
Importance of Estrogenic Signaling and Its Mediated Receptors in Prostate Cancer


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Prostate Cancer Epidemiology
2. Epidemiological and Clinical Evidence of Hormonal Involvements in Prostatic Carcinogenesis
3. Estrogen- and Androgen-Induced Prostatic Lesions in Animals
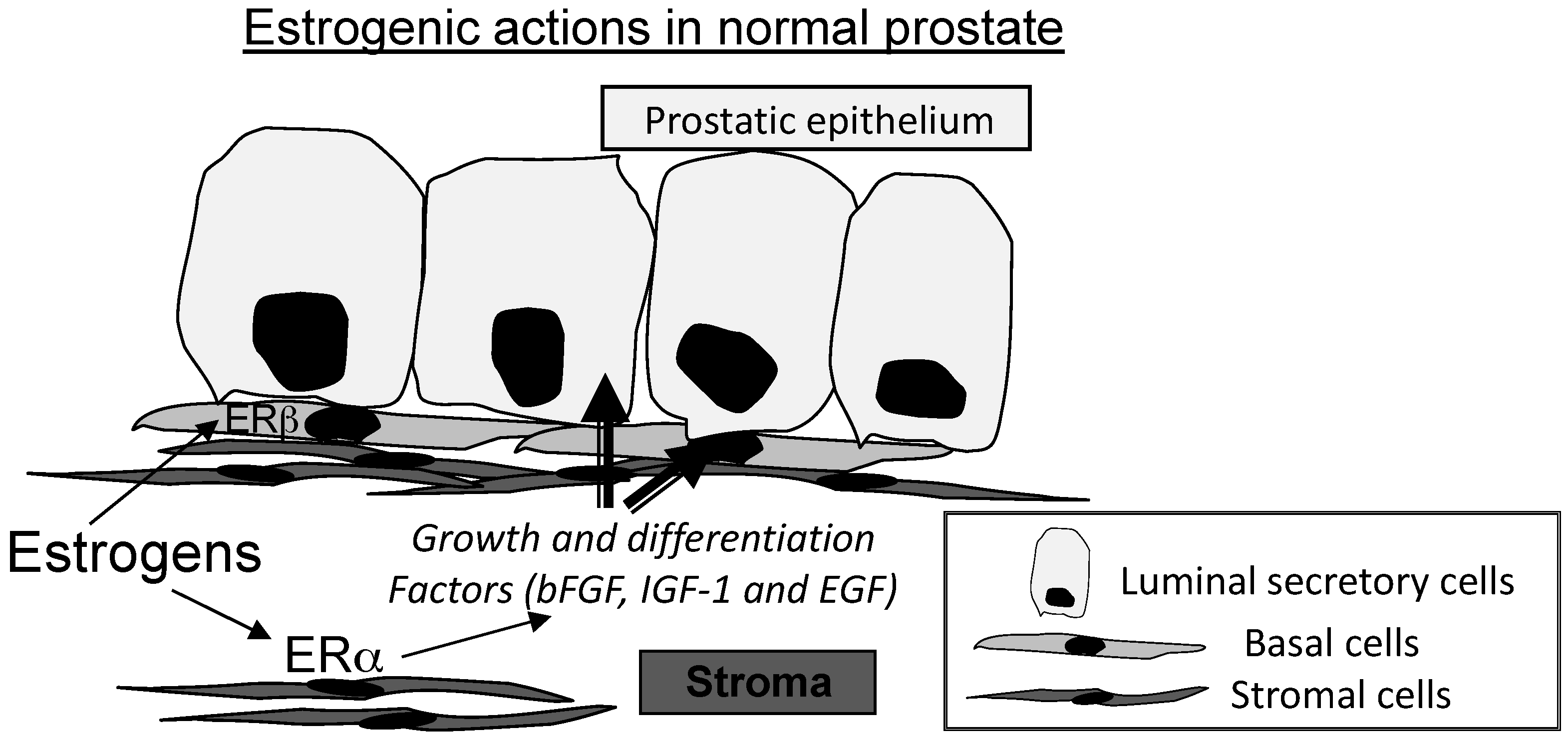
4. Estrogen Receptors (ERs) and Estrogenic Actions in Normal Prostate
5. Estrogen Receptor (ER) Expression and Estrogen Actions in PCa
6. Mechanistic Insights of ERβ- and ERα-Mediated Signals
7. Biological Functions of ERα and ERβ Derived from Their Knockout Mice Models
8. Estrogen Therapy for Prostate Cancer
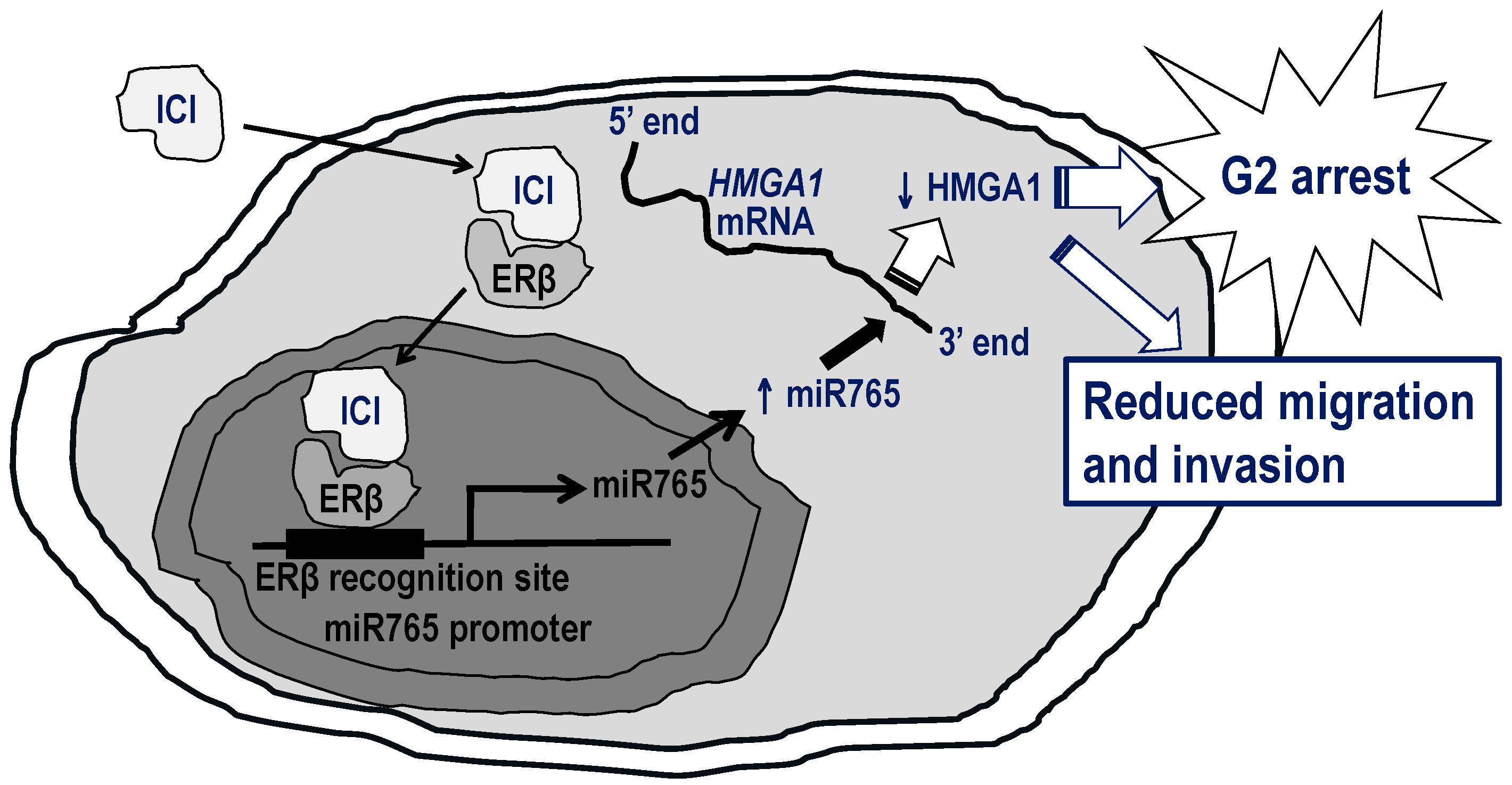
9. Potential of Antiestrogens for PCa Therapy
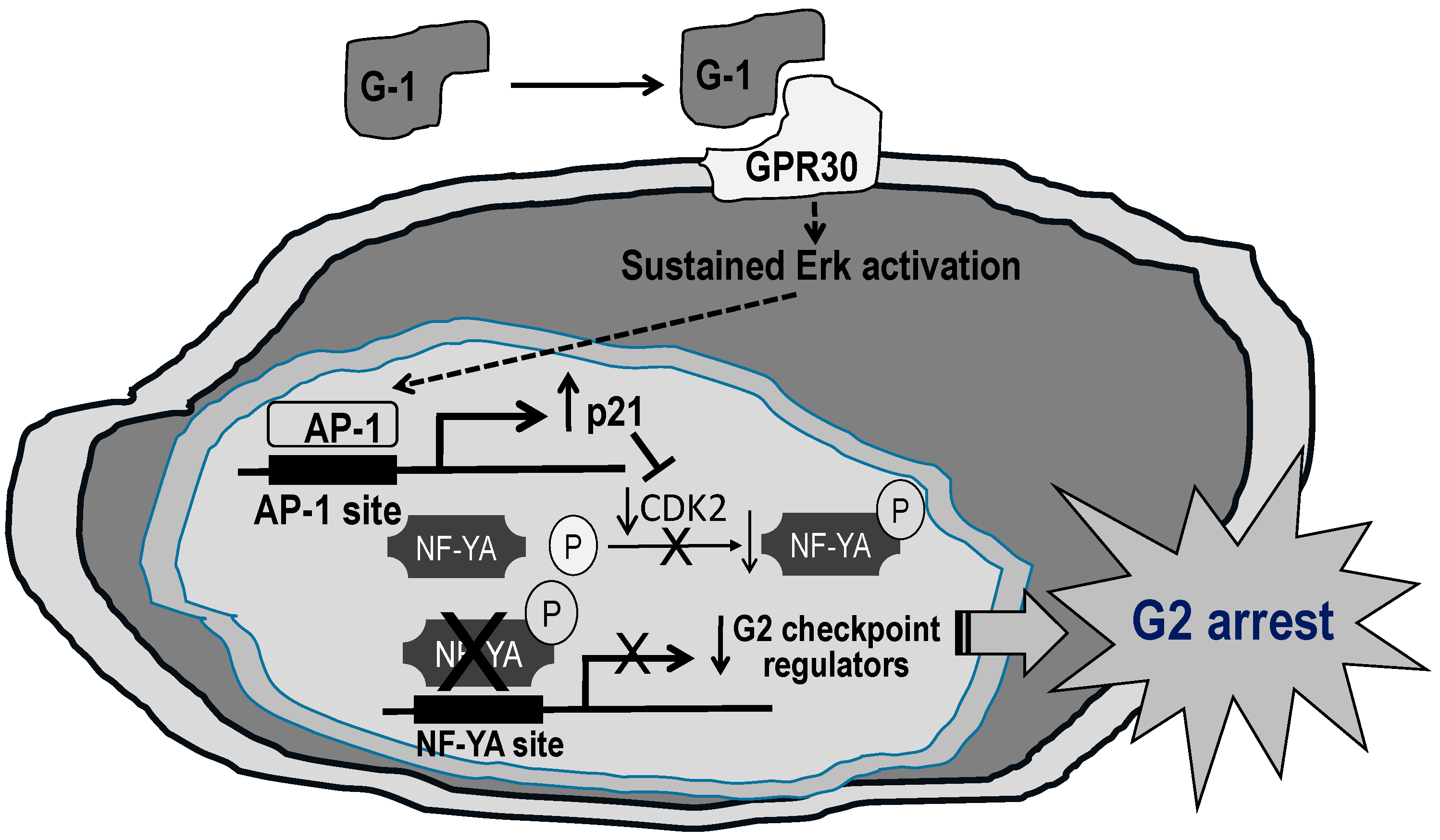
10. Significance of a G-Protein-Coupled Receptor GPR30 and Its Therapeutic Potential in PCa
11. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- American Cancer Society—Cancer Statistics Center. Prostate. Available online: https://cancerstatisticscenter.cancer.org/#/cancer-site/Prostate (assessed on 18 February 2016).
