
The Potential of Targeting Ribosome Biogenesis in High-Grade Serous Ovarian Cancer



Abstract
:
1. Introduction
2. Current Diagnostic and Standard Therapeutic Approach for High-Grade Serous Ovarian Cancer (HGSOC)
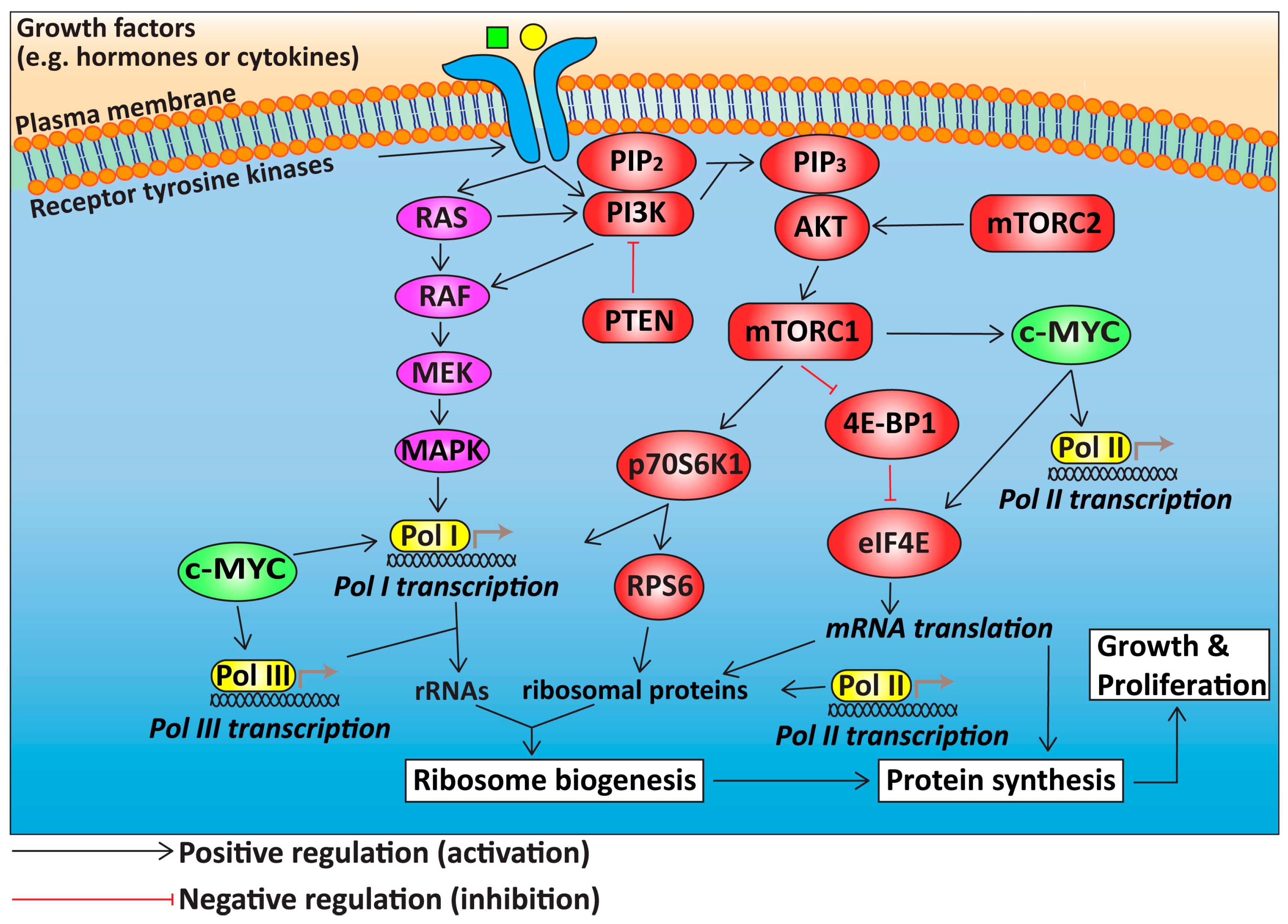
3. Deregulation of Growth Signalling Pathways Upstream of Ribosome Biogenesis in HGSOC
3.1. The PI3K/AKT/mTOR Signalling Pathway in HGSOC
3.2. RAS/RAF/MEK/MAPK Signalling in HGSOC
3.3. c-MYC in HGSOC
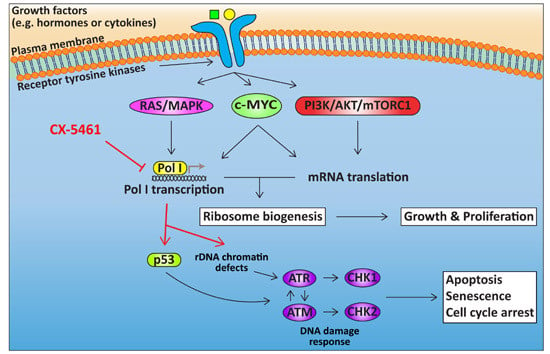
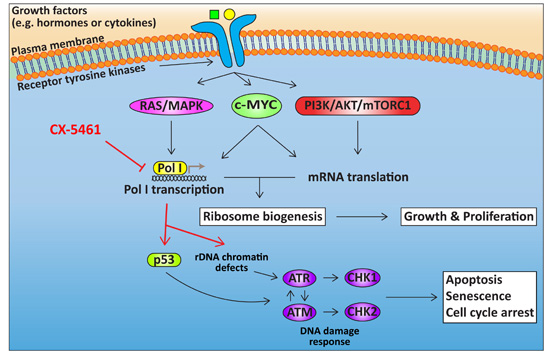
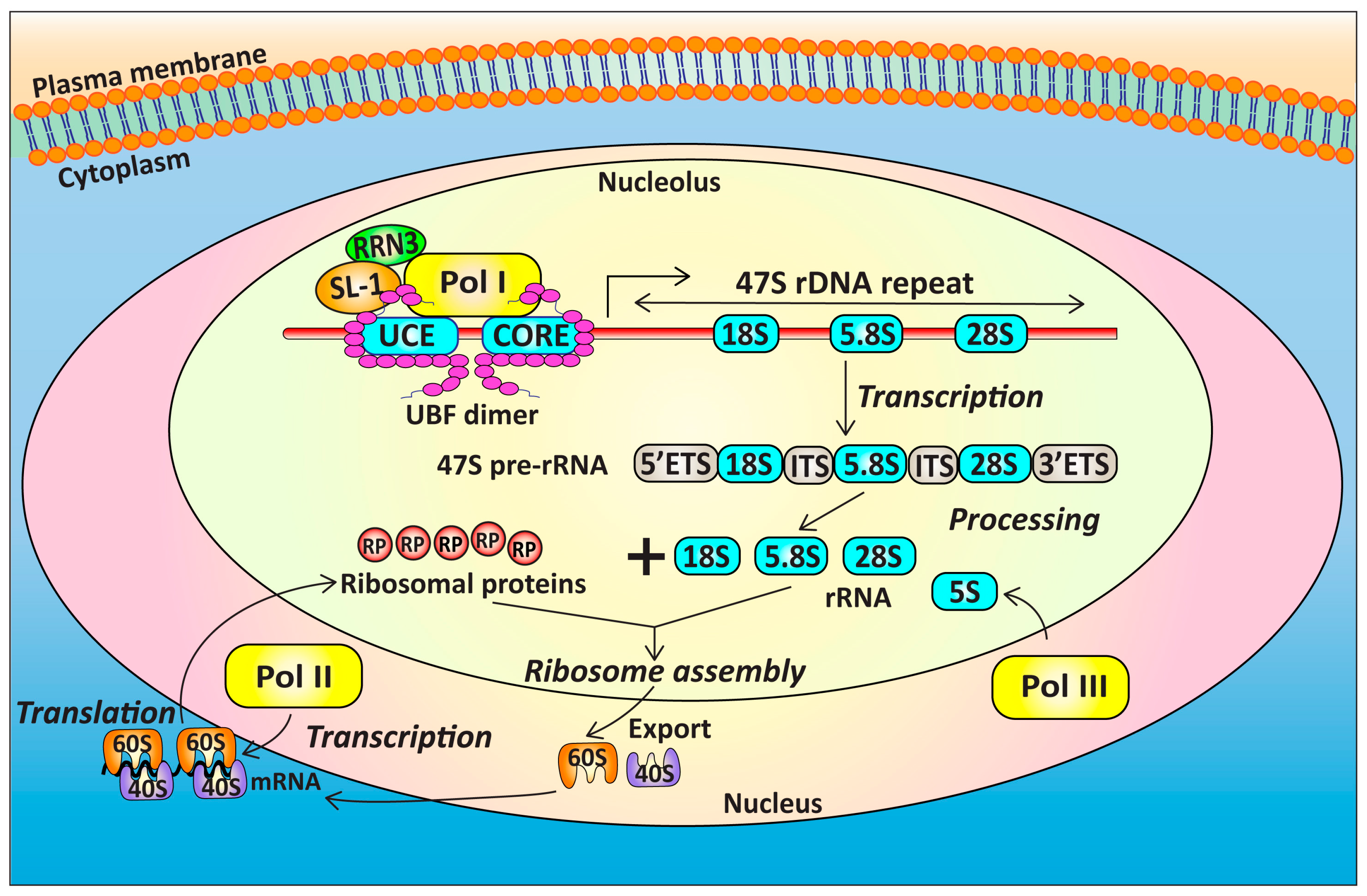
4. Targeting Ribosome Biogenesis Is a Novel Approach for Cancer Treatment
4.1. Targeting Pol I Transcription
4.2. Cellular Response to CX-5461
5. The Potential of Targeting Ribosome Biogenesis in HGSOC
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ALL | acute lymphoblastic leukemia |
| ATM | ataxia telangiectasia mutated |
| ATR | ataxia telangiectasia and rad3-related protein |
| BER | base-excision repair |
| BRCA | breast-related cancer antigens |
| CA-125 | cancer antigen 125 |
| DDR | DNA damage response |
| DSBs | double-strand breaks |
| eIF4E | eukaryotic translation initiation factor 4E |
| EOC | epithelial ovarian cancer |
| ER | estrogen receptor |
| HER2 | human epidermal growth factor receptor 2 |
| HGSOC | high-grade serous ovarian cancer |
| HR | homologous recombination DNA repair |
| HRD | homologous recombination DNA repair deficiency |
