
Mechanism Governing Human Kappa-Opioid Receptor Expression under Desferrioxamine-Induced Hypoxic Mimic Condition in Neuronal NMB Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
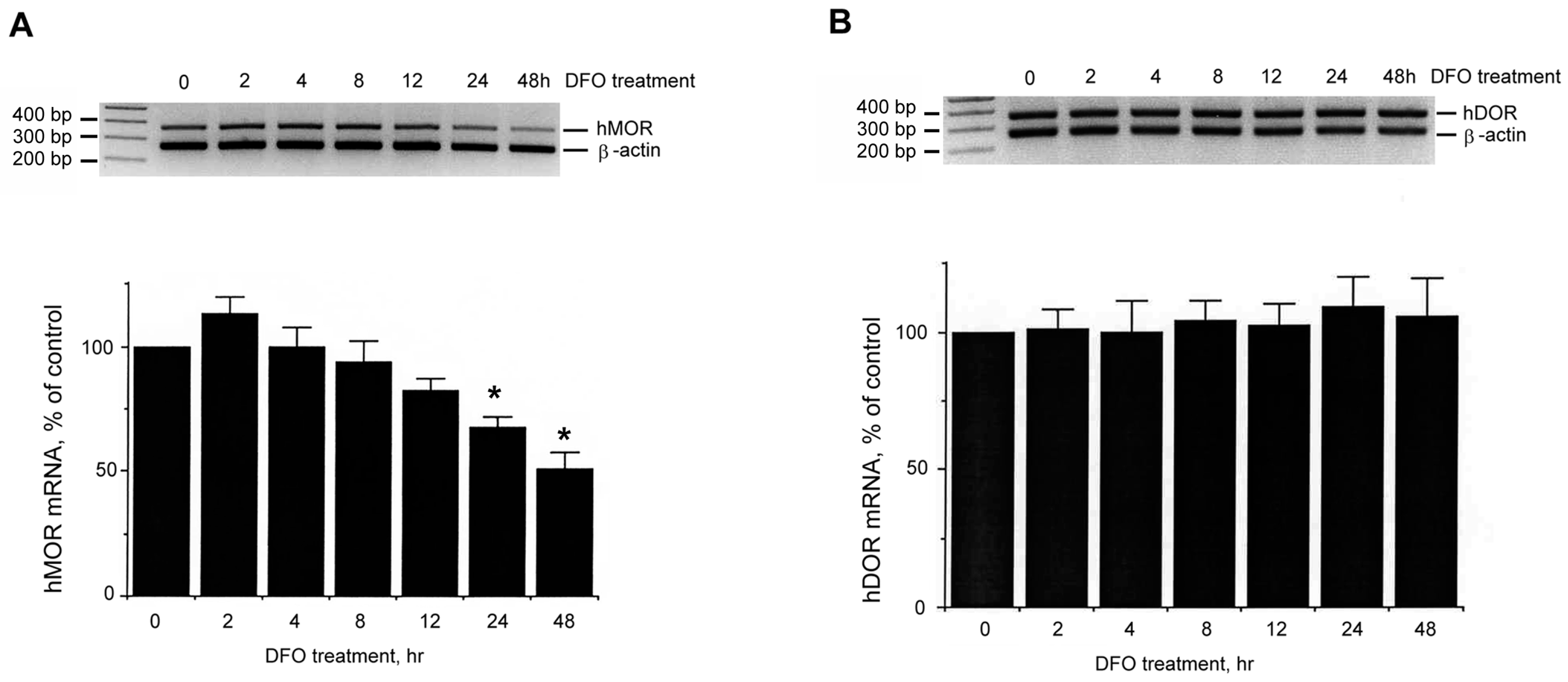
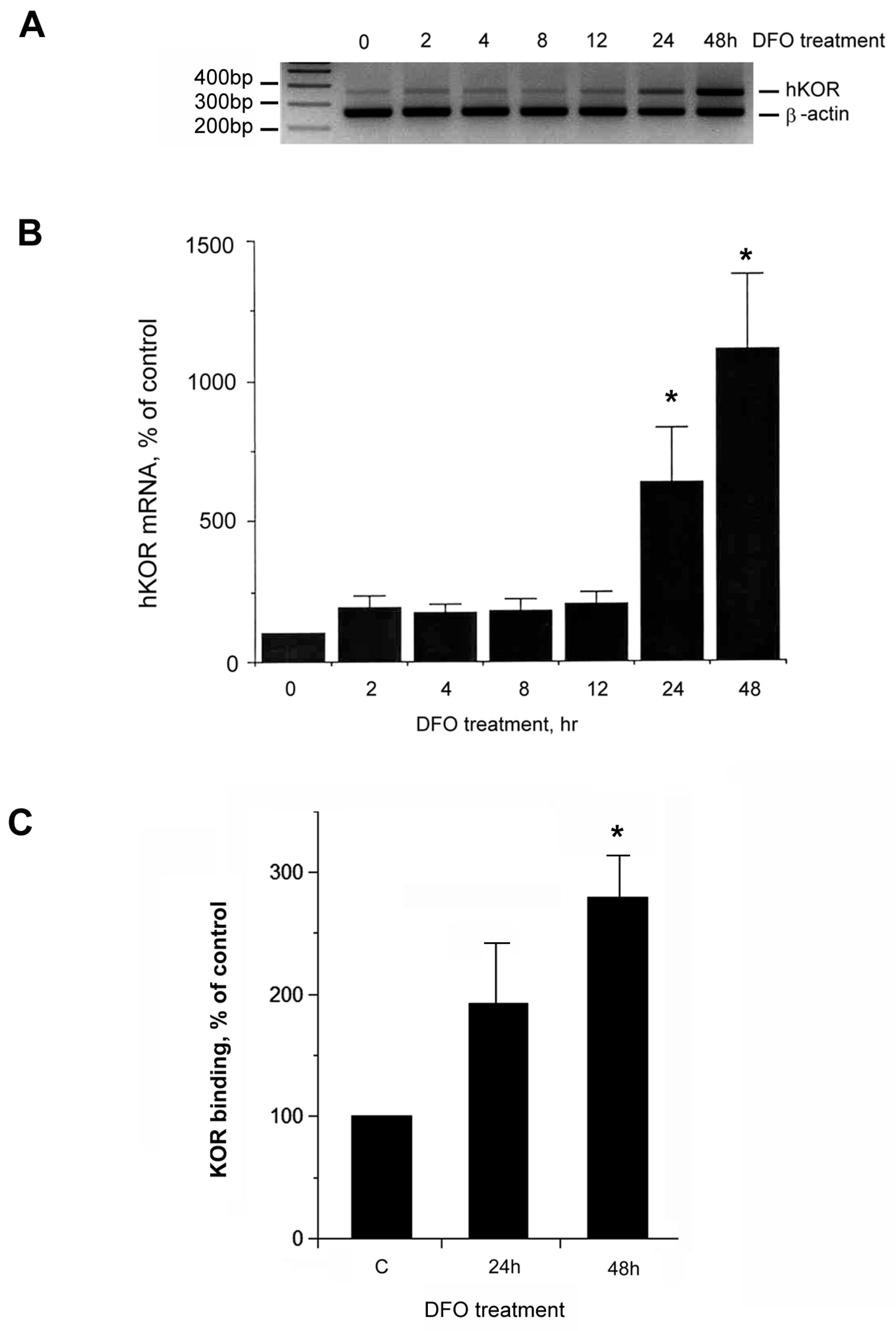
2.1. Increase of Neuronal hKOR Gene Expression under DFO Challenge
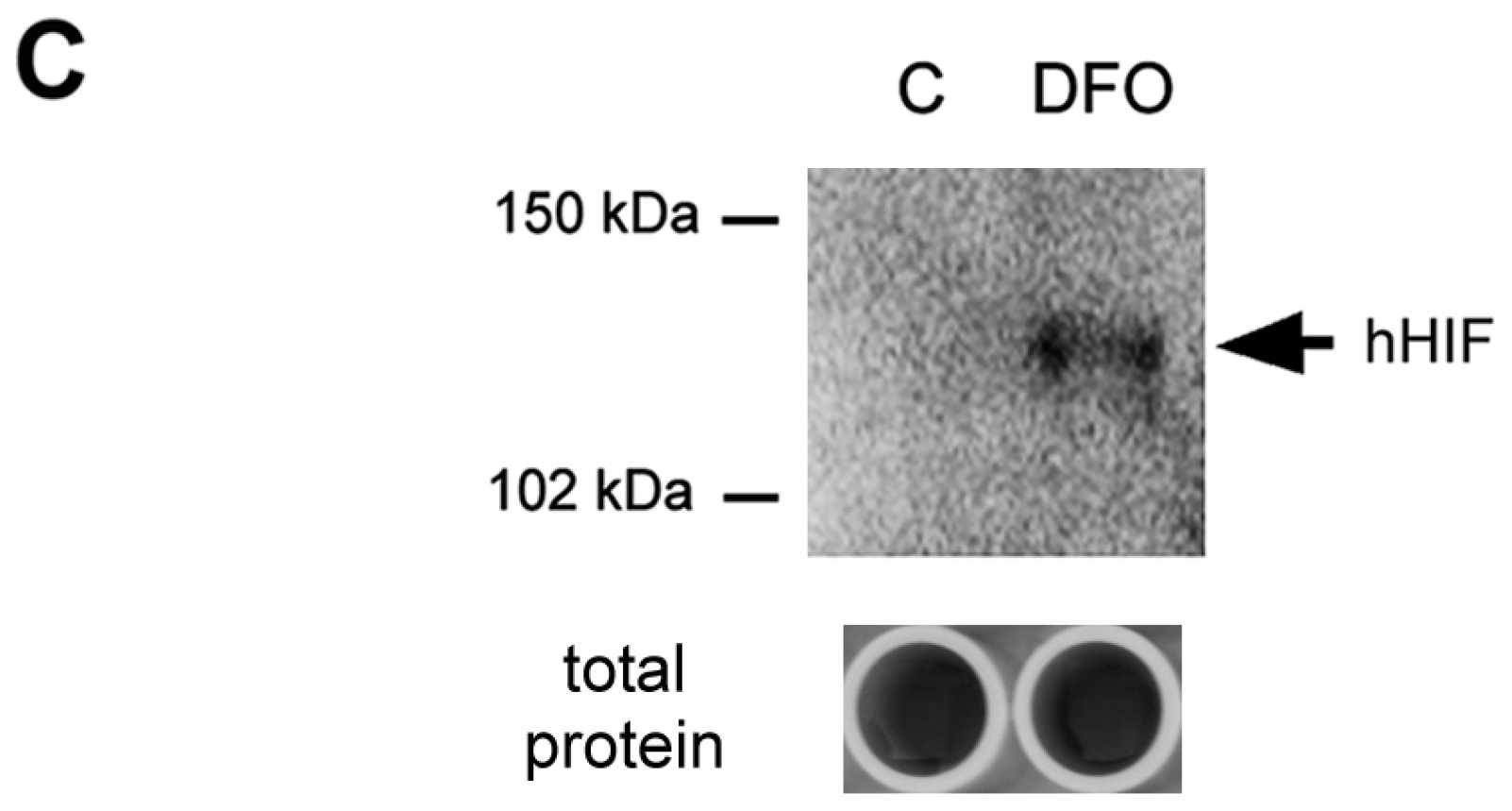
2.2. Increase of HIF-1α Amount in the Nucleus of Survival Cells under DFO Challenge
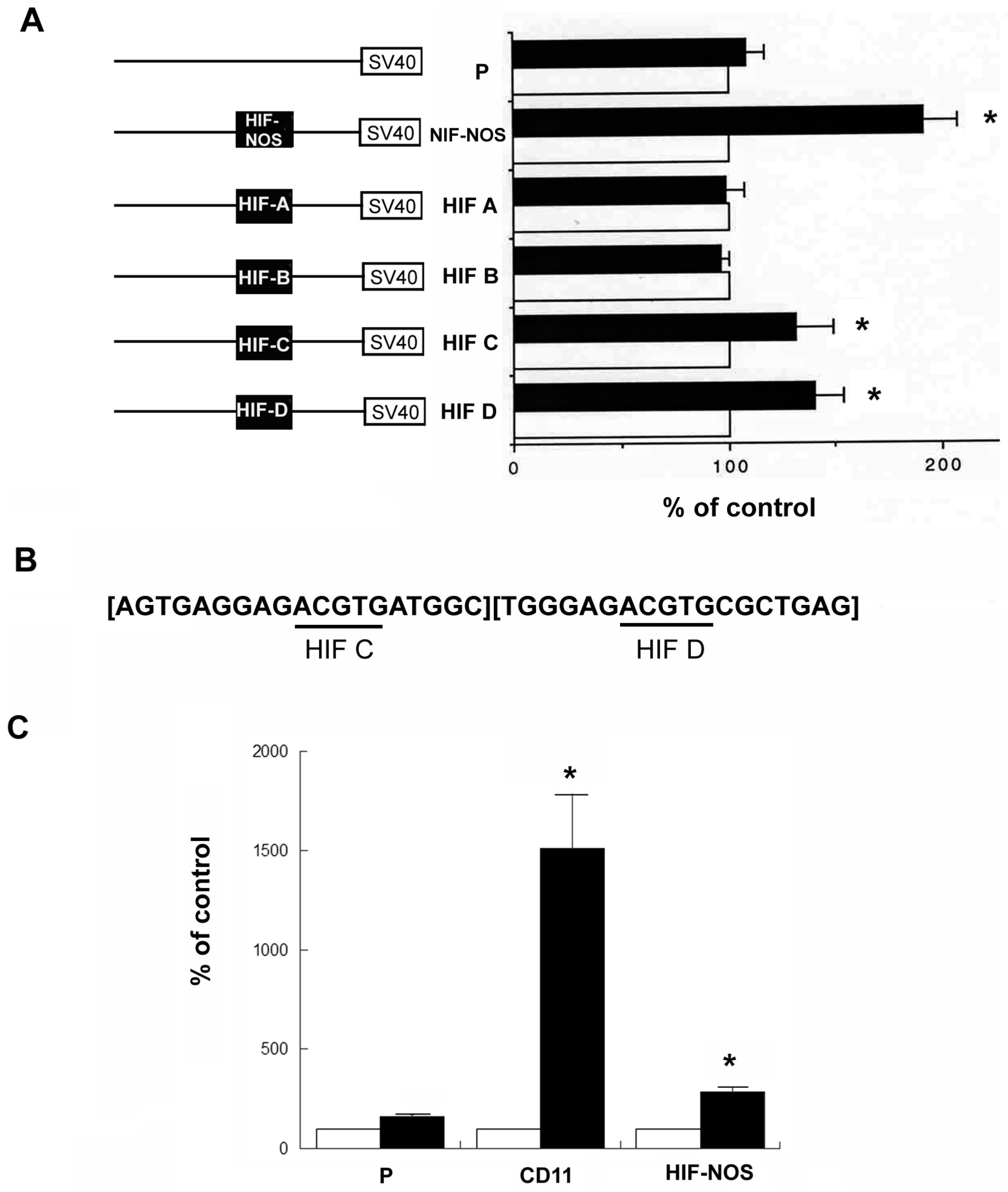
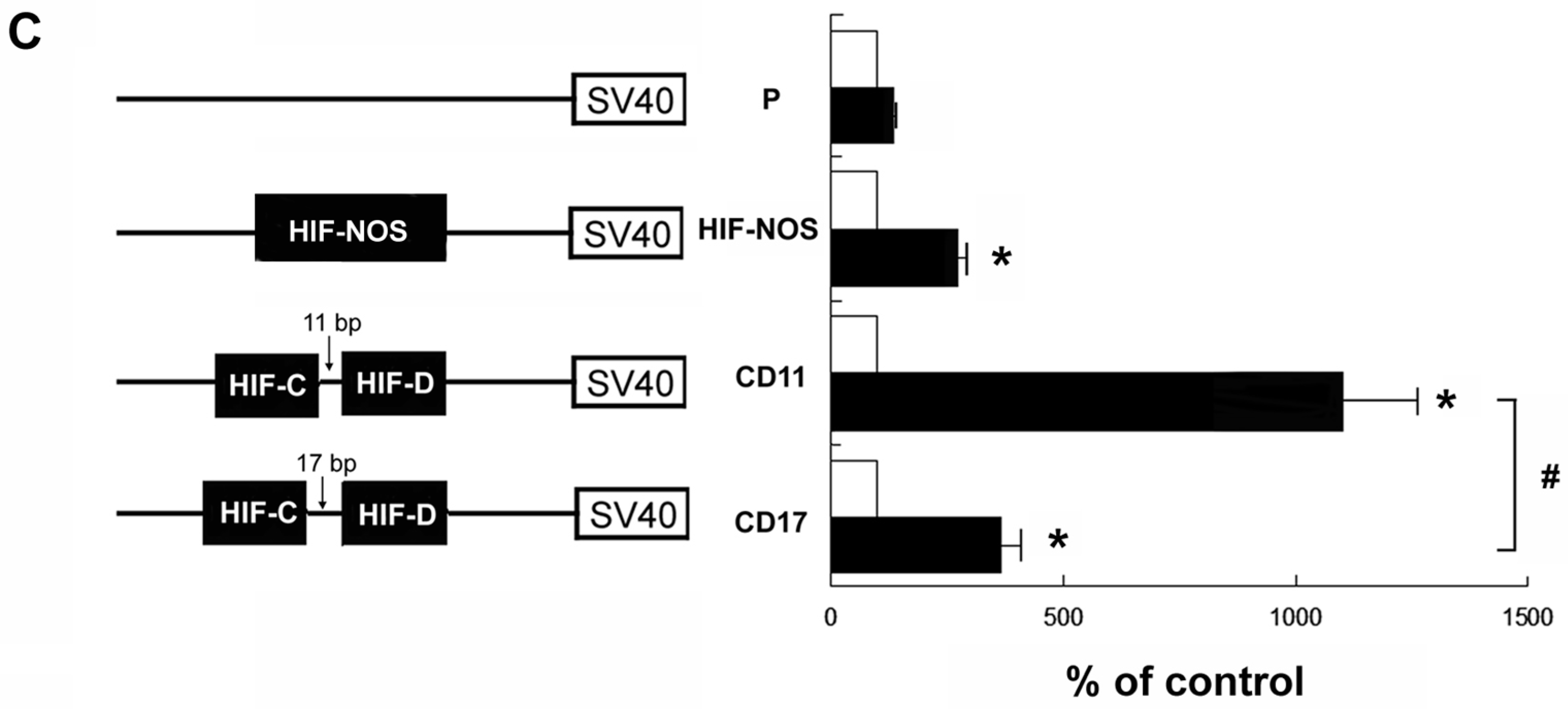
2.3. Effect of HIF Response Elements of hKOR Gene on the Promoter Activity upon DFO Treatment
2.4. Effect of HIF-1α siRNA on the hKOR Gene Expression under DFO Challenge
2.5. Increase of hKOR Ligand Binding in NMB Cells upon DFO Challenge
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Nuclear Extract Preparation
4.3. SDS-PAGE and Western Blot Analysis
4.4. Generations of Various Constructs
4.5. RNA Isolation and RT-PCR
4.6. SiRNA Transfection
4.7. Transient Transfection and Luciferase Reporter Assay
4.8. Cell Membrane Preparation
4.9. Competitive Opioid Binding Assay
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Won, S.J.; Kim, D.Y.; Gwag, B.J. Cellular and molecular pathways of ischemic neuronal death. J. Biochem. Mol. Biol. 2002, 35, 67–86. [Google Scholar] [CrossRef] [PubMed]
- Nangaku, M.; Kojima, I.; Tanaka, T.; Ohse, T.; Kato, H.; Fujita, T. Novel drugs and the response to hypoxia: HIF stabilizers and prolyl hydroxylase. Recent Pat. Cardiovasc. Drug Discov. 2006, 1, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Buttgereit, F.; Brand, M.D. A hierarchy of ATP-consuming processes in mammalian cells. Biochem. J. 1995, 312, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Michiels, C. Physiological and pathological responses to hypoxia. Am. J. Pathol. 