- Cook, P.J.; Doll, R.; Fellingham, S.A. A mathematical model for the age distribution of cancer in man. Int. J. Cancer 1969, 4, 93–112. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.K. Epidemiology of prostate cancer and bladder cancer: An overview. Cancer Treat Res. 1996, 88, 1–11. [Google Scholar] [PubMed]
- Platz, E.A.; Rimm, E.B.; Willett, W.C.; Kantoff, P.W.; Giovannucci, E. Racial variation in prostate cancer incidence and in hormonal system markers among male health professionals. J. Natl. Cancer Inst. 2000, 92, 2009–2017. [Google Scholar] [CrossRef] [PubMed]
- Farkas, A.; Marcella, S.; Rhoads, G.G. Ethnic and racial differences in prostate cancer incidence and mortality. Ethn. Dis. 2000, 10, 69–75. [Google Scholar] [PubMed]
- Hoffman, R.M.; Gilliland, F.D.; Eley, J.W.; Harlan, L.C.; Stephenson, R.A.; Stanford, J.L.; Albertson, P.C.; Hamilton, A.S.; Hunt, W.C.; Potosky, A.L. Racial and ethnic differences in advanced-stage prostate cancer: The prostate cancer outcomes study. J. Natl. Cancer Inst. 2001, 93, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Fowler, J.E., Jr.; Bigler, S.A.; Lynch, C.; Wilson, S.S.; Farabaugh, P.B. Prospective study of correlations between biopsy-detected high grade prostatic intraepithelial neoplasia, serum prostate specific antigen concentration, and race. Cancer 2001, 91, 1291–1296. [Google Scholar] [CrossRef]
- Powell, I.J.; Meyskens, F.L., Jr. African American men and hereditary/familial prostate cancer: Intermediate-risk populations for chemoprevention trials. Urology 2001, 57, 178–181. [Google Scholar] [CrossRef]
- Du, X.L.; Fang, S.; Coker, A.L.; Sanderson, M.; Aragaki, C.; Cormier, J.N.; Xing, Y.; Gor, B.J.; Chan, W. Racial disparity and socioeconomic status in association with survival in older men with local/regional stage prostate carcinoma: Findings from a large community-based cohort. Cancer 2006, 106, 1276–1285. [Google Scholar] [CrossRef] [PubMed]
- Virnig, B.A.; Baxter, N.N.; Habermann, E.B.; Feldman, R.D.; Bradley, C.J. A matter of race: Early-versus late-stage cancer diagnosis. Health Aff. 2009, 28, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, T.R.; Devesa, S.S.; Chang, B.L.; Bunker, C.H.; Cheng, I.; Cooney, K.; Eeles, R.; Fernandez, P.; Giri, V.N.; Gueye, S.M.; et al. Global patterns of prostate cancer incidence, aggressiveness, and mortality in men of African descent. Prostate Cancer 2013, 2013, 560857. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Nakayama, T.; Shiraishi, T.; Stemmermann, G.N.; Yatani, R. Comparative studies of prostate cancer in japan versus the United States. A review. Urol. Oncol. 2000, 5, 274–283. [Google Scholar] [CrossRef]
- Ellem, S.J.; Risbridger, G.P. Treating prostate cancer: A rationale for targeting local oestrogens. Nat. Rev. Cancer 2007, 7, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Ross, R.K.; Bernstein, L.; Yatani, R.; Henderson, B.E.; Mack, T.M. Cancers of the prostate and breast among Japanese and white immigrants in Los Angeles county. Br. J. Cancer 1991, 63, 963–966. [Google Scholar] [CrossRef] [PubMed]
- McCredie, M.; Coates, M.; Grulich, A. Cancer incidence in migrants to New South Wales (Australia) from the Middle East, 1972–1991. Cancer Causes Control 1994, 5, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Whittemore, A.S.; Kolonel, L.N.; Wu, A.H.; John, E.M.; Gallagher, R.P.; Howe, G.R.; Burch, J.D.; Hankin, J.; Dreon, D.M.; West, D.W.; et al. Prostate cancer in relation to diet, physical activity, and body size in blacks, whites, and Asians in the United States and Canada. J. Natl. Cancer Inst. 1995, 87, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Angwafo, F.F. Migration and prostate cancer: An international perspective. J. Natl. Med. Assoc. 1998, 90, S720–S723. [Google Scholar] [PubMed]
- Guileyardo, J.M.; Johnson, W.D.; Welsh, R.A.; Akazaki, K.; Correa, P. Prevalence of latent prostate carcinoma in two U.S. Populations. J. Natl. Cancer Inst. 1980, 65, 311–316. [Google Scholar] [PubMed]
- Sakr, W.A.; Grignon, D.J.; Haas, G.P.; Schomer, K.L.; Heilbrun, L.K.; Cassin, B.J.; Powell, J.; Montie, J.A.; Pontes, J.E.; Crissman, J.D. Epidemiology of high grade prostatic intraepithelial Neoplasia. Pathol. Res. Pract. 1995, 191, 838–841. [Google Scholar] [CrossRef]
- Sakr, W.A.; Ward, C.; Grignon, D.J.; Haas, G.P. Epidemiology and molecular biology of early prostatic neoplasia. Mol. Urol. 2000, 4, 109–113. [Google Scholar] [PubMed]
- Selman, S.H. “Latent” carcinoma of the prostate: A medical misnomer? Urology 2000, 56, 708–711. [Google Scholar] [CrossRef]
- Rich, A.R. On the frequency of occurrence of occult carcinoma of the prostrate. 1934. Int. J. Epidemiol. 2007, 36, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Andrews, G.S. Latent carcinoma of the prostate. J. Clin. Pathol. 1949, 2, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Franks, L.M. Latent carcinoma of the prostate. J. Pathol. Bacteriol. 1954, 68, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, K. Estrogens and prostatic disease. International prostate health council study group. Prostate 2000, 45, 87–100. [Google Scholar] [CrossRef]
- Hill, P.; Garbaczewski, L.; Walker, A.R. Age, environmental factors and prostatic cancer. Med. Hypotheses 1984, 14, 29–39. [Google Scholar] [CrossRef]
- Henderson, B.E.; Bernstein, L.; Ross, R.K.; Depue, R.H.; Judd, H.L. The early in utero oestrogen and testosterone environment of blacks and whites: Potential effects on male offspring. Br. J. Cancer 1988, 57, 216–218. [Google Scholar] [CrossRef] [PubMed]
- De Jong, F.H.; Oishi, K.; Hayes, R.B.; Bogdanowicz, J.F.; Raatgever, J.W.; van der Maas, P.J.; Yoshida, O.; Schroeder, F.H. Peripheral hormone levels in controls and patients with prostatic cancer or benign prostatic hyperplasia: Results from the Dutch-Japanese case-control study. Cancer Res. 1991, 51, 3445–3450. [Google Scholar] [PubMed]
- Bosland, M.C. The role of steroid hormones in prostate carcinogenesis. J. Natl. Cancer Inst. Monogr. 2000, 27, 39–66. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.; Bernstein, L.; Judd, H.; Hanisch, R.; Pike, M.; Henderson, B. Serum testosterone levels in healthy young black and white men. J. Natl. Cancer Inst. 1986, 76, 45–48. [Google Scholar] [PubMed]
- Rohrmann, S.; Nelson, W.G.; Rifai, N.; Brown, T.R.; Dobs, A.; Kanarek, N.; Yager, J.D.; Platz, E.A. Serum estrogen, but not testosterone, levels differ between black and white men in a nationally representative sample of Americans. J. Clin. Endocrinol. Metab. 2007, 92, 2519–2525. [Google Scholar] [CrossRef] [PubMed]
- Abd Elmageed, Z.Y.; Moroz, K.; Srivastav, S.K.; Fang, Z.; Crawford, B.E.; Moparty, K.; Thomas, R.; Abdel-Mageed, A.B. High circulating estrogens and selective expression of Erβ in prostate tumors of Americans: Implications for racial disparity of prostate cancer. Carcinogenesis 2013, 34, 2017–2023. [Google Scholar] [CrossRef] [PubMed]
- Nomura, A.; Heilbrun, L.K.; Stemmermann, G.N.; Judd, H.L. Prediagnostic serum hormones and the risk of prostate cancer. Cancer Res. 1988, 48, 3515–3517. [Google Scholar] [PubMed]
- Hsing, A.W.; Comstock, G.W. Serological precursors of cancer: Serum hormones and risk of subsequent prostate cancer. Cancer Epidemiol. Biomark. Prev. 1993, 2, 27–32. [Google Scholar]
- Comstock, G.W.; Gordon, G.B.; Hsing, A.W. The relationship of serum dehydroepiandrosterone and its sulfate to subsequent cancer of the prostate. Cancer Epidemiol. Biomark. Prev. 1993, 2, 219–221. [Google Scholar]
- Gann, P.H.; Hennekens, C.H.; Ma, J.; Longcope, C.; Stampfer, M.J. Prospective study of sex hormone levels and risk of prostate cancer. J. Natl. Cancer Inst. 1996, 88, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- Dorgan, J.F.; Albanes, D.; Virtamo, J.; Heinonen, O.P.; Chandler, D.W.; Galmarini, M.; McShane, L.M.; Barrett, M.J.; Tangrea, J.; Taylor, P.R. Relationships of serum androgens and estrogens to prostate cancer risk: Results from a prospective study in Finland. Cancer Epidemiol. Biomark. Prev. 1998, 7, 1069–1074. [Google Scholar]
- Barrett-Connor, E.; Garland, C.; McPhillips, J.B.; Khaw, K.T.; Wingard, D.L. A prospective, population-based study of androstenedione, estrogens, and prostatic cancer. Cancer Res. 1990, 50, 169–173. [Google Scholar] [PubMed]