| IGF-R | insulin-like growth factor receptor |
| LGSOC | low-grade serous ovarian cancer |
| MAPK | mitogen-activated protein kinase |
| MEK | MAPK/ERK kinase |
| mTOR | mammalian target of rapamycin |
| mTORC1 | mammalian target of rapamycin complex 1 |
| MDM2 | murine double minute 2 |
| MYC | v-myc avian myelocytomatosis viral oncogene homolog |
| OC | ovarian cancer |
| PARP | poly ADP-ribose polymerase |
| PDX | patient-derived xenograft |
| PI3K | phosphatidylinositol-3-kinase |
| Pol I | RNA polymerase I |
| Pol II | RNA polymerase II |
| Pol III | RNA polymerase III |
| PTEN | phosphatase and tensin homolog |
| RAS | retrovirus-associated DNA sequences |
| rDNA | ribosomal DNA |
| rRNA | ribosomal RNA |
| RPs | ribosomal proteins |
| SL-1 | selectivity factor 1 |
| TERT | telomerase reverse transcriptase |
| UBF | upstream binding factor |
| VEGF | vascular endothelial growth factor |
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in globocan 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Lokadasan, R.; James, F.V.; Narayanan, G.; Prabhakaran, P.K. Targeted agents in epithelial ovarian cancer: Review on emerging therapies and future developments. Ecancermedicalscience 2016, 10, 626. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Surveillance, Epidemiology and End Results Program. Available online: https://seer.cancer.gov (accessed on 1 December 2016).