2004, 164, 1875–1882. [Google Scholar] [CrossRef]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Cook, R.; Karch, C.; Nahar, P.; Rivera, A.; Ko, J.L. Effects of desferoxamine-induced hypoxia on neuronal human mu-opioid receptor gene expression. Biochem. Biophys. Res. Commun. 2010, 398, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Carmelit, P.; Dor, Y.; Herbert, J.; Fukumura, D.; Brusselmans, K.; Dewerchin, M.; Neeman, M.; Bono, F.; Abramovitch, R.; Maxwell, P.; et al. Role of HIF-1 α in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Lett. Nat. 1998, 394, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Ryan, H.E.; Poloni, M.; McNulty, W.; Elson, D.; Gassmann, M.; Arbeit, J.M.; Johnson, R.S. Hypoxia-inducible factor-1 α is a positive factor in solid tumor growth. Cancer Res. 2000, 60, 4010–4015. [Google Scholar] [PubMed]
- Koivunen, P.; Hirsila, M.; Remes, A.M.; Hassinen, I.E.; Kivirikko, K.I.; Myllyharju, J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates. J. Biol. Chem. 2007, 282, 4524–4532. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Miyake, M.; Liu, K.J.; Shi, H. Specific inhibition of hypoxia inducible factor 1 exaggerates cell injury induced by in vitro ischemia through deteriorating cellular redox environment. J. Neurochem. 2009, 108, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- Bruick, R.K.; McKnight, S.L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 2001, 294, 1337–1340. [Google Scholar] [CrossRef] [PubMed]
- Epstein, A.C.R.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef]
