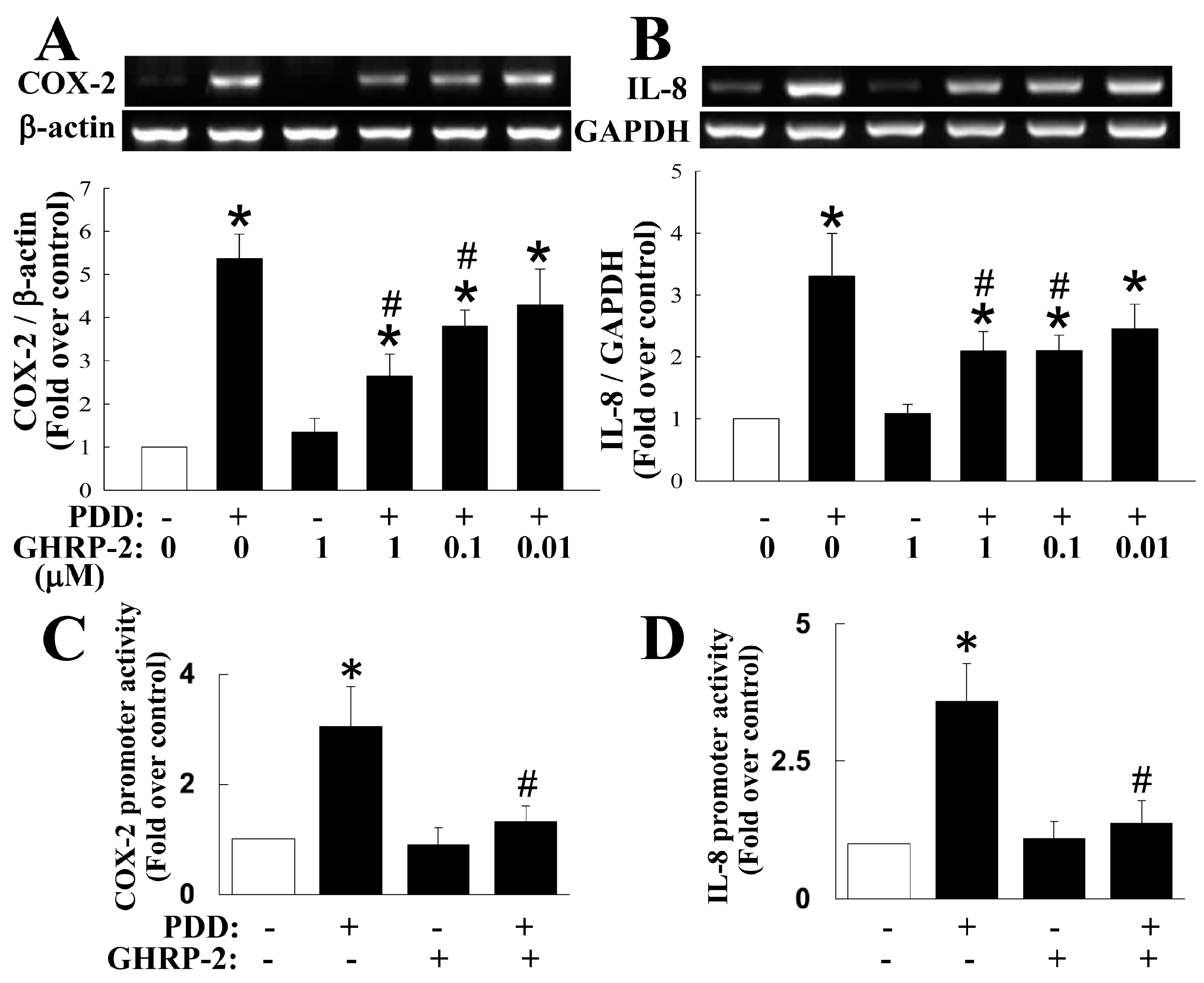
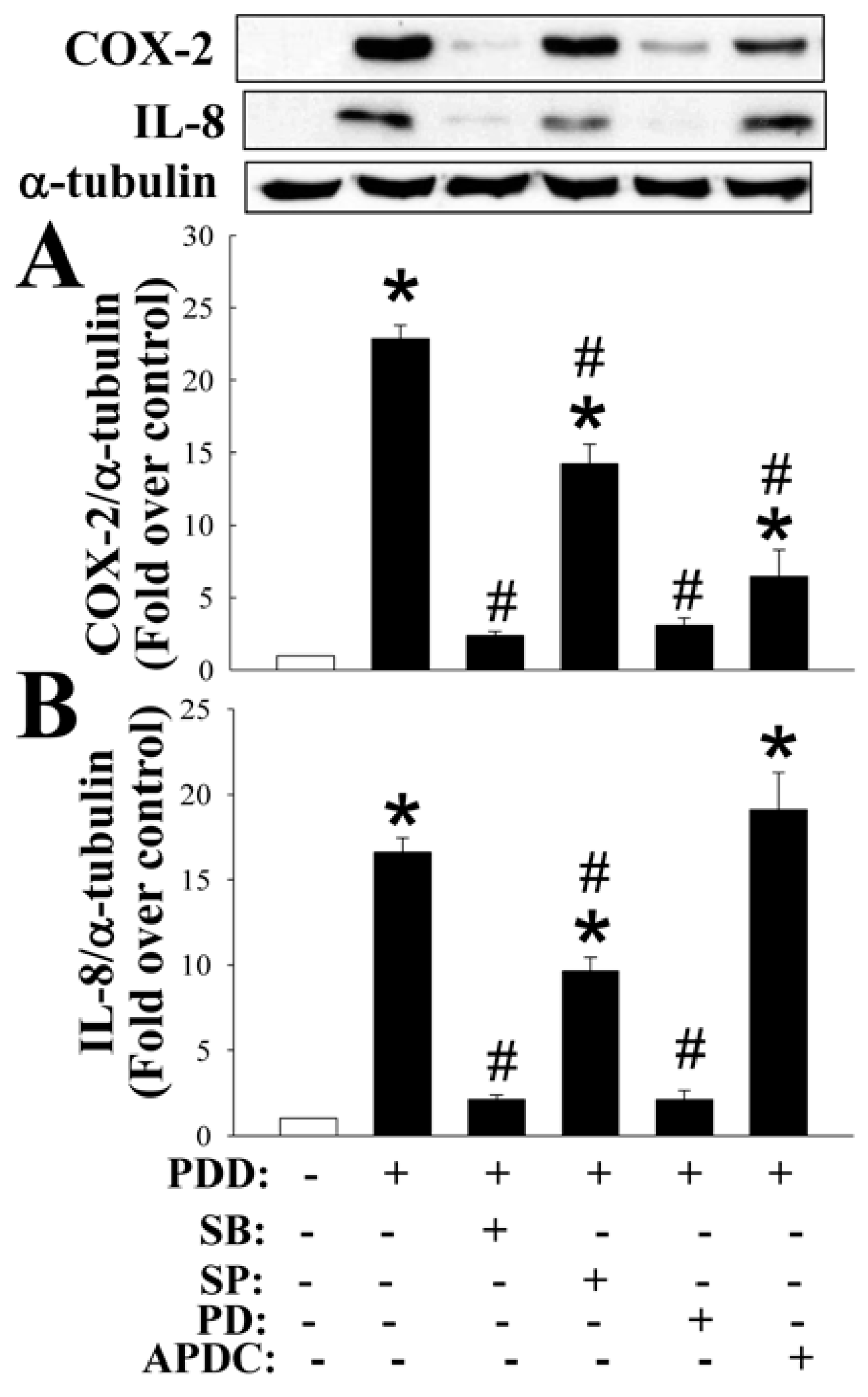
2.1. GHRP-2 Inhibition of the PKC-Induced Expression of COX-2 and IL-8 and Secretion of PGE2 and IL-8
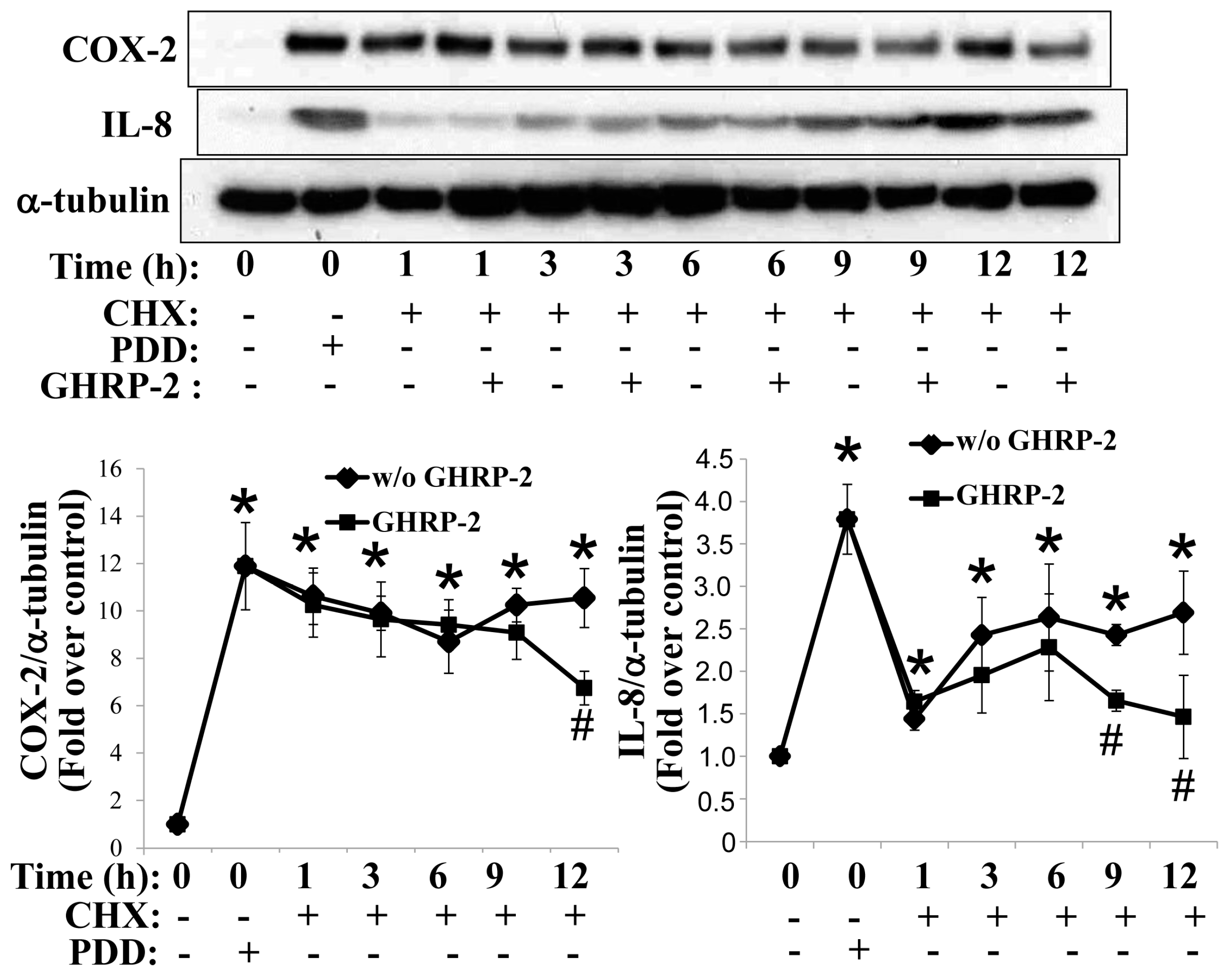
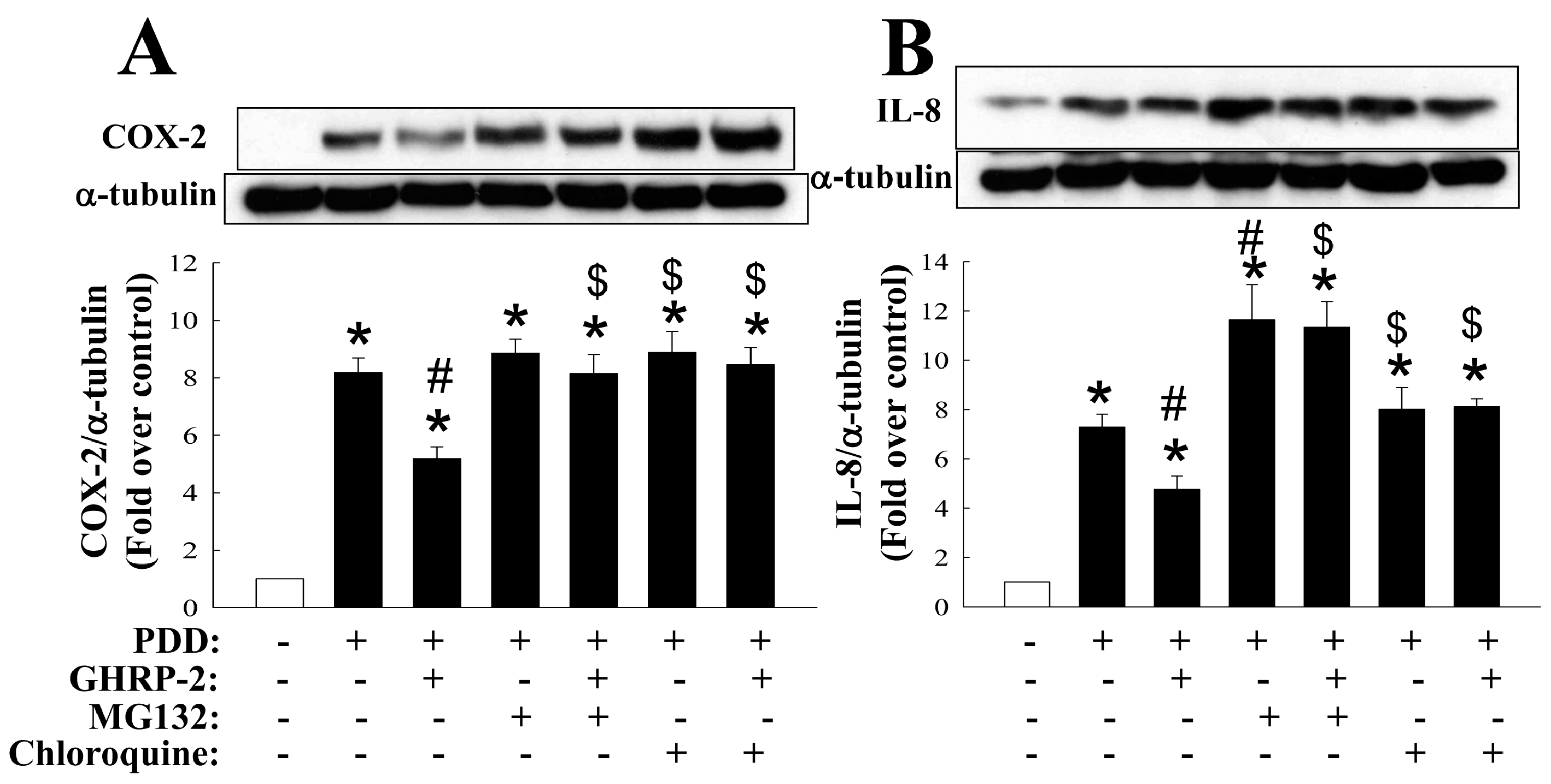
2.2. GHRP-2 Promotion of the Degradation of PKC-Induced COX-2 and IL-8 Proteins via Both Proteasomal and Lysosomal Pathways
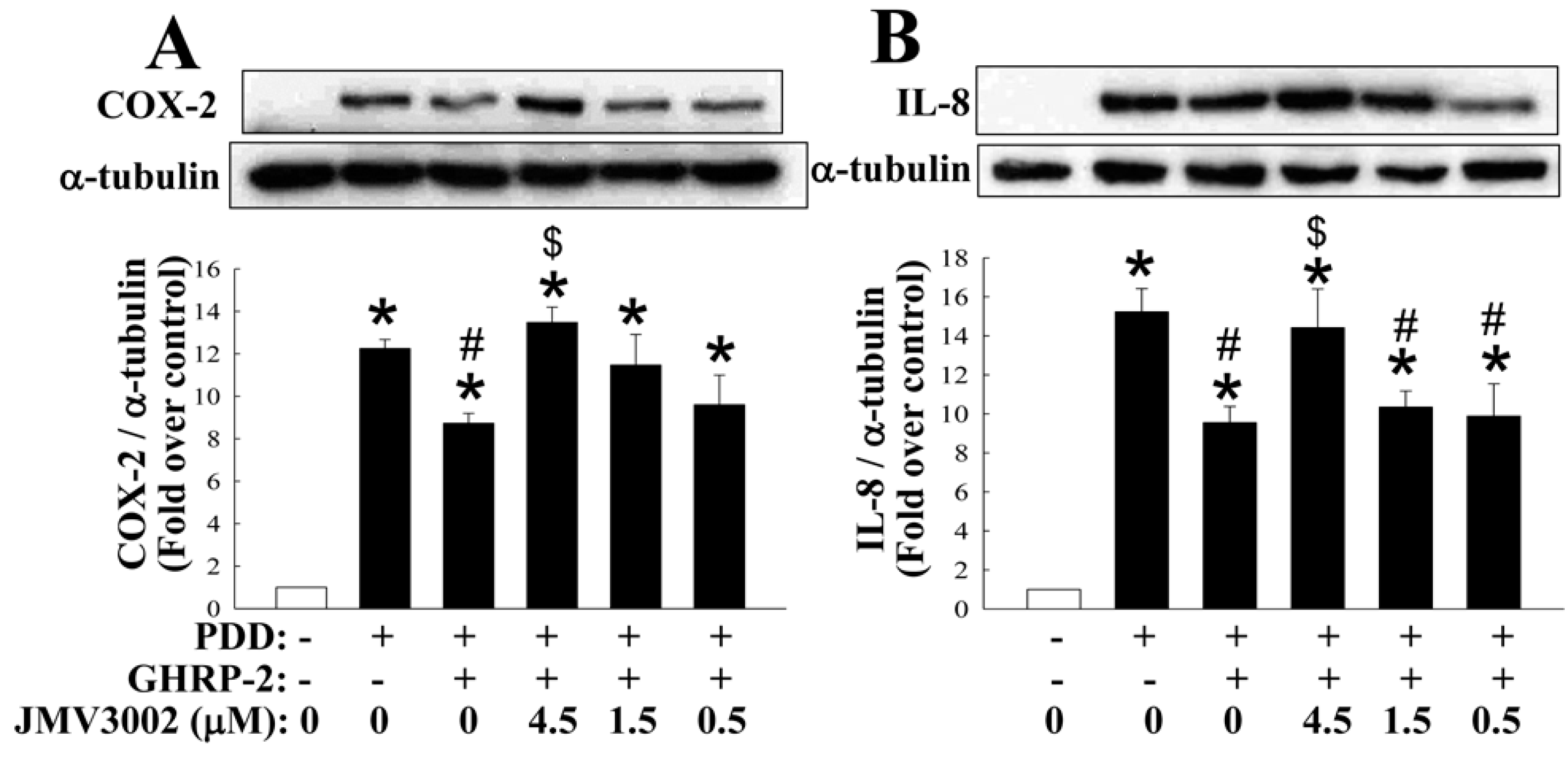
2.3. GHRP-2 Attenuation of the Induction of COX-2 and IL-8 Transcription by PKC
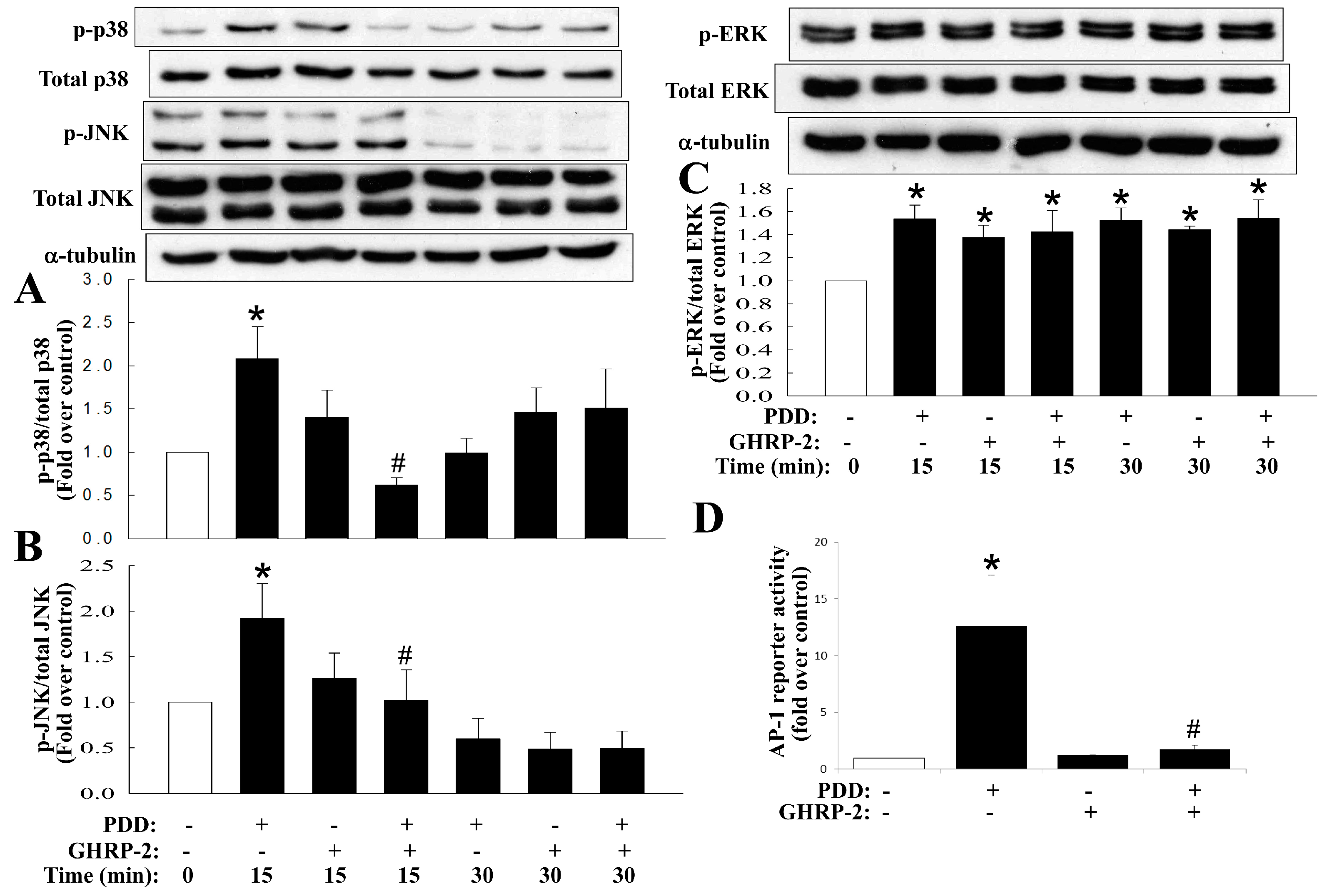
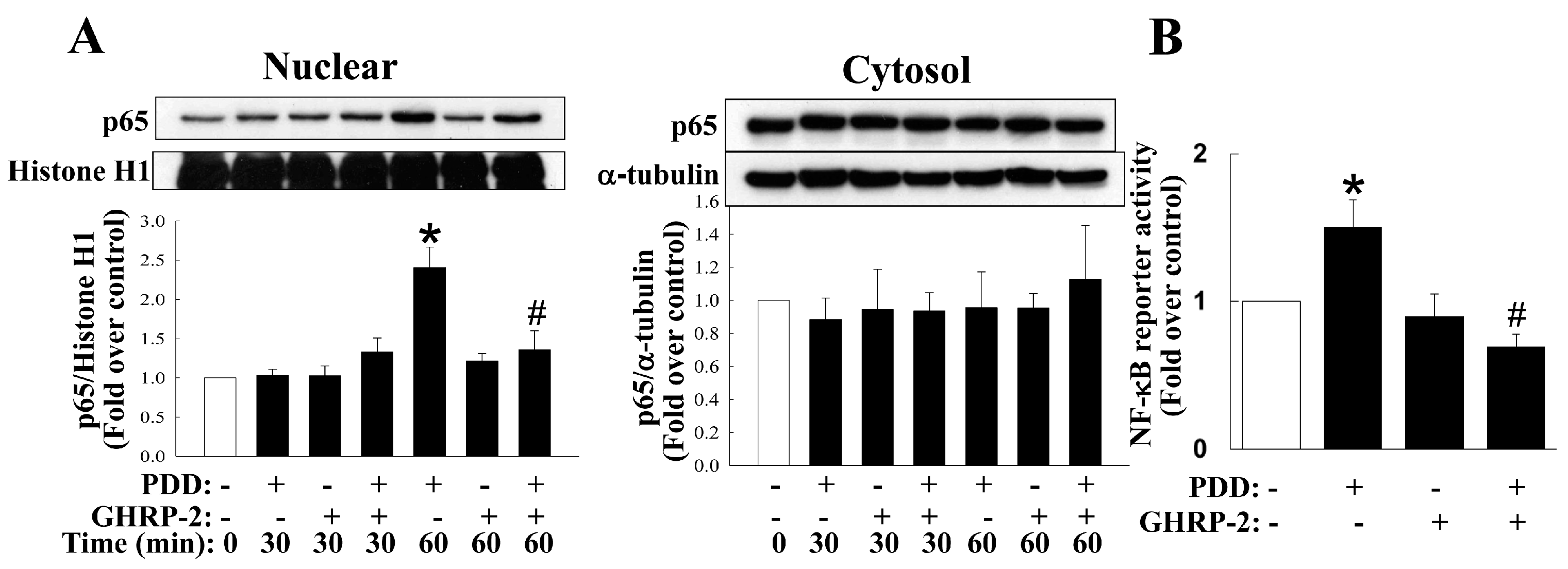
2.4. Inhibition by GHRP-2 of PKC-Mediated Activation of Various MAPKs and NF-κB
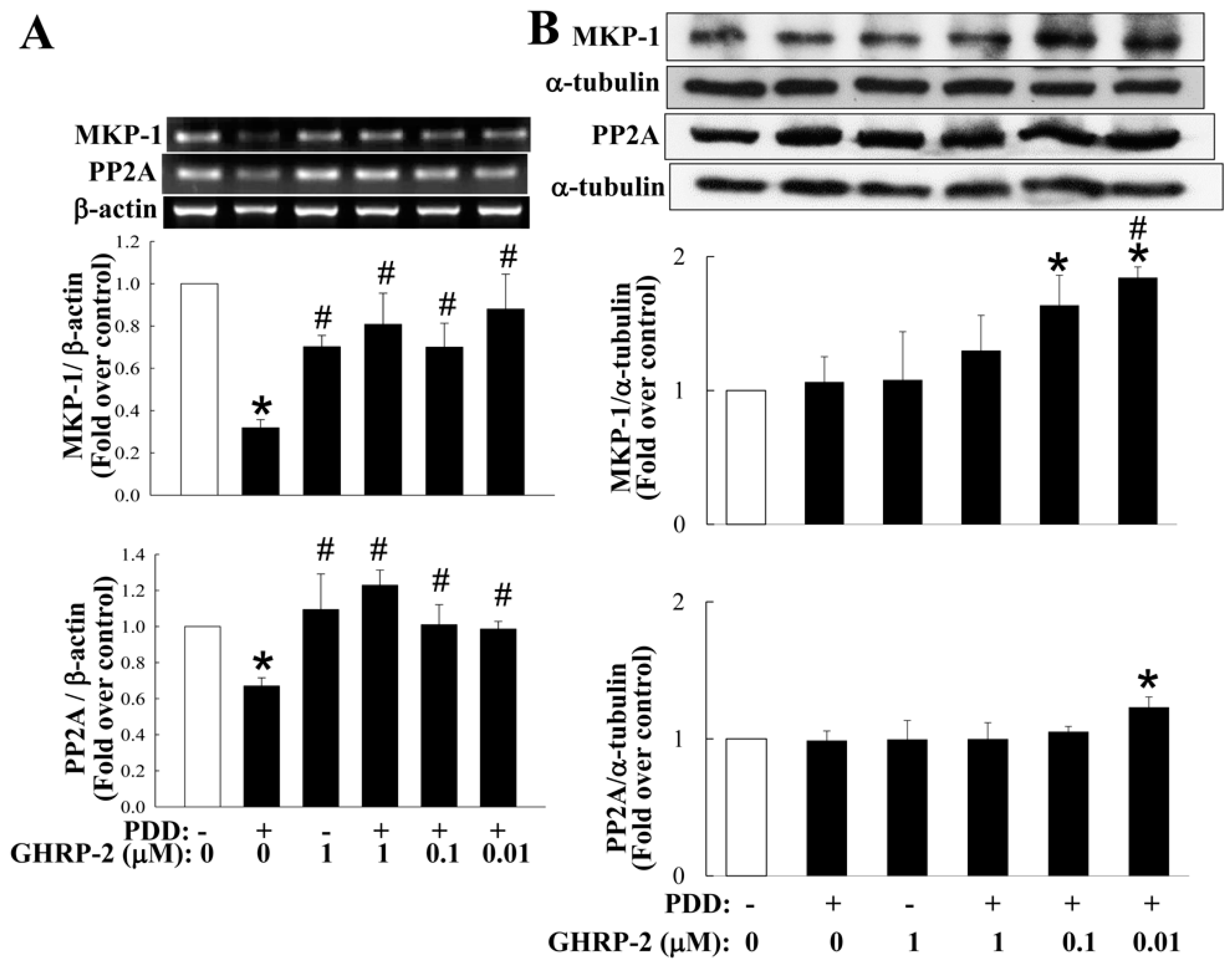
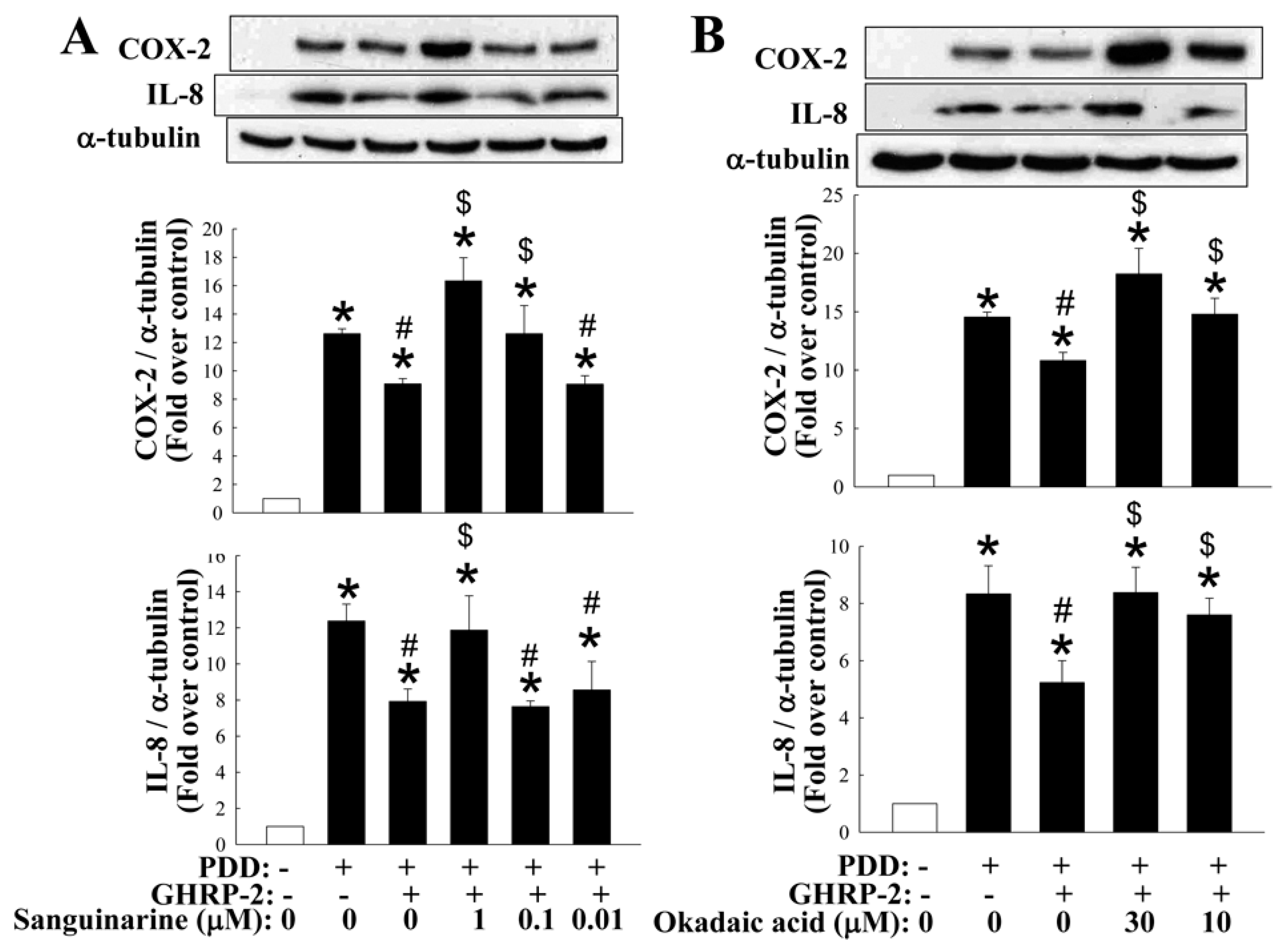
2.5. Involvement of MKP-1 and PP2A in the Inhibitory Effect of GHRP-2 on PKC-Mediated COX-2 and IL-8 Expression
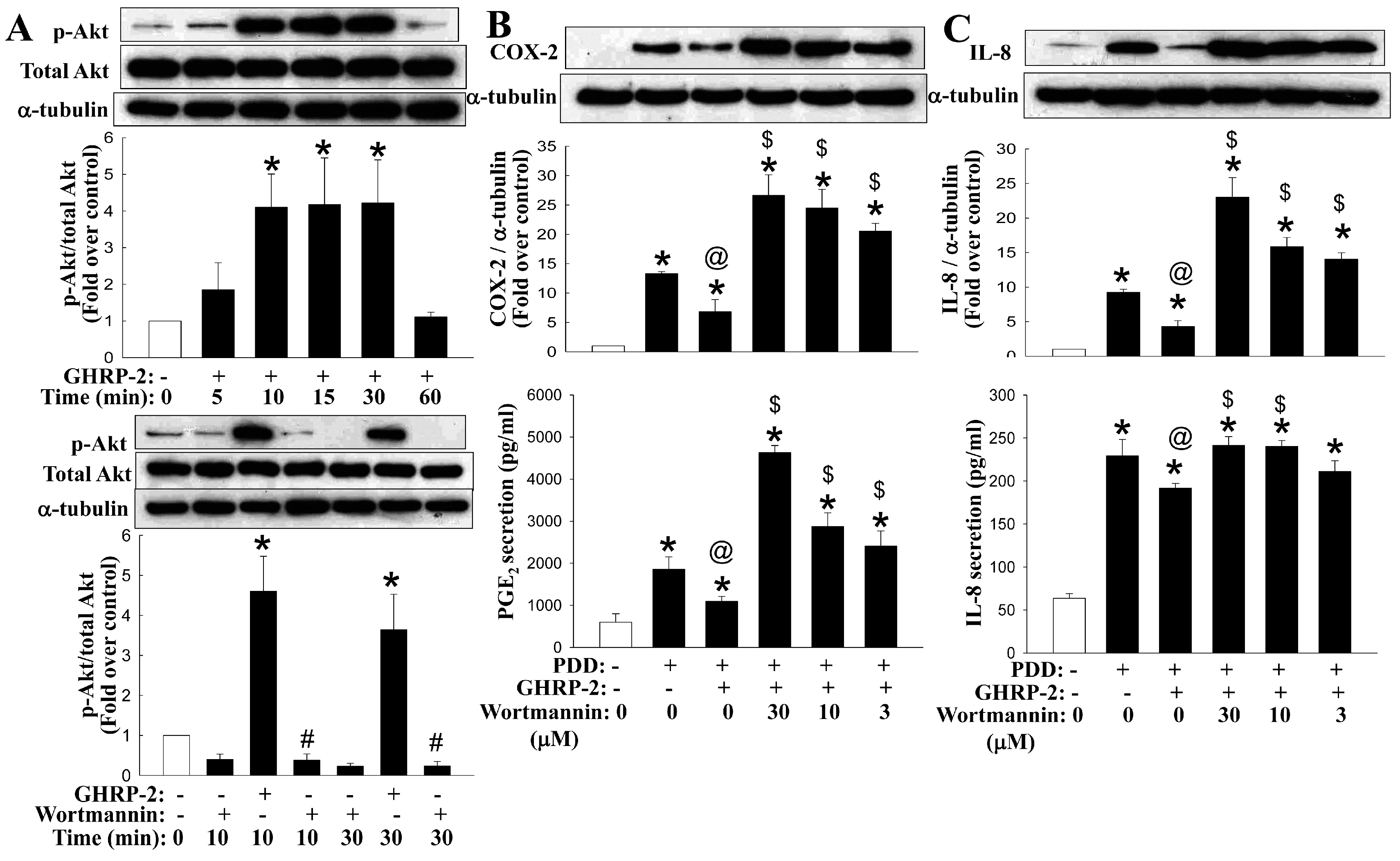
2.6. Involvement of the Akt Pathway in the Regulation by GHRP-2 of the PKC-Mediated Production of COX-2 and IL-8
2.7. Inhibition of PKC-Induced COX-2 Expression by GHRP-2 in Primary Rat Ovarian Granulosa Cells
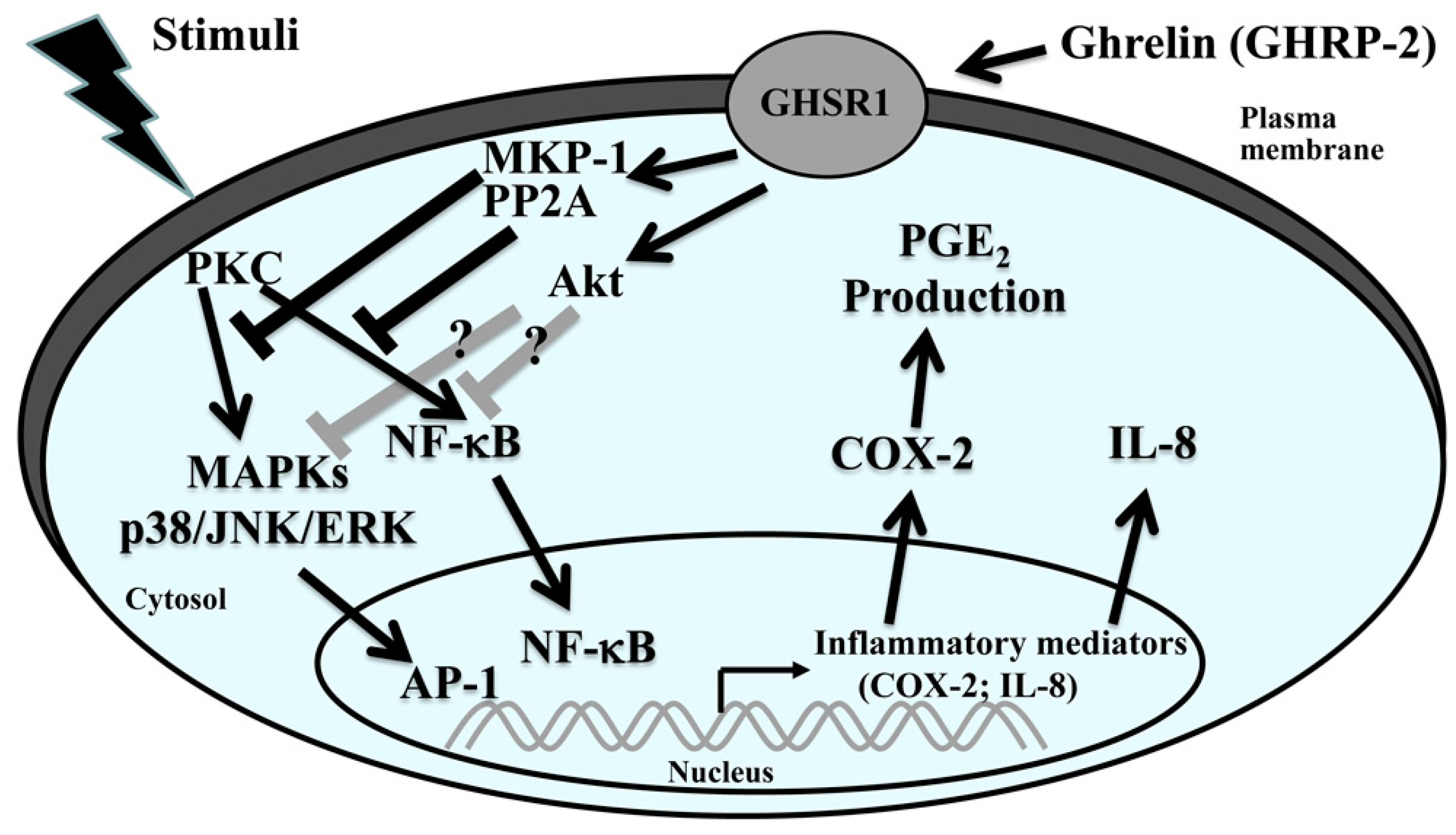
3. Discussion
4. Experimental Section
4.1. Chemicals and Reagents
4.2. Cell Culture
4.3. Western Blotting Analysis
4.4. Enzyme-Linked Immunosorbent Assay (ELISA)
4.5. Semi-Quantitative Reverse Transcription Polymerase Chain Reaction (RT-PCR)
4.6. Transfection and Analysis of COX-2 and IL-8 Promoter Activity Levels as well as NF-κB and Activator Protein-1 (AP-1) Reporter Activity Levels
4.7. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hanada, T.; Yoshimura, A. Regulation of cytokine signaling and inflammation. Cytokine Growth Factor Rev. 2002, 13, 413–421. [Google Scholar] [CrossRef]
- Rodriguez-Vita, J.; Lawrence, T. The resolution of inflammation and cancer. Cytokine Growth Factor Rev. 2010, 21, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Gershon, E.; Hourvitz, A.; Reikhav, S.; Maman, E.; Dekel, N. Low expression of COX-2, reduced cumulus expansion, and impaired ovulation in SULT1E1-deficient mice. FASEB J. 2007, 21, 1893–1901. [Google Scholar] [CrossRef] [PubMed]
- Szukiewicz, D.; Pyzlak, M.; Klimkiewicz, J.; Szewczyk, G.; Maslinska, D. Mast cell-derived interleukin-8 may be involved in the ovarian mechanisms of follicle growth and ovulation. Inflamm. Res. 2007, 56, S35–S36. [Google Scholar] [CrossRef] [PubMed]