- Vermeulen, A.; Rubens, R.; Verdonck, L. Testosterone secretion and metabolism in male senescence. J. Clin. Endocrinol. Metab. 1972, 34, 730–735. [Google Scholar] [CrossRef] [PubMed]
- Gray, A.; Feldman, H.A.; McKinlay, J.B.; Longcope, C. Age, disease, and changing sex hormone levels in middle-aged men: Results of the Massachusetts male aging study. J. Clin. Endocrinol. Metab. 1991, 73, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Gray, A.; Berlin, J.A.; McKinlay, J.B.; Longcope, C. An examination of research design effects on the association of testosterone and male aging: Results of a meta-analysis. J. Clin. Epidemiol. 1991, 44, 671–684. [Google Scholar] [CrossRef]
- Kaufman, J.M.; Kaufman, J.L.; Borges, F.D. Immediate salvage procedure for infected penile prosthesis. J. Urol. 1998, 159, 816–818. [Google Scholar] [CrossRef]
- Vermeulen, A.; Kaufman, J.M.; Goemaere, S.; van Pottelberg, I. Estradiol in elderly men. Aging Male 2002, 5, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Krieg, M.; Nass, R.; Tunn, S. Effect of aging on endogenous level of 5 α-dihydrotestosterone, testosterone, estradiol, and estrone in epithelium and stroma of normal and hyperplastic human prostate. J. Clin. Endocrinol. Metab. 1993, 77, 375–381. [Google Scholar] [PubMed]
- Bartsch, G.; Rittmaster, R.S.; Klocker, H. Dihydrotestosterone and the concept of 5 α-reductase inhibition in human benign prostatic hyperplasia. Eur. Urol. 2000, 37, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Tunn, S.; Hochstrate, H.; Grunwald, I.; Fluchter, S.H.; Krieg, M. Effect of aging on kinetic parameters of 5 α-reductase in epithelium and stroma of normal and hyperplastic human prostate. J. Clin. Endocrinol. Metab. 1988, 67, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Zumoff, B.; Strain, G.W.; Kream, J.; O’Connor, J.; Rosenfeld, R.S.; Levin, J.; Fukushima, D.K. Age variation of the 24-hour mean plasma concentrations of androgens, estrogens, and gonadotropins in normal adult men. J. Clin. Endocrinol. Metab. 1982, 54, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Leav, I.; Merk, F.B.; Ofner, P.; Goodrich, G.; Kwan, P.W.; Stein, B.M.; Sar, M.; Stumpf, W.E. Bipotentiality of response to sex hormones by the prostate of castrated or hypophysectomized dogs. Direct effects of estrogen. Am. J. Pathol. 1978, 93, 69–92. [Google Scholar] [PubMed]
- Merk, F.B.; Warhol, M.J.; Kwan, P.W.; Leav, I.; Alroy, J.; Ofner, P.; Pinkus, G.S. Multiple phenotypes of prostatic glandular cells in castrated dogs after individual or combined treatment with androgen and estrogen. Morphometric, ultrastructural, and cytochemical distinctions. Lab. Investig. 1986, 54, 442–456. [Google Scholar] [PubMed]
- Levine, A.C.; Ren, M.; Huber, G.K.; Kirschenbaum, A. The effect of androgen, estrogen, and growth factors on the proliferation of cultured fibroblasts derived from human fetal and adult prostates. Endocrinology 1992, 130, 2413–2419. [Google Scholar] [PubMed]
- Bruengger, A.; Mariotti, A.; Rohr, H.P.; Bartsch, G.; Stahel, W.; Wiederkehr, P.; Carmichael, S.W.; Mawhinney, M.G. Androgen and estrogen effect on guinea pig seminal vesicle muscle: A combined stereological and biochemical study. Prostate 1986, 9, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.C.; Wong, Y.C.; White, F.H.; Fowler, J.P. Morphometric and stereological study of the glandular epithelium of the lateral prostate of the intact and castrated guinea pig. Prostate 1991, 19, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.C.; Wong, Y.C. Ultrastructural study of the effects of 17 β-oestradiol on the lateral prostate and seminal vesicle of the castrated guinea pig. Acta Anat. 1991, 141, 51–62. [Google Scholar] [PubMed]
- Ricciardelli, C.; Horsfall, D.J.; Sykes, P.J.; Marshall, V.R.; Tilley, W.D. Effects of oestradiol-17 β and 5 α-dihydrotestosterone on guinea-pig prostate smooth muscle cell proliferation and steroid receptor expression in vitro. J. Endocrinol. 1994, 140, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Risbridger, G.P.; Wang, H.; Frydenberg, M.; Cunha, G. The metaplastic effects of estrogen on mouse prostate epithelium: Proliferation of cells with basal cell phenotype. Endocrinology 2001, 142, 2443–2450. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Mori, T.; Suzuki, Y.; Bern, H.A. Long-term effects of perinatal exposure to sex steroids and diethylstilbestrol on the reproductive system of male mammals. Int. Rev. Cytol. 1983, 84, 235–268. [Google Scholar] [PubMed]
- Vorherr, H.; Messer, R.H.; Vorherr, U.F.; Jordan, S.W.; Kornfeld, M. Teratogenesis and carcinogenesis in rat offspring after transplacental and transmammary exposure to diethylstilbestrol. Biochem. Pharmacol. 1979, 28, 1865–1877. [Google Scholar] [CrossRef]
- Arai, Y.; Suzuki, Y.; Nishizuka, Y. Hyperplastic and metaplastic lesions in the reproductive tract of male rats induced by neonatal treatment with diethylstilbestrol. Virchows Arch. A Pathol. Anat. Histol. 1977, 376, 21–28. [Google Scholar] [CrossRef] [PubMed]
- McLachlan, J.A.; Newbold, R.R.; Bullock, B. Reproductive tract lesions in male mice exposed prenatally to diethylstilbestrol. Science 1975, 190, 991–992. [Google Scholar] [CrossRef] [PubMed]
- Pylkkanen, L.; Santti, R.; Newbold, R.; McLachlan, J.A. Regional differences in the prostate of the neonatally estrogenized mouse. Prostate 1991, 18, 117–129. [Google Scholar] [CrossRef] [PubMed]
- vom Saal, F.S.; Timms, B.G.; Montano, M.M.; Palanza, P.; Thayer, K.A.; Nagel, S.C.; Dhar, M.D.; Ganjam, V.K.; Parmigiani, S.; Welshons, W.V. Prostate enlargement in mice due to fetal exposure to low doses of estradiol or diethylstilbestrol and opposite effects at high doses. Proc. Natl. Acad. Sci. USA 1997, 94, 2056–2061. [Google Scholar] [CrossRef] [PubMed]
- Rajfer, J.; Coffey, D.S. Sex steroid imprinting of the immature prostate. Long-term effects. Investig. Urol. 1978, 16, 186–190. [Google Scholar]
- Prins, G.S. Neonatal estrogen exposure induces lobe-specific alterations in adult rat prostate androgen receptor expression. Endocrinology 1992, 130, 3703–3714. [Google Scholar] [CrossRef] [PubMed]
- Prins, G.S.; Ho, S.M. Early-life estrogens and prostate cancer in an animal model. J. Dev. Orig. Health Dis. 2010, 1, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Prins, G.S.; Marmer, M.; Woodham, C.; Chang, W.; Kuiper, G.; Gustafsson, J.A.; Birch, L. Estrogen receptor-β messenger ribonucleic acid ontogeny in the prostate of normal and neonatally estrogenized rats. Endocrinology 1998, 139, 874–883. [Google Scholar] [CrossRef] [PubMed]
- Leav, I.; Merk, F.B.; Kwan, P.W.; Ho, S.M. Androgen-supported estrogen-enhanced epithelial proliferation in the prostates of intact noble rats. Prostate 1989, 15, 23–40. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.M.; Yu, M. Hormonal regulation of nuclear type II estrogen binding sites in the dorsolateral prostate of noble rats. J. Steroid Biochem. Mol. Biol. 1995, 52, 233–238. [Google Scholar] [CrossRef]
- Noble, R.L. Sex steroids as a cause of adenocarcinoma of the dorsal prostate in NB rats, and their influence on the growth of transplants. Oncology 1977, 34, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Noble, R.L. The development of prostatic adenocarcinoma in NB rats following prolonged sex hormone administration. Cancer Res. 1977, 37, 1929–1933. [Google Scholar] [PubMed]
- Drago, J.R. The induction of NB rat prostatic carcinomas. Anticancer Res. 1984, 4, 255–256. [Google Scholar] [PubMed]
- Ho, S.M.; Leav, I.; Merk, F.B.; Yu, M.; Kwan, P.W.; Ziar, J. Induction of atypical hyperplasia, apoptosis, and type II estrogen-binding sites in the ventral prostates of noble rats treated with testosterone and pharmacologic doses of estradiol-17 β. Lab. Investig. 1995, 73, 356–365. [Google Scholar] [PubMed]
- Bosland, M.C.; Ford, H.; Horton, L. Induction at high incidence of ductal prostate adenocarcinomas in NBL/CR and sprague-dawley HSD:SD rats treated with a combination of testosterone and estradiol-17 β or diethylstilbestrol. Carcinogenesis 1995, 16, 1311–1317. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Wang, Y.Z.; Tam, N.N. The prostate gland and prostate carcinogenesis. Ital. J. Anat. Embryol. 1998, 103, 237–252. [Google Scholar] [PubMed]