- Semenza, G.L. Oxygen sensing, homeostasis, and disease. N. Engl. J. Med. 2011, 365, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Semenza, G.L. Desferrioxamine induces erythropoietin gene expression and hypoxia-inducible factor 1 DNA-binding activity: Implications for models of hypoxia signal transduction. Blood 1993, 82, 3610–3615. [Google Scholar] [PubMed]
- Yeom, C.J.; Chung, J.K.; Kang, J.H.; Jeon, Y.H.; Kim, K.I.; Jin, Y.N.; Lee, Y.M.; Jeong, J.M.; Lee, D.S. Visualization of hypoxia-inducible factor-1 transcriptional activation in C6 glioma using luciferase and sodium iodide symporter genes. J. Nucl. Med. 2008, 49, 1489–1497. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Bodily, J.M.; Beglin, M.; Kyo, S.; Inoue, M.; Laimins, L.A. Hypoxia-specific stabilization of HIF-1α by human papillomaviruses. Virology 2009, 387, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Herlitz, J.; Hjalmarson, A.; Waagstein, F. Treatment of pain in acute myocardial infarction. Br. Heart J. 1989, 61, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.; Fox, C.A.; Akil, H.; Watson, S.J. Opioid-receptor mRNA expression in the rat CNS. Trends Neurosci. 1995, 18, 22–29. [Google Scholar] [CrossRef]
- Kieffer, B.L.; Gaveriaux-Ruff, C. Exploring opioid system by gene knockout. Prog. Neurobiol. 2002, 66, 285–306. [Google Scholar] [CrossRef]
- Baumhaker, Y.; Ben-Dor, T.; Bar-Hamburger, R.; Sarne, Y. Characterization of a triple opioid system in the human neuroblastoma NMB cell line. Brain Res. 1994, 665, 94–100. [Google Scholar] [CrossRef]
- Coulet, F.; Nadaud, S.; Agrapart, M.; Soubrier, F. Identification of hypoxia response element in the human endothelial nitric oxide synthase gene promoter. J. Biol. Chem. 2003, 278, 46230–46240. [Google Scholar] [CrossRef] [PubMed]