- Ujioka, T.; Matsukawa, A.; Tanaka, N.; Matsuura, K.; Yoshinaga, M.; Okamura, H. Interleukin-8 as an essential factor in the human chorionic gonadotropin-induced rabbit ovulatory process: Interleukin-8 induces neutrophil accumulation and activation in ovulation. Biol. Reprod. 1998, 58, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Trushin, S.A.; Pennington, K.N.; Carmona, E.M.; Asin, S.; Savoy, D.N.; Billadeau, D.D.; Paya, C.V. Protein kinase Cα (PKCα) acts upstream of PKCθ to activate IκB kinase and NF-κB in T lymphocytes. Mol. Cell. Biol. 2003, 23, 7068–7081. [Google Scholar] [CrossRef] [PubMed]
- Dobkin-Bekman, M.; Naidich, M.; Pawson, A.J.; Millar, R.P.; Seger, R.; Naor, Z. Activation of mitogen-activated protein kinase (MAPK) by GnRH is cell-context dependent. Mol. Cell. Endocrinol. 2006, 252, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Chou, W.Y.; Chuang, K.H.; Sun, D.; Lee, Y.H.; Kao, P.H.; Lin, Y.Y.; Wang, H.W.; Wu, Y.L. Inhibition of PKC-Induced COX-2 and IL-8 expression in human breast cancer cells by glucosamine. J. Cell. Physiol. 2015, 230, 2240–2251. [Google Scholar] [CrossRef] [PubMed]
- Diaz, F.J.; Anderson, L.E.; Wu, Y.L.; Rabot, A.; Tsai, S.J.; Wiltbank, M.C. Regulation of progesterone and prostaglandin F2alpha production in the CL. Mol. Cell. Endocrinol. 2002, 191, 65–80. [Google Scholar] [CrossRef]
- Hua, V.K.; Fleming, S.D.; Illingworth, P. Effects of protein kinase A and C inhibitors on follicular inhibin and activin during ovulation. Reprod. Biomed. Online 2008, 17, 642–651. [Google Scholar] [CrossRef]
- Lorenzi, T.; Meli, R.; Marzioni, D.; Morroni, M.; Baragli, A.; Castellucci, M.; Gualillo, O.; Muccioli, G. Ghrelin: A metabolic signal affecting the reproductive system. Cytokine Growth Factor Rev. 2009, 20, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Howard, A.D.; Feighner, S.D.; Cully, D.F.; Arena, J.P.; Liberator, P.A.; Rosenblum, C.I.; Hamelin, M.; Hreniuk, D.L.; Palyha, O.C.; Anderson, J.; et al. A receptor in pituitary and hypothalamus that functions in growth hormone release. Science 1996, 273, 974–977. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Kangawa, N.; Kinouchi, A.; Sakamoto, Y.; Kimura, C.; Horikawa, R.; Shigematsu, Y.; Itakura, M.; Ogata, T.; Fujieda, K. Identification and functional analysis of novel human growth hormone secretagogue receptor (GHSR) gene mutations in Japanese subjects with short stature. J. Clin. Endocrinol. Metab. 2011, 96, E373–E378. [Google Scholar] [CrossRef] [PubMed]
- Zigman, J.M.; Jones, J.E.; Lee, C.E.; Saper, C.B.; Elmquist, J.K. Expression of ghrelin receptor mRNA in the rat and the mouse brain. J. Comp. Neurol. 2006, 494, 528–548. [Google Scholar] [CrossRef] [PubMed]