- Bosland, M.C. Animal models for the study of prostate carcinogenesis. J. Cell. Biochem. Suppl. 1992, 16H, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Bostwick, D.G.; Montironi, R. Prostatic intraepithelial neoplasia and the origins of prostatic carcinoma. Pathol. Res. Pract. 1995, 191, 828–832. [Google Scholar] [CrossRef]
- Shin, H.J.; Ro, J.Y. Prostatic intraepithelial neoplasia: A potential precursor lesion of prostatic adenocarcinoma. Yonsei Med. J. 1995, 36, 215–231. [Google Scholar] [CrossRef] [PubMed]
- Haggman, M.J.; Macoska, J.A.; Wojno, K.J.; Oesterling, J.E. The relationship between prostatic intraepithelial neoplasia and prostate cancer: Critical issues. J. Urol. 1997, 158, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Zlotta, A.R.; Schulman, C.C. Clinical evolution of prostatic intraepithelial neoplasia. Eur. Urol. 1999, 35, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Alcaraz, A.; Barranco, M.A.; Corral, J.M.; Ribal, M.J.; Carrio, A.; Mallofre, C.; Llopis, J.; Cetina, A.; Alvarez-Vijande, R. High-grade prostate intraepithelial neoplasia shares cytogenetic alterations with invasive prostate cancer. Prostate 2001, 47, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Nelson, W.G.; De Marzo, A.M.; Isaacs, W.B. Prostate cancer. N. Engl. J. Med. 2003, 349, 366–381. [Google Scholar] [CrossRef] [PubMed]
- Merrimen, J.L.; Jones, G.; Walker, D.; Leung, C.S.; Kapusta, L.R.; Srigley, J.R. Multifocal high grade prostatic intraepithelial neoplasia is a significant risk factor for prostatic adenocarcinoma. J. Urol. 2009, 182, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hayward, S.W.; Donjacour, A.A.; Young, P.; Jacks, T.; Sage, J.; Dahiya, R.; Cardiff, R.D.; Day, M.L.; Cunha, G.R. Sex hormone-induced carcinogenesis in RB-deficient prostate tissue. Cancer Res. 2000, 60, 6008–6017. [Google Scholar] [PubMed]
- Nicholson, T.M.; Ricke, E.A.; Marker, P.C.; Miano, J.M.; Mayer, R.D.; Timms, B.G.; vom Saal, F.S.; Wood, R.W.; Ricke, W.A. Testosterone and 17β-estradiol induce glandular prostatic growth, bladder outlet obstruction, and voiding dysfunction in male mice. Endocrinology 2012, 153, 5556–5565. [Google Scholar] [CrossRef] [PubMed]
- Ricke, W.A.; McPherson, S.J.; Bianco, J.J.; Cunha, G.R.; Wang, Y.; Risbridger, G.P. Prostatic hormonal carcinogenesis is mediated by in situ estrogen production and estrogen receptor alpha signaling. FASEB J. 2008, 22, 1512–1520. [Google Scholar] [CrossRef] [PubMed]
- Ellem, S.J.; Schmitt, J.F.; Pedersen, J.S.; Frydenberg, M.; Risbridger, G.P. Local aromatase expression in human prostate is altered in malignancy. J. Clin. Endocrinol. Metab. 2004, 89, 2434–2441. [Google Scholar] [CrossRef] [PubMed]
- Celhay, O.; Yacoub, M.; Irani, J.; Dore, B.; Cussenot, O.; Fromont, G. Expression of estrogen related proteins in hormone refractory prostate cancer: Association with tumor progression. J. Urol. 2010, 184, 2172–2178. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.; Wallace, J.; Jeltsch, J.M.; Berry, M. The 5′ flanking region of the human PS2 gene mediates its transcriptional activation by estrogen in mcf-7 cells. Biochem. Biophys. Res. Commun. 1988, 151, 306–313. [Google Scholar] [CrossRef]
- Clarke, C.L. Cell-specific regulation of progesterone receptor in the female reproductive system. Mol. Cell. Endocrinol. 1990, 70, 29–33. [Google Scholar] [CrossRef]
- Webb, P.; Lopez, G.N.; Uht, R.M.; Kushner, P.J. Tamoxifen activation of the estrogen receptor/AP-1 pathway: Potential origin for the cell-specific estrogen-like effects of antiestrogens. Mol. Endocrinol. 1995, 9, 443–456. [Google Scholar] [PubMed]
- Paech, K.; Webb, P.; Kuiper, G.G.; Nilsson, S.; Gustafsson, J.; Kushner, P.J.; Scanlan, T.S. Differential ligand activation of estrogen receptors ERα and ERβ at AP1 sites. Science 1997, 277, 1508–1510. [Google Scholar] [CrossRef] [PubMed]
- Saville, B.; Wormke, M.; Wang, F.; Nguyen, T.; Enmark, E.; Kuiper, G.; Gustafsson, J.A.; Safe, S. Ligand-, cell-, and estrogen receptor subtype (α/β)-dependent activation at GC-rich (SP1) promoter elements. J. Biol. Chem. 2000, 275, 5379–5387. [Google Scholar] [CrossRef] [PubMed]
- Swaneck, G.E.; Alvarez, J.M.; Sufrin, G. Multiple species of estrogen binding sites in the nuclear fraction of the rat prostate. Biochem. Biophys. Res. Commun. 1982, 106, 1441–1447. [Google Scholar] [CrossRef]
- Yu, M.; Cates, J.; Leav, I.; Ho, S.M. Heterogeneity of [3H]estradiol binding sites in the rat prostate: Properties and distribution of type I and type II sites. J. Steroid Biochem. 1989, 33, 449–457. [Google Scholar] [CrossRef]
- Ekman, P.; Barrack, E.R.; Greene, G.L.; Jensen, E.V.; Walsh, P.C. Estrogen receptors in human prostate: Evidence for multiple binding sites. J. Clin. Endocrinol. Metab. 1983, 57, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, B.J.; Lakey, W.H.; McBlain, W.A. Estrogen receptor in human benign prostatic hyperplasia. J. Urol. 1983, 130, 183–187. [Google Scholar] [PubMed]
- Markaverich, B.M.; Alejandro, M.A. Type II [3H]estradiol binding site antagonists: Inhibition of normal and malignant prostate cell growth and proliferation. Int. J. Oncol. 1998, 12, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Shoulars, K.; Brown, T.; Alejandro, M.A.; Crowley, J.; Markaverich, B.M. Identification of nuclear type II [(3)H]estradiol binding sites as histone H4. Biochem. Biophys. Res. Commun. 2002, 296, 1083–1090. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Enmark, E.; Pelto-Huikko, M.; Nilsson, S.; Gustafsson, J.A. Cloning of a novel receptor expressed in rat prostate and ovary. Proc. Natl. Acad. Sci. USA 1996, 93, 5925–5930. [Google Scholar] [CrossRef] [PubMed]
- Mosselman, S.; Polman, J.; Dijkema, R. Er β: Identification and characterization of a novel human estrogen receptor. FEBS Lett. 1996, 392, 49–53. [Google Scholar] [CrossRef]
- Seitz, G.; Wernert, N. Immunohistochemical estrogen receptor demonstration in the prostate and prostate cancer. Pathol. Res. Pract. 1987, 182, 792–796. [Google Scholar] [CrossRef]
- Konishi, N.; Nakaoka, S.; Hiasa, Y.; Kitahori, Y.; Ohshima, M.; Samma, S.; Okajima, E. Immunohistochemical evaluation of estrogen receptor status in benign prostatic hypertrophy and in prostate carcinoma and the relationship to efficacy of endocrine therapy. Oncology 1993, 50, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Ehara, H.; Koji, T.; Deguchi, T.; Yoshii, A.; Nakano, M.; Nakane, P.K.; Kawada, Y. Expression of estrogen receptor in diseased human prostate assessed by non-radioactive in situ hybridization and immunohistochemistry. Prostate 1995, 27, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.Y.; Leav, I.; Lau, K.M.; Ho, S.M.; Pflueger, S.M. Expression of estrogen receptor β in the fetal, neonatal, and prepubertal human prostate. Prostate 2002, 52, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Chung, L.W. Implications of stromal-epithelial interaction in human prostate cancer growth, progression and differentiation. Semin. Cancer Biol. 1993, 4, 183–192. [Google Scholar] [PubMed]
- Farnsworth, W.E. Roles of estrogen and SHBG in prostate physiology. Prostate 1996, 28, 17–23. [Google Scholar] [CrossRef]
- Chung, L.W.; Davies, R. Prostate epithelial differentiation is dictated by its surrounding stroma. Mol. Biol. Rep. 1996, 23, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Bacher, M.; Rausch, U.; Goebel, H.W.; Polzar, B.; Mannherz, H.G.; Aumuller, G. Stromal and epithelial cells from rat ventral prostate during androgen deprivation and estrogen treatment—Regulation of transcription. Exp. Clin. Endocrinol. 1993, 101, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Gupta, C. The role of estrogen receptor, androgen receptor and growth factors in diethylstilbestrol-induced programming of prostate differentiation. Urol. Res. 2000, 28, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Marengo, S.R.; Chung, L.W. An orthotopic model for the study of growth factors in the ventral prostate of the rat: Effects of epidermal growth factor and basic fibroblast growth factor. J. Androl. 1994, 15, 277–286. [Google Scholar] [PubMed]