- Erickson, B.K.; Conner, M.G.; Landen, C.N., Jr. The role of the fallopian tube in the origin of ovarian cancer. Am. J. Obstet. Gynecol. 2013, 209, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Bowtell, D.D. The genesis and evolution of high-grade serous ovarian cancer. Nat. Rev. Cancer 2010, 10, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Koulouris, C.R.; Penson, R.T. Ovarian stromal and germ cell tumors. Semin. Oncol. 2009, 36, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Landen, C.N., Jr.; Birrer, M.J.; Sood, A.K. Early events in the pathogenesis of epithelial ovarian cancer. J. Clin. Oncol. 2008, 26, 995–1005. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [Green Version]
- Vang, R.; Shih Ie, M.; Kurman, R.J. Ovarian low-grade and high-grade serous carcinoma: Pathogenesis, clinicopathologic and molecular biologic features, and diagnostic problems. Adv. Anat. Pathol. 2009, 16, 267–282. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih Ie, M. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer—Shifting the paradigm. Hum. Pathol. 2011, 42, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Bowtell, D.D.; Bohm, S.; Ahmed, A.A.; Aspuria, P.J.; Bast, R.C., Jr.; Beral, V.; Berek, J.S.; Birrer, M.J.; Blagden, S.; Bookman, M.A.; et al. Rethinking ovarian cancer ii: Reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 2015, 15, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Seidman, J.D.; Horkayne-Szakaly, I.; Haiba, M.; Boice, C.R.; Kurman, R.J.; Ronnett, B.M. The histologic type and stage distribution of ovarian carcinomas of surface epithelial origin. Int. J. Gynecol. Pathol. 2004, 23, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.Z.; Miow, Q.H.; Huang, R.Y.; Wong, M.K.; Ye, J.; Lau, J.A.; Wu, M.C.; Bin Abdul Hadi, L.H.; Soong, R.; Choolani, M.; et al. Functional genomics identifies five distinct molecular subtypes with clinical relevance and pathways for growth control in epithelial ovarian cancer. EMBO Mol. Med. 2013, 5, 1051–1066. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.A.; Etemadmoghadam, D.; Temple, J.; Lynch, A.G.; Riad, M.; Sharma, R.; Stewart, C.; Fereday, S.; Caldas, C.; Defazio, A.; et al. Driver mutations in tp53 are ubiquitous in high grade serous carcinoma of the ovary. J. Pathol. 2010, 221, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Pal, T.; Permuth-Wey, J.; Betts, J.A.; Krischer, J.P.; Fiorica, J.; Arango, H.; LaPolla, J.; Hoffman, M.; Martino, M.A.; Wakeley, K.; et al. BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer 2005, 104, 2807–2816. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous recombination deficiency: Exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef] [PubMed]
- Alsop, K.; Fereday, S.; Meldrum, C.; deFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. BRCA mutation frequency and patterns of treatment response in brca mutation-positive women with ovarian cancer: A report from the australian ovarian cancer study group. J. Clin. Oncol. 2012, 30, 2654–2663. [Google Scholar] [CrossRef] [PubMed]
- Bast, R.C., Jr.; Klug, T.L.; St John, E.; Jenison, E.; Niloff, J.M.; Lazarus, H.; Berkowitz, R.S.; Leavitt, T.; Griffiths, C.T.; Parker, L.; et al. A radioimmunoassay using a monoclonal antibody to monitor the course of epithelial ovarian cancer. N. Engl. J. Med. 1983, 309, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Menon, U.; Gentry-Maharaj, A.; Hallett, R.; Ryan, A.; Burnell, M.; Sharma, A.; Lewis, S.; Davies, S.; Philpott, S.; Lopes, A.; et al. Sensitivity and specificity of multimodal and ultrasound screening for ovarian cancer, and stage distribution of detected cancers: Results of the prevalence screen of the uk collaborative trial of ovarian cancer screening (UKCTOCS). Lancet Oncol. 2009, 10, 327–340. [Google Scholar] [CrossRef]
- Kulasingam, V.; Pavlou, M.P.; Diamandis, E.P. Integrating high-throughput technologies in the quest for effective biomarkers for ovarian cancer. Nat. Rev. Cancer 2010, 10, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Bast, R.C., Jr.; Hennessy, B.; Mills, G.B. The biology of ovarian cancer: New opportunities for translation. Nat. Rev. Cancer 2009, 9, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [PubMed]
- Cepeda, V.; Fuertes, M.A.; Castilla, J.; Alonso, C.; Quevedo, C.; Perez, J.M. Biochemical mechanisms of cisplatin cytotoxicity. Anticancer Agents Med. Chem. 2007, 7, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Heintz, A.P.; Odicino, F.; Maisonneuve, P.; Quinn, M.A.; Benedet, J.L.; Creasman, W.T.; Ngan, H.Y.; Pecorelli, S.; Beller, U. Carcinoma of the ovary. Figo 26th annual report on the results of treatment in gynecological cancer. Int. J. Gynaecol. Obstet. 2006, 95 (Suppl. S1), S161–S192. [Google Scholar] [CrossRef]