- Belaiba, R.S.; Bonello, S.; Zähringer, C.; Schmidt, S.; Hess, J.; Kietzmann, T.; Görlach, A. Hypoxia up-regulates hypoxia-inducible factor-1 α transcription by involving phosphatidylinositol 3-kinase and nuclear factor κB in pulmonary artery smooth muscle cells. Mol. Biol. Cell 2007, 18, 4691–4697. [Google Scholar] [CrossRef] [PubMed]
- Formisano, L.; Noh, K.; Miyawaki, T.; Mashiko, T.; Bennett, M.V.; Zukin, R.S. Ischemic insults promote epigenetic reprogramming of mu opioid receptor expression in hippocampual neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 4170–4175. [Google Scholar] [CrossRef] [PubMed]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.J.; Iyer, S.; Sataur, A.; Covello, K.L.; Chodosh, L.A.; Simon, M.C. Differential regulation of the transcriptional activities of hypoxia-inducible factor 1 α (HIF-1α) and HIF-2α in stem cells. Mol. Cell. Biol. 2006, 26, 3514–3526. [Google Scholar] [CrossRef] [PubMed]
- Chaston, T.B.; Richardson, D.R. Iron chelators for the treatment of iron overload disease: Relationship between structure, redox activity, and toxicity. Am. J. Hematol. 2003, 73, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Mobarra, N.; Shanaki, M.; Ehteram, H.; Nasiri, H.; Sahmani, M.; Saedi, M.; Goudarzi, M.; Pourkarim, H.; Azad, M. A review of iron chelators in treatment of iron overload syndromes. Int. J. Hematol. Oncol. Stem Cell Res. 2016, 10, 239–247. [Google Scholar] [PubMed]
- Nyholm, S.; Mann, G.J.; Johansson, A.G.; Bergeron, R.J.; Graslund, A.; Thelander, L. Role of ribonucleotide reductase in inhibition of mammalian growth by potent iron chelators. J. Biol. Chem. 1993, 268, 26200–26205. [Google Scholar] [PubMed]