- Udayakumar, T.S.; Jeyaraj, D.A.; Rajalakshmi, M.; Sharma, R.S. Culture of prostate epithelial cells of the rhesus monkey on extracellular matrix substrate: Influence of steroids and insulin-like growth factors. J. Endocrinol. 1999, 162, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Torring, N.; Vinter-Jensen, L.; Pedersen, S.B.; Sorensen, F.B.; Flyvbjerg, A.; Nexo, E. Systemic administration of insulin-like growth factor I (IGF-I) causes growth of the rat prostate. J. Urol. 1997, 158, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Eddy, E.M.; Washburn, T.F.; Bunch, D.O.; Goulding, E.H.; Gladen, B.C.; Lubahn, D.B.; Korach, K.S. Targeted disruption of the estrogen receptor gene in male mice causes alteration of spermatogenesis and infertility. Endocrinology 1996, 137, 4796–4805. [Google Scholar] [PubMed]
- Donaldson, K.M.; Tong, S.Y.; Washburn, T.; Lubahn, D.B.; Eddy, E.M.; Hutson, J.M.; Korach, K.S. Morphometric study of the gubernaculum in male estrogen receptor mutant mice. J. Androl. 1996, 17, 91–95. [Google Scholar] [PubMed]
- Bonkhoff, H.; Fixemer, T.; Hunsicker, I.; Remberger, K. Estrogen receptor expression in prostate cancer and premalignant prostatic lesions. Am. J. Pathol. 1999, 155, 641–647. [Google Scholar] [CrossRef]
- Leav, I.; Lau, K.M.; Adams, J.Y.; McNeal, J.E.; Taplin, M.E.; Wang, J.; Singh, H.; Ho, S.M. Comparative studies of the estrogen receptors β and α and the androgen receptor in normal human prostate glands, dysplasia, and in primary and metastatic carcinoma. Am. J. Pathol. 2001, 159, 79–92. [Google Scholar] [CrossRef]
- Royuela, M.; de Miguel, M.P.; Bethencourt, F.R.; Sanchez-Chapado, M.; Fraile, B.; Arenas, M.I.; Paniagua, R. Estrogen receptors α and β in the normal, hyperplastic and carcinomatous human prostate. J. Endocrinol. 2001, 168, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Bashirelahi, N.; Young, J.D.; Shida, K.; Yamanaka, H.; Ito, Y.; Harada, M. Androgen, estrogen, and progesterone receptors in peripheral and central zones of human prostate with adenocarcinoma. Urology 1983, 21, 530–535. [Google Scholar] [CrossRef]
- Bowman, S.P.; Barnes, D.M.; Blacklock, N.J.; Sullivan, P.J. Regional variation of cytosol androgen receptors throughout the diseased human prostate gland. Prostate 1986, 8, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Sciarra, F.; Monti, S.; Adamo, M.V.; Palma, E.; Toscano, V.; d’Eramo, G.; di Silverio, F. Regional distribution of epidermal growth factor, testosterone and dihydrotestosterone in benign prostatic hyperplasia tissue. Urol. Res. 1995, 23, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Greene, D.R.; Fitzpatrick, J.M.; Scardino, P.T. Anatomy of the prostate and distribution of early prostate cancer. Semin. Surg. Oncol. 1995, 11, 9–22. [Google Scholar] [CrossRef] [PubMed]
- McNeal, J.E.; Redwine, E.A.; Freiha, F.S.; Stamey, T.A. Zonal distribution of prostatic adenocarcinoma. Correlation with histologic pattern and direction of spread. Am. J. Surg. Pathol. 1988, 12, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Wernert, N.; Gerdes, J.; Loy, V.; Seitz, G.; Scherr, O.; Dhom, G. Investigations of the estrogen (ER-ICA-test) and the progesterone receptor in the prostate and prostatic carcinoma on immunohistochemical basis. Virchows Arch. A Pathol. Anat. Histopathol. 1988, 412, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Hobisch, A.; Hittmair, A.; Daxenbichler, G.; Wille, S.; Radmayr, C.; Hobisch-Hagen, P.; Bartsch, G.; Klocker, H.; Culig, Z. Metastatic lesions from prostate cancer do not express oestrogen and progesterone receptors. J. Pathol. 1997, 182, 356–361. [Google Scholar] [CrossRef]
- Bodker, A.; Bruun, J.; Balslev, E.; Iversen, H.G.; Meyhoff, H.H.; Andersson, K.E. Estrogen receptors in the human male prostatic urethra and prostate in prostatic cancer and benign prostatic hyperplasia. Scand. J. Urol. Nephrol. 1999, 33, 237–242. [Google Scholar] [PubMed]
- Carruba, G.; Pfeffer, U.; Fecarotta, E.; Coviello, D.A.; D’Amato, E.; Lo Castro, M.; Vidali, G.; Castagnetta, L. Estradiol inhibits growth of hormone-nonresponsive PC3 human prostate cancer cells. Cancer Res. 1994, 54, 1190–1193. [Google Scholar] [PubMed]
- Castagnetta, L.A.; Miceli, M.D.; Sorci, C.M.; Pfeffer, U.; Farruggio, R.; Oliveri, G.; Calabro, M.; Carruba, G. Growth of LNCAP human prostate cancer cells is stimulated by estradiol via its own receptor. Endocrinology 1995, 136, 2309–2319. [Google Scholar] [PubMed]
- Ye, Q.; Chung, L.W.; Cinar, B.; Li, S.; Zhau, H.E. Identification and characterization of estrogen receptor variants in prostate cancer cell lines. J. Steroid Biochem. Mol. Biol. 2000, 75, 21–31. [Google Scholar] [CrossRef]
- Latil, A.; Bieche, I.; Vidaud, D.; Lidereau, R.; Berthon, P.; Cussenot, O.; Vidaud, M. Evaluation of androgen, estrogen (ER α and ER β), and progesterone receptor expression in human prostate cancer by real-time quantitative reverse transcription-polymerase chain reaction assays. Cancer Res. 2001, 61, 1919–1926. [Google Scholar] [PubMed]
- Horvath, L.G.; Henshall, S.M.; Lee, C.S.; Head, D.R.; Quinn, D.I.; Makela, S.; Delprado, W.; Golovsky, D.; Brenner, P.C.; O’Neill, G.; et al. Frequent loss of estrogen receptor-β expression in prostate cancer. Cancer Res. 2001, 61, 5331–5335. [Google Scholar] [PubMed]
- Mak, P.; Leav, I.; Pursell, B.; Bae, D.; Yang, X.; Taglienti, C.A.; Gouvin, L.M.; Sharma, V.M.; Mercurio, A.M. ERbeta impedes prostate cancer EMT by destabilizing HIF-1α and inhibiting VEGF-mediated snail nuclear localization: Implications for Gleason grading. Cancer Cell 2010, 17, 319–332. [Google Scholar] [PubMed]
- Lai, J.S.; Brown, L.G.; True, L.D.; Hawley, S.J.; Etzioni, R.B.; Higano, C.S.; Ho, S.M.; Vessella, R.L.; Corey, E. Metastases of prostate cancer express estrogen receptor-β. Urology 2004, 64, 814–820. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, T. Cellular and molecular mechanisms of breast and prostate cancer metastasis to bone. Eur. J. Cancer 1998, 34, 240–245. [Google Scholar] [CrossRef]
- Moore, J.T.; McKee, D.D.; Slentz-Kesler, K.; Moore, L.B.; Jones, S.A.; Horne, E.L.; Su, J.L.; Kliewer, S.A.; Lehmann, J.M.; Willson, T.M. Cloning and characterization of human estrogen receptor β isoforms. Biochem. Biophys. Res. Commun. 1998, 247, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Leung, Y.K.; Mak, P.; Hassan, S.; Ho, S.M. Estrogen receptor (ER)-β isoforms: A key to understanding er-beta signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 13162–13167. [Google Scholar] [CrossRef] [PubMed]
- Leung, Y.K.; Lam, H.M.; Wu, S.; Song, D.; Levin, L.; Cheng, L.; Wu, C.L.; Ho, S.M. Estrogen receptor β 2 and β 5 are associated with poor prognosis in prostate cancer, and promote cancer cell migration and invasion. Endocr. Relat. Cancer 2010, 17, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Cotrim, C.Z.; Fabris, V.; Doria, M.L.; Lindberg, K.; Gustafsson, J.A.; Amado, F.; Lanari, C.; Helguero, L.A. Estrogen receptor beta growth-inhibitory effects are repressed through activation of MAPK and PI3K signalling in mammary epithelial and breast cancer cells. Oncogene 2013, 32, 2390–2402. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Matthews, J.; Tujague, M.; Wan, J.; Strom, A.; Toresson, G.; Lam, E.W.; Cheng, G.; Gustafsson, J.A.; Dahlman-Wright, K. Estrogen receptor β 2 negatively regulates the transactivation of estrogen receptor α in human breast cancer cells. Cancer Res. 2007, 67, 3955–3962. [Google Scholar] [CrossRef] [PubMed]
- Greene, G.L.; Gilna, P.; Waterfield, M.; Baker, A.; Hort, Y.; Shine, J. Sequence and expression of human estrogen receptor complementary DNA. Science 1986, 231, 1150–1154. [Google Scholar] [CrossRef] [PubMed]
- Green, S.; Walter, P.; Kumar, V.; Krust, A.; Bornert, J.M.; Argos, P.; Chambon, P. Human oestrogen receptor cDNA: Sequence, expression and homology to V-ERB-A. Nature 1986, 320, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Enmark, E.; Kainu, T.; Pelto-Huikko, M.; Gustafsson, J.A. Identification of a novel member of the nuclear receptor superfamily which is closely related to REV-ERBA. Biochem. Biophys. Res. Commun. 1994, 204, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, G.B.; Tremblay, A.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Labrie, F.; Giguere, V. Cloning, chromosomal localization, and functional analysis of the murine estrogen receptor β. Mol. Endocrinol. 1997, 11, 353–365. [Google Scholar] [PubMed]