- Gaytan, F.; Barreiro, M.L.; Chopin, L.K.; Herington, A.C.; Morales, C.; Pinilla, L.; Casanuev, F.F.; Aguilar, E.; Diéguez, C.; Tena-Sempere, M. Immunolocalization of ghrelin and its functional receptor, the type 1a growth hormone secretagogue receptor, in the cyclic human ovary. J. Clin. Endocrinol. Metab. 2003, 88, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.W.; Harrison, J.L.; Brown, Y.A.; Doyle, U.; Lindsay, A.; Adam, C.L.; Lea, R.G. Immunohistochemical evidence for an endocrine/paracrine role for ghrelin in the reproductive tissues of sheep. Reprod. Biol. Endocrinol. 2005, 3, 60. [Google Scholar] [CrossRef] [PubMed]
- Gnanapavan, S.; Kola, B.; Bustin, S.A.; Morris, D.G.; McGee, P.; Fairclough, P.; Bhattacharya, S.; Carpenter, R.; Grossman, A.B.; Korbonits, M. The tissue distribution of the mRNA of ghrelin and subtypes of its receptor, GHS-R, in humans. J. Clin. Endocrinol. Metab. 2002, 87, 2988. [Google Scholar] [CrossRef] [PubMed]
- Rak-Mardyla, A. Ghrelin role in hypothalamus-pituitary-ovarian axis. J. Physiol. Pharmacol. 2013, 64, 695–704. [Google Scholar] [PubMed]
- Dafopoulos, K.; Messini, C.I.; Anifandis, G.; Georgoulias, P.; Sourlas, D.; Messinis, I.E. Blood ghrelin, adiponectin and resistin levels during controlled ovarian stimulation in IVF cycles. Physiol. Res. 2016, in press. [Google Scholar]
- Messini, C.I.; Dafopoulos, K.; Chalvatzas, N.; Georgoulias, P.; Anifandis, G.; Messinis, I.E. Effect of ghrelin and metoclopramide on prolactin secretion in normal women. J. Endocrinol. Investig. 2011, 34, 276–279. [Google Scholar] [CrossRef]
- Messini, C.I.; Dafopoulos, K.; Malandri, M.; Georgoulias, P.; Anifandis, G.; Messinis, I.E. Growth hormone response to submaximal doses of ghrelin remains unchanged during the follicular phase of the cycle. Reprod. Biol. Endocrinol. 2013, 11. [Google Scholar] [CrossRef] [PubMed]
- Messini, C.I.; Dafopoulos, K.; Malandri, M.; Georgoulias, P.; Anifandis, G.; Messinis, I.E. Inhibitory effect of submaximal doses of ghrelin on gonadotropin secretion in women. Horm. Metab. Res. 2014, 46, 36–40. [Google Scholar] [PubMed]
- Dovolou, E.; Chadio, S.; Messinis, I.E.; Rekkas, C.A.; Deligiannis, C.; Kalogiannis, D.; Amiridis, G.S. Human ghrelin decreases pituitary response to GnRH in superovulated ewes. Theriogenology 2013, 80, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Dovolou, E.; Periquesta, E.; Messinis, I.E.; Tsiligianni, T.; Dafopoulos, K.; Gutierrez-Adan, A.; Amiridis, G.S. Daily supplementation with ghrelin improves in vitro bovine blastocysts formation rate and alters gene expression related to embryo quality. Theriogenology 2014, 81, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Dovolou, E.; Messinis, I.E.; Periquesta, E.; Dafopoulos, K.; Gutierrez-Adan, A.; Amiridis, G.S. Ghrelin