- Hatcher, H.C.; Singh, R.N.; Torti, F.M.; Torti, S.V. Synthetic and natural iron chelators: Therapeutic potential and clinical use. Future Med. Chem. 2009, 1, 643–1670. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.M.; Novak, A.E.; Elliott, J.P. The role of HIF in cobalt-induced ischemic tolerance. Neuroscience 2013, 252, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Hilliard, G.; Ferguson, T.; Millhorn, D.E. Cobalt Inhibits the interaction between hypoxia-inducible factor-α and von Hippel-Lindau protein by direct binding to hypoxia-inducible factors-α. J. Biol. Chem. 2003, 278, 15911–15916. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.Y.; Powis, G. Passing the baton: The HIF switch. Trends Biochem. Sci. 2012, 37, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.C.; Baranova, O.; Lin, J.; Pichiule, P. The transcriptional activator hypoxia inducible factor 2 (HIF-2/EPAS-1) regulates the oxygen-dependent expression of erythropoietin in cortical astrocytes. J. Neurosci. 2006, 26, 9471–9481. [Google Scholar] [CrossRef] [PubMed]
- Bruchas, M.R.; Land, B.B.; Chavkin, C. The dynorphin/kappa-opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Res. 2010, 1314, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Chartoff, E.; Sawyer, A.; Rachlin, A.; Potter, D.; Pliakas, A.; Carlezon, W.A. Blockade of kappa-opioid receptors attenuates the development of depressive-like behaviors induced by cocaine withdrawal in rats. Neuropharmacology 2012, 62, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Cunha, T.M.; Souza, G.R.; Domingues, A.C.; Carreira, E.U.; Lotufo, C.M.; Funez, M.I.; Verri, W.A.; Chunha, F.Q.; Ferreira, S.H. Stimulation of peripheral kappa-opioid receptors inhibits inflammatory hyperalgesia via activation of the PI3kγ/AKT/nNOS/NO signaling