- O’Lone, R.; Frith, M.C.; Karlsson, E.K.; Hansen, U. Genomic targets of nuclear estrogen receptors. Mol. Endocrinol. 2004, 18, 1859–1875. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, G.G.; Carlsson, B.; Grandien, K.; Enmark, E.; Haggblad, J.; Nilsson, S.; Gustafsson, J.A. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors α and β. Endocrinology 1997, 138, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Pace, P.; Taylor, J.; Suntharalingam, S.; Coombes, R.C.; Ali, S. Human estrogen receptor β binds DNA in a manner similar to and dimerizes with estrogen receptor α. J. Biol. Chem. 1997, 272, 25832–25838. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, K.; Grandien, K.; Kuiper, G.G.; Gustafsson, J.A. Mouse estrogen receptor beta forms estrogen response element-binding heterodimers with estrogen receptor α. Mol. Endocrinol. 1997, 11, 1486–1496. [Google Scholar] [PubMed]
- Ogawa, S.; Inoue, S.; Watanabe, T.; Hiroi, H.; Orimo, A.; Hosoi, T.; Ouchi, Y.; Muramatsu, M. The complete primary structure of human estrogen receptor β (HER β) and its heterodimerization with ER α in vivo and in vitro. Biochem. Biophys. Res. Commun. 1998, 243, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Kushner, P.J.; Agard, D.A.; Greene, G.L.; Scanlan, T.S.; Shiau, A.K.; Uht, R.M.; Webb, P. Estrogen receptor pathways to AP-1. J. Steroid Biochem. Mol. Biol. 2000, 74, 311–317. [Google Scholar] [CrossRef]
- Zhao, C.; Dahlman-Wright, K.; Gustafsson, J.A. Estrogen signaling via estrogen receptor β. J. Biol. Chem. 2010, 285, 39575–39579. [Google Scholar] [CrossRef] [PubMed]
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998, 95, 927–937. [Google Scholar] [CrossRef]
- Pike, A.C.; Brzozowski, A.M.; Hubbard, R.E.; Bonn, T.; Thorsell, A.G.; Engstrom, O.; Ljunggren, J.; Gustafsson, J.A.; Carlquist, M. Structure of the ligand-binding domain of oestrogen receptor β in the presence of a partial agonist and a full antagonist. EMBO J. 1999, 18, 4608–4618. [Google Scholar] [CrossRef] [PubMed]
- Brzozowski, A.M.; Pike, A.C.; Dauter, Z.; Hubbard, R.E.; Bonn, T.; Engstrom, O.; Ohman, L.; Greene, G.L.; Gustafsson, J.A.; Carlquist, M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997, 389, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Endoh, H.; Masuhiro, Y.; Kitamoto, T.; Uchiyama, S.; Sasaki, H.; Masushige, S.; Gotoh, Y.; Nishida, E.; Kawashima, H.; et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science 1995, 270, 1491–1494. [Google Scholar] [CrossRef] [PubMed]
- Bunone, G.; Briand, P.A.; Miksicek, R.J.; Picard, D. Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. EMBO J. 1996, 15, 2174–2183. [Google Scholar] [PubMed]
- Tremblay, A.; Tremblay, G.B.; Labrie, F.; Giguere, V. Ligand-independent recruitment of SRC-1 to estrogen receptor β through phosphorylation of activation function AF-1. Mol. Cell 1999, 3, 513–519. [Google Scholar] [CrossRef]
- Webb, P.; Nguyen, P.; Shinsako, J.; Anderson, C.; Feng, W.; Nguyen, M.P.; Chen, D.; Huang, S.M.; Subramanian, S.; McKinerney, E.; et al. Estrogen receptor activation function 1 works by binding P160 coactivator proteins. Mol. Endocrinol. 1998, 12, 1605–1618. [Google Scholar] [CrossRef] [PubMed]
- Gaub, M.P.; Bellard, M.; Scheuer, I.; Chambon, P.; Sassone-Corsi, P. Activation of the ovalbumin gene by the estrogen receptor involves the FOS-JUN complex. Cell 1990, 63, 1267–1276. [Google Scholar] [CrossRef]
- Umayahara, Y.; Kawamori, R.; Watada, H.; Imano, E.; Iwama, N.; Morishima, T.; Yamasaki, Y.; Kajimoto, Y.; Kamada, T. Estrogen regulation of the insulin-like growth factor I gene transcription involves an AP-1 enhancer. J. Biol. Chem. 1994, 269, 16433–16442. [Google Scholar] [PubMed]
- Webb, P.; Nguyen, P.; Valentine, C.; Lopez, G.N.; Kwok, G.R.; McInerney, E.; Katzenellenbogen, B.S.; Enmark, E.; Gustafsson, J.A.; Nilsson, S.; et al. The estrogen receptor enhances AP-1 activity by two distinct mechanisms with different requirements for receptor transactivation functions. Mol. Endocrinol. 1999, 13, 1672–1685. [Google Scholar] [CrossRef] [PubMed]
- Jackson, T.A.; Richer, J.K.; Bain, D.L.; Takimoto, G.S.; Tung, L.; Horwitz, K.B. The partial agonist activity of antagonist-occupied steroid receptors is controlled by a novel hinge domain-binding coactivator l7/SPA and the corepressors N-COR or SMRT. Mol. Endocrinol. 1997, 11, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Lavinsky, R.M.; Jepsen, K.; Heinzel, T.; Torchia, J.; Mullen, T.M.; Schiff, R.; Del-Rio, A.L.; Ricote, M.; Ngo, S.; Gemsch, J.; et al. Diverse signaling pathways modulate nuclear receptor recruitment of N-COR and SMRT complexes. Proc. Natl. Acad. Sci. USA 1998, 95, 2920–2925. [Google Scholar] [CrossRef] [PubMed]
- Montano, M.M.; Katzenellenbogen, B.S. The quinone reductase gene: A unique estrogen receptor-regulated gene that is activated by antiestrogens. Proc. Natl. Acad. Sci. USA 1997, 94, 2581–2586. [Google Scholar] [CrossRef] [PubMed]
- Montano, M.M.; Jaiswal, A.K.; Katzenellenbogen, B.S. Transcriptional regulation of the human quinone reductase gene by antiestrogen-liganded estrogen receptor-α and estrogen receptor-β. J. Biol. Chem. 1998, 273, 25443–25449. [Google Scholar] [CrossRef] [PubMed]
- Piette, J.; Hirai, S.; Yaniv, M. Constitutive synthesis of activator protein 1 transcription factor after viral transformation of mouse fibroblasts. Proc. Natl. Acad. Sci. USA 1988, 85, 3401–3405. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Pickett, C.B. Regulation of rat glutathione S-transferase ya subunit gene expression. DNA-protein interaction at the antioxidant responsive element. J. Biol. Chem. 1992, 267, 13535–13539. [Google Scholar] [PubMed]
- Favreau, L.V.; Pickett, C.B. Transcriptional regulation of the rat NAD(P)H:Quinone reductase gene. Characterization of a DNA-protein interaction at the antioxidant responsive element and induction by 12-O-tetradecanoylphorbol 13-acetate. J. Biol. Chem. 1993, 268, 19875–19881. [Google Scholar] [PubMed]
- Montano, M.M.; Wittmann, B.M.; Bianco, N.R. Identification and characterization of a novel factor that regulates quinone reductase gene transcriptional activity. J. Biol. Chem. 2000, 275, 34306–34313. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, N.; Ngam, C.R.; Berdis, A.J.; Montano, M.M. The exonuclease activity of HPMC2 is required for transcriptional regulation of the QR gene and repair of estrogen-induced abasic sites. Oncogene 2011, 30, 4731–4739. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Forman, H.J.; Schonthal, A.H.; Cadenas, E. Induction of p21 mediated by reactive oxygen species formed during the metabolism of aziridinylbenzoquinones by hct116 cells. J. Biol. Chem. 1996, 271, 31915–31921. [Google Scholar] [CrossRef] [PubMed]
- Lau, K.M.; LaSpina, M.; Long, J.; Ho, S.M. Expression of estrogen receptor (ER)-α and ER-β in normal and malignant prostatic epithelial cells: Regulation by methylation and involvement in growth regulation. Cancer Res. 2000, 60, 3175–3182. [Google Scholar] [PubMed]
- Leung, Y.K.; Gao, Y.; Lau, K.M.; Zhang, X.; Ho, S.M. Ici 182,780-regulated gene expression in du145 prostate cancer cells is mediated by estrogen receptor-β/NFκB crosstalk. Neoplasia 2006, 8, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Lubahn, D.B.; Moyer, J.S.; Golding, T.S.; Couse, J.F.; Korach, K.S.; Smithies, O. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc. Natl. Acad. Sci. USA 1993, 90, 11162–11166. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Krust, A.; Gansmuller, A.; Dierich, A.; Chambon, P.; Mark, M. Effect of single and compound knockouts of estrogen receptors α (Erα) and β (Erβ) on mouse reproductive phenotypes. Development 2000, 127, 4277–4291. [Google Scholar] [PubMed]
- Krege, J.H.; Hodgin, J.B.; Couse, J.F.; Enmark, E.; Warner, M.; Mahler, J.F.; Sar, M.; Korach, K.S.; Gustafsson, J.A.; Smithies, O. Generation and reproductive phenotypes of mice lacking estrogen receptor β. Proc. Natl. Acad. Sci. USA 1998, 95, 15677–15682. [Google Scholar] [CrossRef] [PubMed]