accelerates in vitro maturation of bovine oocytes. Reprod. Domest. Anim. 2014, 49, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Chouzouris, T.M.; Dovolou, E.; Dafopoulos, K.; Georgoulias, P.; Vasileiou, N.G.; Fthenakis, G.C.; Anifandis, G.; Amiridis, G.S. Ghrelin suppresses the GnRH-induced preovulatory gonadotropin surge in dairy heifers. Theriogenology 2016. [Google Scholar] [CrossRef] [PubMed]
- Otero, M.; Nogueiras, R.; Lago, F.; Dieguez, C.; Gomez-Reino, J.J.; Gualillo, O. Chronic inflammation modulates ghrelin levels in humans and rats. Rheumatology 2004, 43, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Granado, M.; Martin, A.I.; Lopez-Menduina, M.; Lopez-Calderon, A.; Villanua, M.A. GH-releasing peptide-2 administration prevents liver inflammatory response in endotoxemia. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E131–E141. [Google Scholar] [CrossRef] [PubMed]
- Granado, M.; Priego, T.; Martin, A.I.; Villanua, M.A.; Lopez-Calderon, A. Anti-inflammatory effect of the ghrelin agonist growth hormone-releasing peptide-2 (GHRP-2) in arthritic rats. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E486–E492. [Google Scholar] [CrossRef] [PubMed]
- Myung, J.; Kim, K.B.; Crews, C.M. The ubiquitin-proteasome pathway and proteasome inhibitors. Med. Res. Rev. 2001, 21, 245–273. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T.; Fukuda, M. Ubiquitin and breast cancer. Oncogene 2004, 23, 2079–2088. [Google Scholar] [CrossRef] [PubMed]
- Baer, R.; Ludwig, T. The BRCA1/BARD1 heterodimer, a tumor suppressor complex with ubiquitin E3 ligase activity. Curr. Opin. Genet. Dev. 2002, 12, 86–91. [Google Scholar] [CrossRef]
- Dhanasekaran, N.; Moudgal, N.R. Gonadotropin regulation of rat ovarian lysosomes: Existence of a hormone specific dual control mechanism. Biosci. Rep. 1988, 8, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Sagne, C.; Agulhon, C.; Ravassard, P.; Darmon, M.; Hamon, M.; El Mestikawy, S.; Gasnier, B.; Giros, B. Identification and characterization of a lysosomal transporter for small neutral amino acids. Proc. Natl. Acad. Sci. USA 2001, 98, 7206–7211. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B. Inflammatory signaling in macrophages: Transitions from acute to tolerant and alternative activation states. Eur. J. Immunol. 2011, 41, 2477–2481. [Google Scholar] [CrossRef] [PubMed]