- Chetrit, A.; Hirsh-Yechezkel, G.; Ben-David, Y.; Lubin, F.; Friedman, E.; Sadetzki, S. Effect of brca1/2 mutations on long-term survival of patients with invasive ovarian cancer: The national israeli study of ovarian cancer. J. Clin. Oncol. 2008, 26, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Bolton, K.L.; Chenevix-Trench, G.; Goh, C.; Sadetzki, S.; Ramus, S.J.; Karlan, B.Y.; Lambrechts, D.; Despierre, E.; Barrowdale, D.; McGuffog, L.; et al. Association between BRCA1 and BRCA2 mutations and survival in women with invasive epithelial ovarian cancer. JAMA 2012, 307, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Carden, C.P.; Kaye, S.B. Beyond chemotherapy: Targeted therapies in ovarian cancer. Nat. Rev. Cancer 2009, 9, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Kaye, S.B. Ovarian cancer: Strategies for overcoming resistance to chemotherapy. Nat. Rev. Cancer 2003, 3, 502–516. [Google Scholar] [CrossRef] [PubMed]
- Swisher, E.M.; Sakai, W.; Karlan, B.Y.; Wurz, K.; Urban, N.; Taniguchi, T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008, 68, 2581–2586. [Google Scholar] [CrossRef] [PubMed]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Cooke, S.L.; Brenton, J.D. Evolution of platinum resistance in high-grade serous ovarian cancer. Lancet Oncol. 2011, 12, 1169–1174. [Google Scholar] [CrossRef]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Markman, M.; Bookman, M.A. Second-line treatment of ovarian cancer. Oncologist 2000, 5, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Luvero, D.; Milani, A.; Ledermann, J.A. Treatment options in recurrent ovarian cancer: Latest evidence and clinical potential. Ther. Adv. Med. Oncol. 2014, 6, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Pujade-Lauraine, E.; Hilpert, F.; Weber, B.; Reuss, A.; Poveda, A.; Kristensen, G.; Sorio, R.; Vergote, I.; Witteveen, P.; Bamias, A.; et al. Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: The aurelia open-label randomized phase iii trial. J. Clin. Oncol. 2014, 32, 1302–1308. [Google Scholar] [CrossRef] [PubMed]
- Perren, T.J.; Swart, A.M.; Pfisterer, J.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; Kurzeder, C.; et al. A phase 3 trial of bevacizumab in ovarian cancer. N. Engl. J. Med. 2011, 365, 2484–2496. [Google Scholar] [CrossRef] [PubMed]
- Oza, A.M.; Cook, A.D.; Pfisterer, J.; Embleton, A.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; et al. Standard chemotherapy with or without bevacizumab for women with newly diagnosed ovarian cancer (ICON7): Overall survival results of a phase 3 randomised trial. Lancet Oncol. 2015, 16, 928–936. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in brca mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.A.; Drew, Y.; Kristeleit, R.S. Homologous recombination deficiency and ovarian cancer. Eur. J. Cancer 2016, 60, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.L.; Swisher, E.M.; Kaufmann, S.H. Poly (ADP-ribose) polymerase inhibitors: Recent advances and future development. J. Clin. Oncol. 2015, 33, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.M.; Moshfegh, A.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. Mutant p53 reactivation by prima-1met induces multiple signaling pathways converging on apoptosis. Oncogene 2010, 29, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.B.; Soderqvist, M.; Segerback, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. Prima-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Fransson, A.; Glaessgen, D.; Alfredsson, J.; Wiman, K.G.; Bajalica-Lagercrantz, S.; Mohell, N. Strong synergy with APR-246 and DNA-damaging drugs in primary cancer cells from patients with tp53 mutant high-grade serous ovarian cancer. J. Ovarian Res. 2016, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Cheaib, B.; Auguste, A.; Leary, A. The PI3K/Akt/mTOR pathway in ovarian cancer: Therapeutic opportunities and challenges. Chin. J. Cancer 2015, 34, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, K.E.; Cullinane, C.; Hannan, K.M.; Wall, M.; Chan, J.; Barber, F.; Foo, J.; Cameron, D.; Neilsen, A.; Ng, P.; et al. Synergistic inhibition of ovarian cancer cell growth by combining selective PI3K/mTOR and RAS/ERK pathway inhibitors. Eur. J. Cancer 2013, 49, 3936–3944. [Google Scholar] [CrossRef] [PubMed]
- Kinross, K.M.; Brown, D.V.; Kleinschmidt, M.; Jackson, S.; Christensen, J.; Cullinane, C.; Hicks, R.J.; Johnstone, R.W.; McArthur, G.A. In vivo activity of combined PI3K/mTOR and mek inhibition in a kras(G12D);pten deletion mouse model of ovarian cancer. Mol. Cancer Ther. 2011, 10, 1440–1449. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Altomare, D.A.; Wang, H.Q.; Skele, K.L.; De Rienzo, A.; Klein-Szanto, A.J.; Godwin, A.K.; Testa, J.R. Akt and mtor phosphorylation is frequently detected in ovarian cancer and can be targeted to disrupt ovarian tumor cell growth. Oncogene 2004, 23, 5853–5857. [Google Scholar] [CrossRef] [PubMed]
- Kuo, K.T.; Mao, T.L.; Jones, S.; Veras, E.; Ayhan, A.; Wang, T.L.; Glas, R.; Slamon, D.; Velculescu, V.E.; Kuman, R.J.; et al. Frequent activating mutations of pik3ca in ovarian clear cell carcinoma. Am. J. Pathol. 2009, 174, 1597–1601. [Google Scholar] [CrossRef] [PubMed]