- Couse, J.F.; Hewitt, S.C.; Bunch, D.O.; Sar, M.; Walker, V.R.; Davis, B.J.; Korach, K.S. Postnatal sex reversal of the ovaries in mice lacking estrogen receptors α and β. Science 1999, 286, 2328–2331. [Google Scholar] [CrossRef] [PubMed]
- Couse, J.F.; Korach, K.S. Estrogen receptor null mice: What have we learned and where will they lead us? Endocr. Rev. 1999, 20, 358–417. [Google Scholar] [CrossRef] [PubMed]
- Prins, G.S.; Korach, K.S. The role of estrogens and estrogen receptors in normal prostate growth and disease. Steroids 2008, 73, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Byar, D.P. Proceedings: The veterans administration cooperative urological research group’s studies of cancer of the prostate. Cancer 1973, 32, 1126–1130. [Google Scholar] [CrossRef]
- Pitts, W.R., Jr. Diethylstilbesterol: First-line hormonal therapy for prostate cancer? Urology 1999, 53, 660–661. [Google Scholar] [PubMed]
- Seidenfeld, J.; Samson, D.J.; Aronson, N.; Albertson, P.C.; Bayoumi, A.M.; Bennett, C.; Brown, A.; Garber, A.; Gere, M.; Hasselblad, V.; et al. Relative effectiveness and cost-effectiveness of methods of androgen suppression in the treatment of advanced prostate cancer. Evid. Rep. Technol. Assess. 1999, 4, 1–246. [Google Scholar]
- Kitahara, S.; Umeda, H.; Yano, M.; Koga, F.; Sumi, S.; Moriguchi, H.; Hosoya, Y.; Honda, M.; Yoshida, K. Effects of intravenous administration of high dose-diethylstilbestrol diphosphate on serum hormonal levels in patients with hormone-refractory prostate cancer. Endocr. J. 1999, 46, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Bayoumi, A.M.; Brown, A.D.; Garber, A.M. Cost-effectiveness of androgen suppression therapies in advanced prostate cancer. J. Natl. Cancer Inst. 2000, 92, 1731–1739. [Google Scholar] [CrossRef] [PubMed]
- Paulson, D.F. Management of metastatic prostatic cancer. Urology 1985, 25, 49–52. [Google Scholar] [PubMed]
- Brehmer, B.; Marquardt, H.; Madsen, P.O. Growth and hormonal response of cells derived from carcinoma and hyperplasia of the prostate in monolayer cell culture. A possible in vitro model for clinical chemotherapy. J. Urol. 1972, 108, 890–896. [Google Scholar] [PubMed]
- Hartley-Asp, B.; Deinum, J.; Wallin, M. Diethylstilbestrol induces metaphase arrest and inhibits microtubule assembly. Mutat. Res. 1985, 143, 231–235. [Google Scholar] [CrossRef]
- Robertson, C.N.; Roberson, K.M.; Padilla, G.M.; O’Brien, E.T.; Cook, J.M.; Kim, C.S.; Fine, R.L. Induction of apoptosis by diethylstilbestrol in hormone-insensitive prostate cancer cells. J. Natl. Cancer Inst. 1996, 88, 908–917. [Google Scholar] [CrossRef] [PubMed]
- Trapman, J.; Ris-Stalpers, C.; van der Korput, J.A.; Kuiper, G.G.; Faber, P.W.; Romijn, J.C.; Mulder, E.; Brinkmann, A.O. The androgen receptor: Functional structure and expression in transplanted human prostate tumors and prostate tumor cell lines. J. Steroid Biochem. Mol. Biol. 1990, 37, 837–842. [Google Scholar] [CrossRef] [Green Version]
- Veldscholte, J.; Voorhorst-Ogink, M.M.; Bolt-de Vries, J.; van Rooij, H.C.; Trapman, J.; Mulder, E. Unusual specificity of the androgen receptor in the human prostate tumor cell line LNCAP: High affinity for progestagenic and estrogenic steroids. Biochim. Biophys. Acta 1990, 1052, 187–194. [Google Scholar] [CrossRef]
- Noble, R.L. Production of NB rat carcinoma of the dorsal prostate and response of estrogen-dependent transplants to sex hormones and tamoxifen. Cancer Res. 1980, 40, 3547–3550. [Google Scholar] [PubMed]
- Glick, J.H.; Wein, A.; Padavic, K.; Negendank, W.; Harris, D.; Brodovsky, H. Phase II trial of tamoxifen in metastatic carcinoma of the prostate. Cancer 1982, 49, 1367–1372. [Google Scholar] [CrossRef]
- Spremulli, E.; DeSimone, P.; Durant, J. A phase II study nolvadex: Tamoxifen citrate in the treatment of advanced prostatic adenocarcinoma. Am. J. Clin. Oncol. 1982, 5, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Rayter, Z.; Gazet, J.C.; Shephed, J.; Trott, P.; Svensson, W.; A’Hern, R. Effects of tamoxifen on uterus. Lancet 1994, 344, 623–624. [Google Scholar]
- Eells, T.P.; Alpern, H.D.; Grzywacz, C.; MacMillan, R.W.; Olson, J.E. The effect of tamoxifen on cervical squamous maturation in papanicolaou stained cervical smears of post-menopausal women. Cytopathology 1990, 1, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Kedar, R.P.; Bourne, T.H.; Powles, T.J.; Collins, W.P.; Ashley, S.E.; Cosgrove, D.O.; Campbell, S. Effects of tamoxifen on uterus and ovaries of postmenopausal women in a randomised breast cancer prevention trial. Lancet 1994, 343, 1318–1321. [Google Scholar] [CrossRef]
- Horton, J.; Rosenbaum, C.; Cummings, F.J. Tamoxifen in advanced prostate cancer: An ECOG pilot study. Prostate 1988, 12, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Wakeling, A.E.; Dukes, M.; Bowler, J. A potent specific pure antiestrogen with clinical potential. Cancer Res. 1991, 51, 3867–3873. [Google Scholar] [PubMed]
- Osborne, C.K.; Coronado-Heinsohn, E.B.; Hilsenbeck, S.G.; McCue, B.L.; Wakeling, A.E.; McClelland, R.A.; Manning, D.L.; Nicholson, R.I. Comparison of the effects of a pure steroidal antiestrogen with those of tamoxifen in a model of human breast cancer. J. Natl. Cancer Inst. 1995, 87, 746–750. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, R.S.; Krishnan, A.V.; Swami, S.; Feldman, D. Fulvestrant (ICI 182,780) down-regulates androgen receptor expression and diminishes androgenic responses in LNCAP human prostate cancer cells. Mol. Cancer Ther. 2006, 5, 1539–1549. [Google Scholar] [CrossRef] [PubMed]
- Leung, Y.K.; Chan, Q.K.; Ng, C.F.; Ma, F.M.; Tse, H.M.; To, K.F.; Maranchie, J.; Ho, S.M.; Lau, K.M. Hsa-miRNA-765 as a key mediator for inhibiting growth, migration and invasion in fulvestrant-treated prostate cancer. PLoS ONE 2014, 9, e98037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chadha, M.K.; Ashraf, U.; Lawrence, D.; Tian, L.; Levine, E.; Silliman, C.; Escott, P.; Payne, V.; Trump, D.L. Phase II study of fulvestrant (faslodex) in castration resistant prostate cancer. Prostate 2008, 68, 1461–1466. [Google Scholar] [CrossRef] [PubMed]
- Gasent Blesa, J.M.; Alberola Candel, V.; Giner Marco, V.; Giner-Bosch, V.; Provencio Pulla, M.; Laforga Canales, J.B. Experience with fulvestrant acetate in castration-resistant prostate cancer patients. Ann. Oncol. 2010, 21, 1131–1132. [Google Scholar] [CrossRef] [PubMed]
- Di Leo, A.; Jerusalem, G.; Petruzelka, L.; Torres, R.; Bondarenko, I.N.; Khasanov, R.; Verhoeven, D.; Pedrini, J.L.; Smirnova, I.; Lichinitser, M.R.; et al. Final overall survival: Fulvestrant 500 mg vs. 250 mg in the randomized CONFIRM trial. J. Natl. Cancer Inst. 2014, 106, 349–351. [Google Scholar]
- Losel, R.; Wehling, M. Nongenomic actions of steroid hormones. Nat. Rev. Mol. Cell Biol. 2003, 4, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Moriarty, K.; Kim, K.H.; Bender, J.R. Minireview: Estrogen receptor-mediated rapid signaling. Endocrinology 2006, 147, 5557–5563. [Google Scholar] [CrossRef] [PubMed]
- Pietras, R.J.; Szego, C.M. Cell membrane estrogen receptors resurface. Nat. Med. 1999, 5, 1330. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.P.; Lee, J.Y.; Jeong, J.K.; Bae, S.W.; Lee, H.K.; Jo, I. Nongenomic stimulation of nitric oxide release by estrogen is mediated by estrogen receptor α localized in caveolae. Biochem. Biophys. Res. Commun. 1999, 263, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Haynes, M.P.; Russell, K.S.; Bender, J.R. Molecular mechanisms of estrogen actions on the vasculature. J. Nucl. Cardiol. 2000, 7, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Stefano, G.B.; Cadet, P.; Breton, C.; Goumon, Y.; Prevot, V.; Dessaint, J.P.; Beauvillain, J.C.; Roumier, A.S.; Welters, I.; Salzet, M. Estradiol-stimulated nitric oxide release in human granulocytes is dependent on intracellular calcium transients: Evidence of a cell surface estrogen receptor. Blood 2000, 95, 3951–3958. [Google Scholar] [PubMed]