pathway. Mol. Pain 2012, 8, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peart, J.N.; Gross, E.R.; Reichelt, M.E.; Hsu, A.; Headrick, J.P.; Gross, G.J. Activation of kappa-opioid receptors at reperfusion affords cardioprotection in both rat and mouse hearts. Basic Res. Cardiol. 2008, 103, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Kerros, C.; Brood, I.; Sola, B.; Jauzac, P.; Allouche, S. Reduction of cell proliferation and potentiation of Fas-induced apoptosis by the selective kappa-opioid receptor agonist U50 488 in the multiple myeloma LP-1 cells. J. Neuroimmunol. 2010, 220, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.L.; Farooqui, M.; Nguyen, J.; Ansonoff, M.; Pintar, J.E.; Hebbel, R.P.; Gupta, K. Morphine induces mesangial cell proliferation and glomerulopathy via kappa-opioid receptors. Am. J. Ren. Phys. 2008, 294, 1388–1397. [Google Scholar]
- Yamamizu, K.; Hamada, Y.; Narita, M. Kappa-opioid receptor ligands regulate angiogenesis in development and in tumors. Br. J. Pharm. 2015, 172, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.C.; Qian, H.; Ghassemi, F.; Zhao, P.; Xia, Y. Oxygen-sensitive delta-opioid receptor regulated survival and death signals: Novel insights into neuronal preconditioning and protection. J. Biol. Chem. 2005, 280, 16208–16218. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chao, D.; Chen, T.; Sandhu, H.; Xia, Y. Delta-Opioid receptors and inflammatory cytokines in hypoxia: Differential regulation between glial and neuron-like cells. Transl. Stroke Res. 2014, 5, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, X.; Yang, Y.; Chao, D.; Lazarus, L.H.; Xia, Y. Current research on opioid receptor function. Curr. Drug Targets 2012, 13, 230–246. [Google Scholar] [CrossRef] [PubMed]