- Li, J.K.; Nie, L.; Zhao, Y.P.; Zhang, Y.Q.; Wang, X.; Wang, S.S.; Liu, Y.; Zhao, H.; Cheng, L. IL-17 mediates inflammatory reactions via p38/c-Fos and JNK/c-Jun activation in an AP-1-dependent manner in human nucleus pulposus cells. J. Transl. Med. 2016, 14. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shepherd, E.G.; Nelin, L.D. MAPK phosphatases—Regulating the immune response. Nat. Rev. Immunol. 2007, 7, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Farooq, A.; Zhou, M.M. Structure and regulation of MAPK phosphatases. Cell Signal. 2004, 16, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Charles, C.H.; Lau, L.F.; Tonks, N.K. MKP-1 (3CH134), an immediate early gene product, is a dual specificity phosphatase that dephosphorylates MAP kinase in vivo. Cell 1993, 75, 487–493. [Google Scholar] [CrossRef]
- Jacob, A.; Rajan, D.; Pathickal, B.; Balouch, I.; Hartman, A.; Wu, R.; Zhou, M.; Wang, P. The inhibitory effect of ghrelin on sepsis-induced inflammation is mediated by the MAPK phosphatase-1. Int. J. Mol. Med. 2010, 25, 159–164. [Google Scholar] [PubMed]
- Sun, S.C.; Maggirwar, S.B.; Harhaj, E. Activation of NF-kappa B by phosphatase inhibitors involves the phosphorylation of I kappa B alpha at phosphatase 2A-sensitive sites. J. Biol. Chem. 1995, 270, 18347–18351. [Google Scholar] [CrossRef] [PubMed]
- Chabot-Fletcher, M. A role for transcription factor NF-κB in inflammation. Inflamm. Res. 1997, 46, 1–2. [Google Scholar] [PubMed]
- Lodeiro, M.; Theodoropoulou, M.; Pardo, M.; Casanueva, F.F.; Camina, J.P. c-Src regulates Akt signaling in response to ghrelin via beta-arrestin signaling-independent and -dependent mechanisms. PLoS ONE 2009, 4, e4686. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, P.L.; Murray, R.E.; Yeh, A.H.; McNamara, J.F.; Duncan, R.P.; Francis, G.D.; Herington, A.C.; Chopin, L.K. Expression and function of the ghrelin axis, including a novel preproghrelin isoform, in human breast cancer tissues and cell lines. Endocr. Relat. Cancer 2005, 12, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Dangi, S.S.; Singh, G.; Sarkar, M. Expression and localization of ghrelin and its receptor in ovarian follicles during different stages of development and the modulatory effect of ghrelin on granulosa cells function in buffalo. Gen. Comp. Endocrinol. 2015, 210, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Sirotkin, A.V.; Meszarošová, M.; Grossmann, R.; Benčo, A.; Valenzuela, F. Effect of inhibitor and activator of ghrelin receptor (GHS-R1a) on porcine ovarian granulosa cell functions. Gen. Comp. Endocrinol. 2011, 173, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Li, W.G.; Gavrila, D.; Liu, X.; Wang, L.; Gunnlaugsson, S.; Stoll, L.L.; McCormick, M.L.; Sigmund, C.D.; Tang, C.; Weintraub, N.L. Ghrelin inhibits proinflammatory responses and nuclear factor-κB activation in humanendothelial cells. Circulation 2004, 109, 2221–2226. [Google Scholar] [CrossRef] [PubMed]