- Doolan, C.M.; Harvey, B.J. A Gαs protein-coupled membrane receptor, distinct from the classical oestrogen receptor, transduces rapid effects of oestradiol on [Ca2+]i in female rat distal colon. Mol. Cell. Endocrinol. 2003, 199, 87–103. [Google Scholar] [CrossRef]
- Pedram, A.; Razandi, M.; Levin, E.R. Nature of functional estrogen receptors at the plasma membrane. Mol. Endocrinol. 2006, 20, 1996–2009. [Google Scholar] [CrossRef] [PubMed]
- Russell, K.S.; Haynes, M.P.; Sinha, D.; Clerisme, E.; Bender, J.R. Human vascular endothelial cells contain membrane binding sites for estradiol, which mediate rapid intracellular signaling. Proc. Natl. Acad. Sci. USA 2000, 97, 5930–5935. [Google Scholar] [CrossRef] [PubMed]
- Wyckoff, M.H.; Chambliss, K.L.; Mineo, C.; Yuhanna, I.S.; Mendelsohn, M.E.; Mumby, S.M.; Shaul, P.W. Plasma membrane estrogen receptors are coupled to endothelial nitric-oxide synthase through Gα(I). J. Biol. Chem. 2001, 276, 27071–27076. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, X.; Shen, P.; Loggie, B.W.; Chang, Y.; Deuel, T.F. A variant of estrogen receptor-α, HER-α 36: Transduction of estrogen- and antiestrogen-dependent membrane-initiated mitogenic signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 9063–9068. [Google Scholar] [CrossRef] [PubMed]
- Chambliss, K.L.; Yuhanna, I.S.; Anderson, R.G.; Mendelsohn, M.E.; Shaul, P.W. ERβ has nongenomic action in caveolae. Mol. Endocrinol. 2002, 16, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R., Jr. Estrogen-induced activation of ERK-1 and ERK-2 requires the g protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol. Endocrinol. 2000, 14, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.; Pang, Y.; Filardo, E.J.; Dong, J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 2005, 146, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Prossnitz, E.R.; Arterburn, J.B. International union of basic and clinical pharmacology. Xcvii. G protein-coupled estrogen receptor and its pharmacologic modulators. Pharmacol. Rev. 2015, 67, 505–540. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, D.M.; Rasmussen, S.G.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Thomas, P. Gpr30: A seven-transmembrane-spanning estrogen receptor that triggers EGF release. Trends Endocrinol. Metab. 2005, 16, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Vivacqua, A.; Bonofiglio, D.; Albanito, L.; Madeo, A.; Rago, V.; Carpino, A.; Musti, A.M.; Picard, D.; Ando, S.; Maggiolini, M. 17β-estradiol, genistein, and 4-hydroxytamoxifen induce the proliferation of thyroid cancer cells through the G protein-coupled receptor GPR30. Mol. Pharmacol. 2006, 70, 1414–1423. [Google Scholar] [CrossRef] [PubMed]
- Vivacqua, A.; Bonofiglio, D.; Recchia, A.G.; Musti, A.M.; Picard, D.; Ando, S.; Maggiolini, M. The G protein-coupled receptor GPR30 mediates the proliferative effects induced by 17β-estradiol and hydroxytamoxifen in endometrial cancer cells. Mol. Endocrinol. 2006, 20, 631–646. [Google Scholar] [CrossRef] [PubMed]
- Albanito, L.; Madeo, A.; Lappano, R.; Vivacqua, A.; Rago, V.; Carpino, A.; Oprea, T.I.; Prossnitz, E.R.; Musti, A.M.; Ando, S.; et al. G protein-coupled receptor 30 (GPR30) mediates gene expression changes and growth response to 17β-estradiol and selective GPR30 ligand G-1 in ovarian cancer cells. Cancer Res. 2007, 67, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Teng, J.; Wang, Z.Y.; Prossnitz, E.R.; Bjorling, D.E. The g protein-coupled receptor GPR30 inhibits human urothelial cell proliferation. Endocrinology 2008, 149, 4024–4034. [Google Scholar] [CrossRef] [PubMed]
- Chan, Q.K.; Lam, H.M.; Ng, C.F.; Lee, A.Y.; Chan, E.S.; Ng, H.K.; Ho, S.M.; Lau, K.M. Activation of GPR30 inhibits the growth of prostate cancer cells through sustained activation of erk1/2, c-jun/c-fos-dependent upregulation of p21, and induction of G2 cell-cycle arrest. Cell Death Differ. 2010, 17, 1511–1523. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Le May, C.; Wong, W.P.; Ward, R.D.; Clegg, D.J.; Marcelli, M.; Korach, K.S.; Mauvais-Jarvis, F. Importance of extranuclear estrogen receptor-α and membrane G protein-coupled estrogen receptor in pancreatic islet survival. Diabetes 2009, 58, 2292–2302. [Google Scholar] [CrossRef] [PubMed]
- Prossnitz, E.R.; Hathaway, H.J. What have we learned about GPER function in physiology and disease from knockout mice? J. Steroid Biochem. Mol. Biol. 2015, 153, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Quinn, J.A.; Frackelton, A.R., Jr.; Bland, K.I. Estrogen action via the G protein-coupled receptor, GPR30: Stimulation of adenylyl cyclase and CAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol. Endocrinol. 2002, 16, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Bologa, C.G.; Revankar, C.M.; Young, S.M.; Edwards, B.S.; Arterburn, J.B.; Kiselyov, A.S.; Parker, M.A.; Tkachenko, S.E.; Savchuck, N.P.; Sklar, L.A.; et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat. Chem. Biol. 2006, 2, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Vanaja, D.K.; Cheville, J.C.; Iturria, S.J.; Young, C.Y. Transcriptional silencing of zinc finger protein 185 identified by expression profiling is associated with prostate cancer progression. Cancer Res. 2003, 63, 3877–3882. [Google Scholar] [PubMed]
- Varambally, S.; Yu, J.; Laxman, B.; Rhodes, D.R.; Mehra, R.; Tomlins, S.A.; Shah, R.B.; Chandran, U.; Monzon, F.A.; Becich, M.J.; et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell 2005, 8, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Lam, H.M.; Ouyang, B.; Chen, J.; Ying, J.; Wang, J.; Wu, C.L.; Jia, L.; Medvedovic, M.; Vessella, R.L.; Ho, S.M. Targeting GPR30 with G-1: A new therapeutic target for castration-resistant prostate cancer. Endocr. Relat. Cancer 2014, 21, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Kar, S.; Wang, M.; Carr, B.I. Transient and sustained ERK phosphorylation and nuclear translocation in growth control. J. Cell. Physiol. 2002, 192, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.R.; Plotkin, L.I.; Aguirre, J.I.; Han, L.; Jilka, R.L.; Kousteni, S.; Bellido, T.; Manolagas, S.C. Transient versus sustained phosphorylation and nuclear accumulation of ERKS underlie anti-versus pro-apoptotic effects of estrogens. J. Biol. Chem. 2005, 280, 4632–4638. [Google Scholar] [CrossRef] [PubMed]
- Pandey, D.P.; Lappano, R.; Albanito, L.; Madeo, A.; Maggiolini, M.; Picard, D. Estrogenic GPR30 signalling induces proliferation and migration of breast cancer cells through CTGF. EMBO J. 2009, 28, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Graeber, C.T.; Quinn, J.A.; Resnick, M.B.; Giri, D.; DeLellis, R.A.; Steinhoff, M.M.; Sabo, E. Distribution of GPR30, a seven membrane-spanning estrogen receptor, in primary breast cancer and its association with clinicopathologic determinants of tumor progression. Clin. Cancer Res. 2006, 12, 6359–6366. [Google Scholar] [CrossRef] [PubMed]
- Henic, E.; Noskova, V.; Hoyer-Hansen, G.; Hansson, S.; Casslen, B. Estradiol attenuates EGF-induced rapid UPAR mobilization and cell migration via the g-protein-coupled receptor 30 in ovarian cancer cells. Int. J. Gynecol. Cancer 2009, 19, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Zhang, X.; Xie, Y.; Tu, Y.; Wang, D.; Liu, Z.; Wang, Z.Y. Involvement of estrogen receptor variant ER-α36, not GPR30, in nongenomic estrogen signaling. Mol. Endocrinol. 2010, 24, 709–721. [Google Scholar] [CrossRef] [PubMed]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lau, K.-M.; To, K.-F. Importance of Estrogenic Signaling and Its Mediated Receptors in Prostate Cancer. Int. J. Mol. Sci. 2016, 17, 1434. https://doi.org/10.3390/ijms17091434
Lau K-M, To K-F. Importance of Estrogenic Signaling and Its Mediated Receptors in Prostate Cancer. International Journal of Molecular Sciences. 2016; 17(9):1434. https://doi.org/10.3390/ijms17091434
Chicago/Turabian StyleLau, Kin-Mang, and Ka-Fai To. 2016. "Importance of Estrogenic Signaling and Its Mediated Receptors in Prostate Cancer" International Journal of Molecular Sciences 17, no. 9: 1434. https://doi.org/10.3390/ijms17091434
APA StyleLau, K. -M., & To, K. -F. (2016). Importance of Estrogenic Signaling and Its Mediated Receptors in Prostate Cancer. International Journal of Molecular Sciences, 17(9), 1434. https://doi.org/10.3390/ijms17091434