- Grudt, T.; Williams, J.T. κ-Opioid receptors also increase potassium conductance. Proc. Natl. Acad. Sci. USA 1993, 90, 11429–11432. [Google Scholar] [CrossRef] [PubMed]
- Carroll, F.I.; Carlezon, W.A. Development of kappa-opioid receptor antagonists. J. Med. Chem. 2013, 56, 2178–2195. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, R.J.; Shenoy, S.K. Transduction of receptor signals by β-arrestins. Science 2005, 308, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Chavkin, C.; Schattauer, S.S.; Levin, J.R. Arrestin-mediated activation of p38 MAPK: Molecular mechanisms and behavioral consequences. Handb. Exp. Pharmacol. 2013, 219, 281–292. [Google Scholar]
- McLennan, G.P.; Kiss, A.; Miyatake, M.; Belcheva, M.M.; Chambers, K.T.; Pozek, J.J.; Mohabbat, Y.; Moyer, R.A.; Bohn, L.M.; Coscia, C.J. Kappa opioids promote the proliferation of astrocytes via Gβγ and β-arrestin 2-dependent MAPK-mediated pathways. J. Neurochem. 2008, 107, 1753–1765. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chiu, Y.T.; Wu, W.; Huang, P.; Mann, A.; Schulz, S.; Liu-Chen, L.Y. Determination of sites of U50, 488H-promoted phosphorylation of the mouse kappa-opioid receptor (KOPR): Disconnect between KOPR phosphorylation and internalization. Biochem. J. 2016, 473, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Bruchas, M.R.; Macry, T.A.; Lowe, J.D.; Chavkin, C. Mechanisms of signal transduction: Kappa-opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes. J. Biol. Chem. 2006, 281, 18081–18089. [Google Scholar] [CrossRef] [PubMed]