- Deng, B.; Fang, F.; Yang, T.; Yu, Z.; Zhang, B.; Xie, X. Ghrelin inhibits AngII-induced expression of TNF-α, IL-8, MCP-1 in human umbilical vein endothelial cells. Int. J. Clin. Exp. Med. 2015, 8, 579–588. [Google Scholar] [PubMed]
- Titterington, J.S.; Sukhanov, S.; Higashi, Y.; Vaughn, C.; Bowers, C.; Delafontaine, P. Growth hormone-releasing peptide-2 suppresses vascular oxidative stress in ApoE−/− mice but does not reduce atherosclerosis. Endocrinology 2009, 15, 5478–5487. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D. Protein kinase Cdelta-mediated CREB activation regulates ghrelin-induced cyclooxygenase-2 expression and prostaglandin E2 production in human colonic epithelial cells. J. Cell. Biochem. 2007, 102, 1245–1255. [Google Scholar] [CrossRef] [PubMed]
- Tsatsanis, C.; Androulidaki, A.; Venihaki, M.; Margioris, A.N. Signalling networks regulating cyclooxygenase-2. Int. J. Biochem. Cell. Biol. 2006, 38, 1654–1661. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Oh, J.M.; No, J.H.; Bang, Y.J.; Juhnn, Y.S.; Song, Y.S. Involvement of NF-κB and AP-1 in COX-2 upregulation by human papillomavirus 16 E5 oncoprotein. Carcinogenesis 2009, 30, 753–757. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.; Dittrich-Breiholz, O.; Holtmann, H.; Kracht, M. Multiple control of interleukin-8 gene expression. J. Leukoc. Biol. 2002, 72, 847–855. [Google Scholar] [PubMed]
- Zhang, D.; Li, J.; Song, L.; Ouyang, W.; Gao, J.; Huang, C. A JNK1/AP-1-dependent, COX-2 induction is implicated in 12-O-tetradecanoylphorbol-13-acetate-induced cell transformation through regulating cell cycle progression. Mol. Cancer Res. 2008, 6, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Anttila, H.S.; Reitamo, S.; Ceska, M.; Hurme, M. Signal transduction pathways leading to the production of IL-8 by human monocytes are differentially regulated by dexamethasone. Clin. Exp. Immunol. 1992, 89, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Elliott, C.L.; Allport, V.C.; Loudon, J.A.; Wu, G.D.; Bennett, P.R. Nuclear factor-kappa B is essential for up-regulation of interleukin-8 expression in human amnion and cervical epithelial cells. Mol. Hum. Reprod. 2011, 7, 787–790. [Google Scholar] [CrossRef]