- Hafsi, S.; Pezzino, F.M.; Candido, S.; Ligresti, G.; Spandidos, D.A.; Soua, Z.; McCubrey, J.A.; Travali, S.; Libra, M. Gene alterations in the PI3K/pten/akt pathway as a mechanism of drug-resistance (review). Int. J. Oncol. 2012, 40, 639–644. [Google Scholar] [PubMed]
- King, E.R.; Zu, Z.; Tsang, Y.T.; Deavers, M.T.; Malpica, A.; Mok, S.C.; Gershenson, D.M.; Wong, K.K. The insulin-like growth factor 1 pathway is a potential therapeutic target for low-grade serous ovarian carcinoma. Gynecol. Oncol. 2011, 123, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Mabuchi, S.; Kuroda, H.; Takahashi, R.; Sasano, T. The PI3K/Akt/mTOR pathway as a therapeutic target in ovarian cancer. Gynecol. Oncol. 2015, 137, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Pandolfi, P.P. The pten-PI3K pathway: Of feedbacks and cross-talks. Oncogene 2008, 27, 5527–5541. [Google Scholar] [CrossRef] [PubMed]
- Koul, A.; Willen, R.; Bendahl, P.O.; Nilbert, M.; Borg, A. Distinct sets of gene alterations in endometrial carcinoma implicate alternate modes of tumorigenesis. Cancer 2002, 94, 2369–2379. [Google Scholar] [CrossRef] [PubMed]
- Ryland, G.L.; Hunter, S.M.; Doyle, M.A.; Caramia, F.; Li, J.; Rowley, S.M.; Christie, M.; Allan, P.E.; Stephens, A.N.; Bowtell, D.D.; et al. Mutational landscape of mucinous ovarian carcinoma and its neoplastic precursors. Genome Med. 2015, 7, 87. [Google Scholar] [CrossRef] [PubMed]
- Farley, J.; Brady, W.E.; Vathipadiekal, V.; Lankes, H.A.; Coleman, R.; Morgan, M.A.; Mannel, R.; Yamada, S.D.; Mutch, D.; Rodgers, W.H.; et al. Selumetinib in women with recurrent low-grade serous carcinoma of the ovary or peritoneum: An open-label, single-arm, phase 2 study. Lancet Oncol. 2013, 14, 134–140. [Google Scholar] [CrossRef]
- Coward, J.I.; Middleton, K.; Murphy, F. New perspectives on targeted therapy in ovarian cancer. Int. J. Womens Health 2015, 7, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Sill, M.W.; Thaker, P.H.; Bender, D.P.; Street, D.; McGuire, W.P.; Johnston, C.M.; Rotmensch, J. A phase ii evaluation of selumetinib (AZD6244, ARRY-142886), a selective mek-1/2 inhibitor in the treatment of recurrent or persistent endometrial cancer: An nrg oncology/gynecologic oncology group study. Gynecol. Oncol. 2015, 138, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Gershenson, D.M. The life and times of low-grade serous carcinoma of the ovary. Am. Soc. Clin Oncol. Educ. Book 2013. [Google Scholar] [CrossRef] [PubMed]
- Wierstra, I.; Alves, J. The c-MYC promoter: Still mystery and challenge. Adv. Cancer Res. 2008, 99, 113–333. [Google Scholar] [PubMed]
- Pyndiah, S.; Tanida, S.; Ahmed, K.M.; Cassimere, E.K.; Choe, C.; Sakamuro, D. c-MYC suppresses bin1 to release poly(ADP-ribose) polymerase 1: A mechanism by which cancer cells acquire cisplatin resistance. Sci. Signal. 2011, 4, ra19. [Google Scholar] [CrossRef] [PubMed]
- Van Riggelen, J.; Yetil, A.; Felsher, D.W. Myc as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 2010, 10, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Poortinga, G.; Wall, M.; Sanij, E.; Siwicki, K.; Ellul, J.; Brown, D.; Holloway, T.P.; Hannan, R.D.; McArthur, G.A. c-MYC coordinately regulates ribosomal gene chromatin remodeling and Pol I availability during granulocyte differentiation. Nucleic Acids Res. 2011, 39, 3267–3281. [Google Scholar] [CrossRef] [PubMed]
- Poortinga, G.; Quinn, L.M.; Hannan, R.D. Targeting rna polymerase i to treat myc-driven cancer. Oncogene 2014, 34, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Grandori, C.; Gomez-Roman, N.; Felton-Edkins, Z.A.; Ngouenet, C.; Galloway, D.A.; Eisenman, R.N.; White, R.J. c-MYC binds to human ribosomal DNA and stimulates transcription of rrna genes by rna polymerase i. Nat. Cell Biol. 2005, 7, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Arabi, A.; Wu, S.; Ridderstrale, K.; Bierhoff, H.; Shiue, C.; Fatyol, K.; Fahlen, S.; Hydbring, P.; Soderberg, O.; Grummt, I.; et al. c-MYC associates with ribosomal DNA and activates rna polymerase I transcription. Nat. Cell Biol. 2005, 7, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Shiue, C.N.; Berkson, R.G.; Wright, A.P. c-MYC induces changes in higher order rdna structure on stimulation of quiescent cells. Oncogene 2009, 28, 1833–1842. [Google Scholar] [CrossRef] [PubMed]
- Poortinga, G.; Hannan, K.M.; Snelling, H.; Walkley, C.R.; Jenkins, A.; Sharkey, K.; Wall, M.; Brandenburger, Y.; Palatsides, M.; Pearson, R.B.; et al. Mad1 and c-MYC regulate UBF and rDNA transcription during granulocyte differentiation. EMBO J. 2004, 23, 3325–3335. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.V. The role of c-MYC in regulation of translation initiation. Oncogene 2004, 23, 3217–3221. [Google Scholar] [CrossRef] [PubMed]
- Ruggero, D. The role of myc-induced protein synthesis in cancer. Cancer Res. 2009, 69, 8839–8843. [Google Scholar] [CrossRef] [PubMed]