- Bruchas, M.R.; Yang, T.; Schreiber, M.D.; Kwan, S.C.; Li, S.; Chavkin, C. Mechanisms of signal transduction: Long-acting kappa-opioid antagonists disrupt receptor signaling and produce noncompetitive effects by activating c-Jun N-terminal kinase. J. Biol. Chem. 2007, 282, 29803–29811. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.R.; Levant, S.; Bale, A.E. Direct molecular analysis of archival tumor tissue for loss of heterozygosity. BioTechniques 1995, 19, 192–195. [Google Scholar]










© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Babcock, J.; Herrera, A.; Coricor, G.; Karch, C.; Liu, A.H.; Rivera-Gines, A.; Ko, J.L. Mechanism Governing Human Kappa-Opioid Receptor Expression under Desferrioxamine-Induced Hypoxic Mimic Condition in Neuronal NMB Cells. Int. J. Mol. Sci. 2017, 18, 211. https://doi.org/10.3390/ijms18010211
Babcock J, Herrera A, Coricor G, Karch C, Liu AH, Rivera-Gines A, Ko JL. Mechanism Governing Human Kappa-Opioid Receptor Expression under Desferrioxamine-Induced Hypoxic Mimic Condition in Neuronal NMB Cells. International Journal of Molecular Sciences. 2017; 18(1):211. https://doi.org/10.3390/ijms18010211
Chicago/Turabian StyleBabcock, Jennifer, Alberto Herrera, George Coricor, Christopher Karch, Alexander H. Liu, Aida Rivera-Gines, and Jane L. Ko. 2017. "Mechanism Governing Human Kappa-Opioid Receptor Expression under Desferrioxamine-Induced Hypoxic Mimic Condition in Neuronal NMB Cells" International Journal of Molecular Sciences 18, no. 1: 211. https://doi.org/10.3390/ijms18010211
APA StyleBabcock, J., Herrera, A., Coricor, G., Karch, C., Liu, A. H., Rivera-Gines, A., & Ko, J. L. (2017). Mechanism Governing Human Kappa-Opioid Receptor Expression under Desferrioxamine-Induced Hypoxic Mimic Condition in Neuronal NMB Cells. International Journal of Molecular Sciences, 18(1), 211. https://doi.org/10.3390/ijms18010211