- Nanzer, A.M.; Khalaf, S.; Mozid, A.M.; Fowkes, R.C.; Patel, M.V.; Burrin, J.M.; Grossman, A.B.; Korbonits, M. Ghrelin exerts a proliferative effect on a rat pituitary somatotroph cell line via the mitogen-activated protein kinase pathway. Eur. J. Endocrinol. 2004, 151, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Bavaria, M.N.; Jin, S.; Ray, R.M.; Johnson, L.R. The mechanism by which MEK/ERK regulates JNK and p38 activity in polyamine depleted IEC-6 cells during apoptosis. Apoptosis 2014, 19, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Park, K.A.; Li, Y.; Byun, H.S.; Jeon, J.; Lee, Y.; Hong, J.H.; Kim, J.M.; Huang, S.M.; Choi, S.W.; et al. PHF20 regulates NF-κB signalling by disrupting recruitment of PP2A to p65. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.G.; Sun, Y.; Jiang, H.; Albarran-Zeckler, R.; Timchenko, N. Ghrelin receptor (GHS-R1A) agonists show potential as interventive agents during aging. Ann. N. Y. Acad. Sci. 2007, 1119, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.J.; Huang, H.; Huang, C.X. Potential new role of the GHSR-1a-mediated signaling pathway in cardiac remodeling after myocardial infarction (Review). Oncol. Lett. 2014, 8, 969–997. [Google Scholar] [CrossRef] [PubMed]
- Cassatella, M.A.; Guasparri, I.; Ceska, M.; Bazzoni, F.; Rossi, F. Interferon-gamma inhibits interleukin-8 production by human polymorphonuclear leucocytes. Immunology 1993, 78, 177–184. [Google Scholar] [PubMed]
- Green, J.; Khabar, K.S.; Koo, B.C.; Williams, B.R.; Polyak, S.J. Stability of CXCL-8 and related AU-rich mRNAs in the context of hepatitis C virus replication in vitro. J. Infect. Dis. 2006, 193, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Roger, T.; Out, T.; Mukaida, N.; Matsushima, K.; Jansen, H.; Lutter, R. Enhanced AP-1 and NF-kappaB activities and stability of interleukin 8 (IL-8) transcripts are implicated in IL-8 mRNA superinduction in lung epithelial H292 cells. Biochem. J. 1998, 330, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Meier, R.W.; Niklaus, G.; Fey, M.F.; Tobler, A. The induction kinetics of Il-8 messenger RNA in HL60 cells demonstrate the participation of negative-acting gene(s). Leuk. Res. 1995, 19, 449–455. [Google Scholar] [CrossRef]
- Neagoe, P.E.; Dumas, E.; Hajjar, F.; Sirois, M.G. Angiopoietin-1 but not angiopoietin-2 induces IL-8 synthesis and release by human neutrophils. J. Cell. Physiol. 2012, 227, 3099–3110. [Google Scholar] [CrossRef] [PubMed]
- Wilmer, J.L.; Luster, M.I. Chemical induction of interleukin-8, a proinflammatory chemokine, in human epidermal keratinocyte cultures and its relation to cytogenetic toxicity. Cell Biol. Toxicol. 1995, 11, 37–50. [Google Scholar] [PubMed]