- Hannan, K.M.; Sanij, E.; Hein, N.; Hannan, R.D.; Pearson, R.B. Signaling to the ribosome in cancer—It is more than just mtorc1. IUBMB Life 2011, 63, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Gentilella, A.; Kozma, S.C.; Thomas, G. A liaison between mtor signaling, ribosome biogenesis and cancer. Biochim. Biophys. Acta 2015, 1849, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Altomare, D.A.; Testa, J.R. Perturbations of the akt signaling pathway in human cancer. Oncogene 2005, 24, 7455–7464. [Google Scholar] [CrossRef] [PubMed]
- Memmott, R.M.; Dennis, P.A. Akt-dependent and -independent mechanisms of mtor regulation in cancer. Cell Signal. 2009, 21, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.C.; Hannan, K.M.; Riddell, K.; Ng, P.Y.; Peck, A.; Lee, R.S.; Hung, S.; Astle, M.V.; Bywater, M.; Wall, M.; et al. Akt promotes rrna synthesis and cooperates with c-MYC to stimulate ribosome biogenesis in cancer. Sci. Signal 2011, 4, ra56. [Google Scholar] [CrossRef] [PubMed]
- Devlin, J.R.; Hannan, K.M.; Ng, P.Y.; Bywater, M.J.; Shortt, J.; Cullinane, C.; McArthur, G.A.; Johnstone, R.W.; Hannan, R.D.; Pearson, R.B. Akt signalling is required for ribosomal rna synthesis and progression of EMU-MYC B-cell lymphoma in vivo. FEBS J. 2013, 280, 5307–5316. [Google Scholar] [CrossRef] [PubMed]
- Stefanovsky, V.Y.; Pelletier, G.; Hannan, R.; Gagnon-Kugler, T.; Rothblum, L.I.; Moss, T. An immediate response of ribosomal transcription to growth factor stimulation in mammals is mediated by erk phosphorylation of ubf. Mol. Cell 2001, 8, 1063–1073. [Google Scholar] [CrossRef]
- Zhao, J.; Yuan, X.; Frödin, M.; Grummt, I. Erk-dependent phosphorylation of the transcription initiation factor tif-ia is required for rna polymerase i transcription and cell growth. Mol. Cell 2003, 11, 405–413. [Google Scholar] [CrossRef]
- Leary, D.J.; Huang, S. Regulation of ribosome biogenesis within the nucleolus. FEBS Lett. 2001, 509, 145–150. [Google Scholar] [CrossRef]
- Tschochner, H.; Hurt, E. Pre-ribosomes on the road from the nucleolus to the cytoplasm. Trends Cell Biol. 2003, 13, 255–263. [Google Scholar] [CrossRef]
- Schmidt, E.V. The role of c-MYC in cellular growth control. Oncogene 1999, 18, 2988–2996. [Google Scholar] [CrossRef] [PubMed]
- Moss, T.; Langlois, F.; Gagnon-Kugler, T.; Stefanovsky, V. A housekeeper with power of attorney: The rrna genes in ribosome biogenesis. Cell. Mol. Life Sci. 2007, 64, 29–49. [Google Scholar] [CrossRef] [PubMed]
- Moir, R.D.; Willis, I.M. Regulation of pol iii transcription by nutrient and stress signaling pathways. Biochim. Biophys. Acta 2013, 1829, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Hannan, K.M.; Sanij, E.; Rothblum, L.I.; Hannan, R.D.; Pearson, R.B. Dysregulation of rna polymerase I transcription during disease. Biochim. Biophys. Acta 2013, 1829, 342–360. [Google Scholar] [CrossRef] [PubMed]
- Diesch, J.; Hannan, R.D.; Sanij, E. Perturbations at the ribosomal genes loci are at the centre of cellular dysfunction and human disease. Cell Biosci. 2014, 4, 43. [Google Scholar] [CrossRef] [PubMed]
- Ruggero, D. Revisiting the nucleolus: From marker to dynamic integrator of cancer signaling. Sci. Signal 2012, 5, pe38. [Google Scholar] [CrossRef] [PubMed]
- Ruggero, D.; Pandolfi, P.P. Does the ribosome translate cancer? Nat. Rev. Cancer 2003, 3, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Derenzini, M.; Trere, D.; Pession, A.; Govoni, M.; Sirri, V.; Chieco, P. Nucleolar size indicates the rapidity of cell proliferation in cancer tissues. J. Pathol. 2000, 191, 181–186. [Google Scholar] [CrossRef]
- Derenzini, M.; Trere, D.; Pession, A.; Montanaro, L.; Sirri, V.; Ochs, R.L. Nucleolar function and size in cancer cells. Am. J. Pathol. 1998, 152, 1291–1297. [Google Scholar] [PubMed]
- Bywater, M.J.; Pearson, R.B.; McArthur, G.A.; Hannan, R.D. Dysregulation of the basal rna polymerase transcription apparatus in cancer. Nat. Rev. Cancer 2013, 13, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Bywater, M.J.; Poortinga, G.; Sanij, E.; Hein, N.; Peck, A.; Cullinane, C.; Wall, M.; Cluse, L.; Drygin, D.; Anderes, K.; et al. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell 2012, 22, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Burger, K.; Eick, D. Functional ribosome biogenesis is a prerequisite for p53 destabilization: Impact of chemotherapy on nucleolar functions and rna metabolism. Biol. Chem. 2013, 394, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Vlatkovic, N.; Boyd, M.T.; Rubbi, C.P. Nucleolar control of p53: A cellular achilles’ heel and a target for cancer therapy. Cell. Mol. Life Sci. 2014, 71, 771–791. [Google Scholar] [CrossRef] [PubMed]
- Burger, K.; Muhl, B.; Harasim, T.; Rohrmoser, M.; Malamoussi, A.; Orban, M.; Kellner, M.; Gruber-Eber, A.; Kremmer, E.; Holzel, M.; et al. Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J. Biol. Chem. 2010, 285, 12416–12425. [Google Scholar] [CrossRef] [PubMed]
- Hein, N.; Hannan, K.M.; George, A.J.; Sanij, E.; Hannan, R.D. The nucleolus: An emerging target for cancer therapy. Trends Mol. Med. 2013, 19, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Quin, J.E.; Devlin, J.R.; Cameron, D.; Hannan, K.M.; Pearson, R.B.; Hannan, R.D. Targeting the nucleolus for cancer intervention. Biochim. Biophys. Acta 2014, 1842, 802–816. [Google Scholar] [CrossRef] [PubMed]
- Drygin, D.; Rice, W.G.; Grummt, I. The rna polymerase i transcription machinery: An emerging target for the treatment of cancer. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 131–156. [Google Scholar] [CrossRef] [PubMed]
- Peltonen, K.; Colis, L.; Liu, H.; Trivedi, R.; Moubarek, M.S.; Moore, H.M.; Bai, B.; Rudek, M.A.; Bieberich, C.J.; Laiho, M. A targeting modality for destruction of rna polymerase i that possesses anticancer activity. Cancer Cell 2014, 25, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Drygin, D.; Lin, A.; Bliesath, J.; Ho, C.B.; O’Brien, S.E.; Proffitt, C.; Omori, M.; Haddach, M.; Schwaebe, M.K.; Siddiqui-Jain, A.; et al. Targeting rna polymerase I with an oral small molecule cx-5461 inhibits ribosomal rna synthesis and solid tumor growth. Cancer Res. 2011, 71, 1418–1430. [Google Scholar] [CrossRef] [PubMed]
- Devlin, J.R.; Hannan, K.M.; Hein, N.; Cullinane, C.; Kusnadi, E.; Ng, P.Y.; George, A.J.; Shortt, J.; Bywater, M.J.; Poortinga, G.; et al. Combination therapy targeting ribosome biogenesis and mrna translation synergistically extends survival in myc-driven lymphoma. Cancer Discov. 2016, 6, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Rebello, R.J.; Kusnadi, E.; Cameron, D.P.; Pearson, H.B.; Lesmana, A.; Devlin, J.R.; Drygin, D.; Clark, A.K.; Porter, L.; Pedersen, J.; et al. The dual inhibition of rna Pol I transcription and pim kinase as a new therapeutic approach to treat advanced prostate cancer. Clin. Cancer Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Quin, J.; Chan, K.T.; Devlin, J.R.; Cameron, D.P.; Diesch, J.; Cullinane, C.; Ahern, J.; Khot, A.; Hein, N.; George, A.J.; et al. Inhibition of rna polymerase i transcription initiation by CX-5461 activates non-canonical atm/atr signaling. Oncotarget 2016, 7, 49800–49818. [Google Scholar] [CrossRef] [PubMed]
- Woods, S.J.; Hannan, K.M.; Pearson, R.B.; Hannan, R.D. The nucleolus as a fundamental regulator of the p53 response and a new target for cancer therapy. Biochim. Biophys. Acta 2015, 1849, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Boulon, S.; Westman, B.J.; Hutten, S.; Boisvert, F.M.; Lamond, A.I. The nucleolus under stress. Mol. Cell 2010, 40, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lu, H. Signaling to p53: Ribosomal proteins find their way. Cancer Cell 2009, 16, 369–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horn, H.F.; Vousden, K.H. Cooperation between the ribosomal proteins l5 and l11 in the p53 pathway. Oncogene 2008, 27, 5774–5784. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, S.; Ivanenkov, V.V.; Teng, T.; Thomas, G. Suprainduction of p53 by disruption of 40S and 60S ribosome biogenesis leads to the activation of a novel g2/m checkpoint. Genes Dev. 2012, 26, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Horn, H.F.; Vousden, K.H. Coping with stress: Multiple ways to activate p53. Oncogene 2007, 26, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Scala, F.; Brighenti, E.; Govoni, M.; Imbrogno, E.; Fornari, F.; Trere, D.; Montanaro, L.; Derenzini, M. Direct relationship between the level of p53 stabilization induced by rrna synthesis-inhibiting drugs and the cell ribosome biogenesis rate. Oncogene 2016, 35, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Negi, S.S.; Brown, P. Transient rrna synthesis inhibition with CX-5461 is sufficient to elicit growth arrest and cell death in acute lymphoblastic leukemia cells. Oncotarget 2015, 6, 34846–34858. [Google Scholar] [CrossRef] [PubMed]
- Negi, S.S.; Brown, P. Rrna synthesis inhibitor, CX-5461, activates atm/atr pathway in acute lymphoblastic leukemia, arrests cells in G2 phase and induces apoptosis. Oncotarget 2015, 6, 18094–18104. [Google Scholar] [CrossRef] [PubMed]
- Campone, M.; Levy, V.; Bourbouloux, E.; Berton Rigaud, D.; Bootle, D.; Dutreix, C.; Zoellner, U.; Shand, N.; Calvo, F.; Raymond, E. Safety and pharmacokinetics of paclitaxel and the oral mtor inhibitor everolimus in advanced solid tumours. Br. J. Cancer 2009, 100, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Vlahovic, G.; Meadows, K.L.; Uronis, H.E.; Morse, M.A.; Blobe, G.C.; Riedel, R.F.; Zafar, S.Y.; Alvarez-Secord, A.; Gockerman, J.; Starodub, A.N.; et al. A phase I study of bevacizumab, everolimus and panitumumab in advanced solid tumors. Cancer Chemother. Pharmacol. 2012, 70, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Burgess, M.; Puhalla, S. BRCA 1/2-mutation related and sporadic breast and ovarian cancers: More alike than different. Front. Oncol. 2014, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.J.; Khot, A.; Brajanovski, N.; Cameron, D.; Hein, N.; McArthur, G.A.; Lim, J.K.C.; O’Brien, S.; Ryckman, D.M.; Yu, G.I.; et al. A phase 1, open-label, dose escalation, safety, pk and pd study of a first in class pol1 inhibitor (CX-5461) in patients with advanced hematologic malignancies (HM). In Proceedings of the 2015 ASCO Annual Meeting, Chicago, IL, USA, 29 May–2 June 2015.