- Nishi, Y.; Yanase, T.; Mu, Y.; Oba, K.; Ichino, I.; Saito, M.; Nomura, M.; Mukasa, C.; Okabe, T.; Goto, K.; et al. Establishment and characterization of a steroidogenic human granulosa-like tumor cell line, KGN, that expresses functional follicle-stimulating hormone receptor. Endocrinology 2001, 142, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Yokoyama, C.; Hara, S.; Tone, Y.; Tanabe, T. Transcriptional regulation of human prostaglandin-endoperoxide synthase-2 gene by lipopolysaccharide and phorbol ester in vascular endothelial cells: Involvement of both nuclear factor for interleukin-6 expression site and cAMP response element. J. Biol. Chem. 1995, 270, 24965–24971. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequences | Direction | Size (bp) |
|---|---|---|---|
| COX-2 | 5′-GCATCAGTTTTTCAAGACAG-3′ 5′-TCGCATACTCTGTTGTGTTC-3′ | Sense Antisense | 324 |
| IL-8 | 5′-ACTTCCAAGCTGGCCGTGGCT-3′ 5′-TCACTGGCATCTTCACTGATT-3′ | Sense Antisense | 318 |
| PP2A | 5′-AAGGTTCGTTACCGTGAACG-3′ 5′-ACCTCTTGCACGTTGGATTC-3′ | Sense Antisense | 641 |
| MKP-1 | 5′-CCGGAGCTGTGCAGCAAA-3′ 5′-CTCCACAGGGATGCTCTT-3′ | Sense Antisense | 282 |
| GHSR-1a | 5′-AGCGCTACTTCGCCATC-3′ 5′-CCGATGAGACTGTAGAG-3′ | Sense Antisense | 289 |
| GHSR-1b | 5′-TCTTCCTTCCTGTCTTCTGT-3′ 5′-GATAGGACCCGCGAGAGAAA-3′ | Sense Antisense | 179 |
| β-actin | 5′-GGCACCACACCTTCTACAAT-3′ 5′-CGTCATACTCCTGCTTGCTG-3′ | Sense Antisense | 834 |
| GAPDH | 5′-ATCACCATCTTCCAGGAGCG-3′ 5′-CCTGCTTCACCACCTTCTTG-3′ | Sense Antisense | 574 |
| BRCA1 | 5′-ACAGCTGTGTGGTGCTTCTGTG-3′ 5′-CATTGTCCTCTGTCCAGGCATC-3′ | Sense Antisense | 107 |
| Cathepsin D | 5′-CATTGTGGACACAGGCACTTC-3′ 5′-GACACCTTGAGCGTGTAGTCC-3′ | Sense Antisense | 201 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chao, Y.-N.; Sun, D.; Peng, Y.-C.; Wu, Y.-L. Growth Hormone Releasing Peptide-2 Attenuation of Protein Kinase C-Induced Inflammation in Human Ovarian Granulosa Cells. Int. J. Mol. Sci. 2016, 17, 1359. https://doi.org/10.3390/ijms17081359
Chao Y-N, Sun D, Peng Y-C, Wu Y-L. Growth Hormone Releasing Peptide-2 Attenuation of Protein Kinase C-Induced Inflammation in Human Ovarian Granulosa Cells. International Journal of Molecular Sciences. 2016; 17(8):1359. https://doi.org/10.3390/ijms17081359
Chicago/Turabian StyleChao, Yi-Ning, David Sun, Yen-Chun Peng, and Yuh-Lin Wu. 2016. "Growth Hormone Releasing Peptide-2 Attenuation of Protein Kinase C-Induced Inflammation in Human Ovarian Granulosa Cells" International Journal of Molecular Sciences 17, no. 8: 1359. https://doi.org/10.3390/ijms17081359
APA StyleChao, Y. -N., Sun, D., Peng, Y. -C., & Wu, Y. -L. (2016). Growth Hormone Releasing Peptide-2 Attenuation of Protein Kinase C-Induced Inflammation in Human Ovarian Granulosa Cells. International Journal of Molecular Sciences, 17(8), 1359. https://doi.org/10.3390/ijms17081359