{kind=link}
{kind=link}
{kind=link}
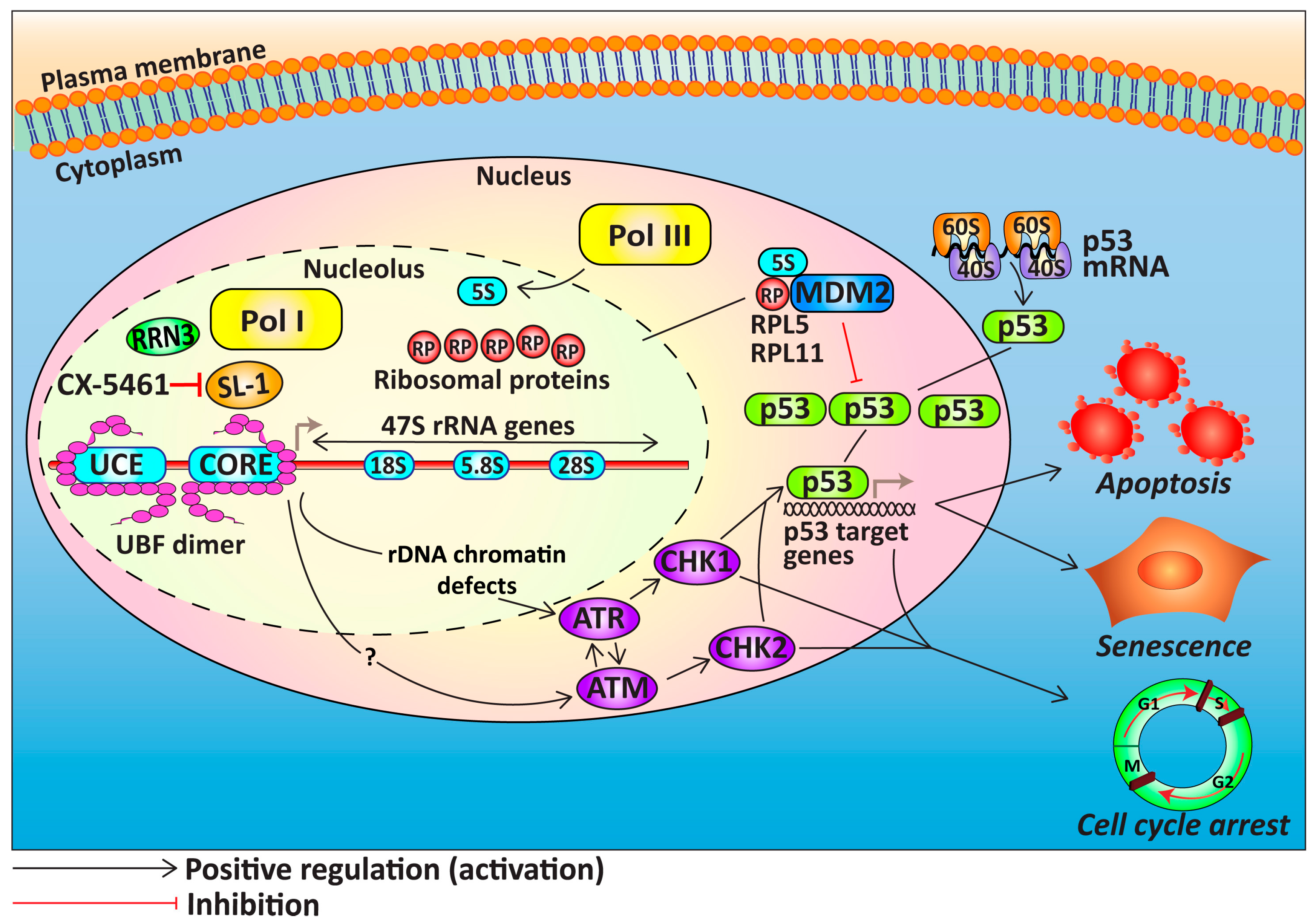{kind=link}
| Epithelial Ovarian Cancer | |||||
|---|---|---|---|---|---|
| Subtypes | High-Grade Serous Ovarian Cancer (Type II) | Low-Grade Serous Ovarian Cancer (Type I) | Clear Cell Ovarian Cancer (Type I) | Endometrioid Ovarian Cancer (Type I) | Mucinous Ovarian Cancer (Type I) |
| Genome Instability | High | Low | Low | Low | Low |
| Frequent Genetic Alternations | TP53 mut (>90%) | TP53 wt | TP53 wt | TP53 wt | TP53 wt |
| BRCA1/2(~15%) HR deficiency (up to 50%) | Uncommon | Uncommon | Uncommon | Uncommon | |
| PI3K pathway (PIK3CA, RICTOR, AKT, RAPTOR, PTEN) RAS pathway (KRAS, MAPK, ERBB2) IGF-1R, EGFR, KIT, CNNE | RAS pathway (KRAS, BRAF) HER2 | PI3K pathway (PIK3CA) MET | PI3K pathway (PIK3CA, PTEN) β-catenin | RAS pathway (KRAS/BRAF) HER2 | |
| 5-year Survival Rate | ~40% | ~70% | >70% | >90% | ~78% |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, S.; Frank, D.; Son, J.; Hannan, K.M.; Hannan, R.D.; Chan, K.T.; Pearson, R.B.; Sanij, E. The Potential of Targeting Ribosome Biogenesis in High-Grade Serous Ovarian Cancer. Int. J. Mol. Sci. 2017, 18, 210. https://doi.org/10.3390/ijms18010210
Yan S, Frank D, Son J, Hannan KM, Hannan RD, Chan KT, Pearson RB, Sanij E. The Potential of Targeting Ribosome Biogenesis in High-Grade Serous Ovarian Cancer. International Journal of Molecular Sciences. 2017; 18(1):210. https://doi.org/10.3390/ijms18010210
Chicago/Turabian StyleYan, Shunfei, Daniel Frank, Jinbae Son, Katherine M. Hannan, Ross D. Hannan, Keefe T. Chan, Richard B. Pearson, and Elaine Sanij. 2017. "The Potential of Targeting Ribosome Biogenesis in High-Grade Serous Ovarian Cancer" International Journal of Molecular Sciences 18, no. 1: 210. https://doi.org/10.3390/ijms18010210
APA StyleYan, S., Frank, D., Son, J., Hannan, K. M., Hannan, R. D., Chan, K. T., Pearson, R. B., & Sanij, E. (2017). The Potential of Targeting Ribosome Biogenesis in High-Grade Serous Ovarian Cancer. International Journal of Molecular Sciences, 18(1), 210. https://doi.org/10.3390/ijms18010210