
Cancer’s Achilles’ Heel: Apoptosis and Necroptosis to the Rescue



Abstract
:
1. Introduction
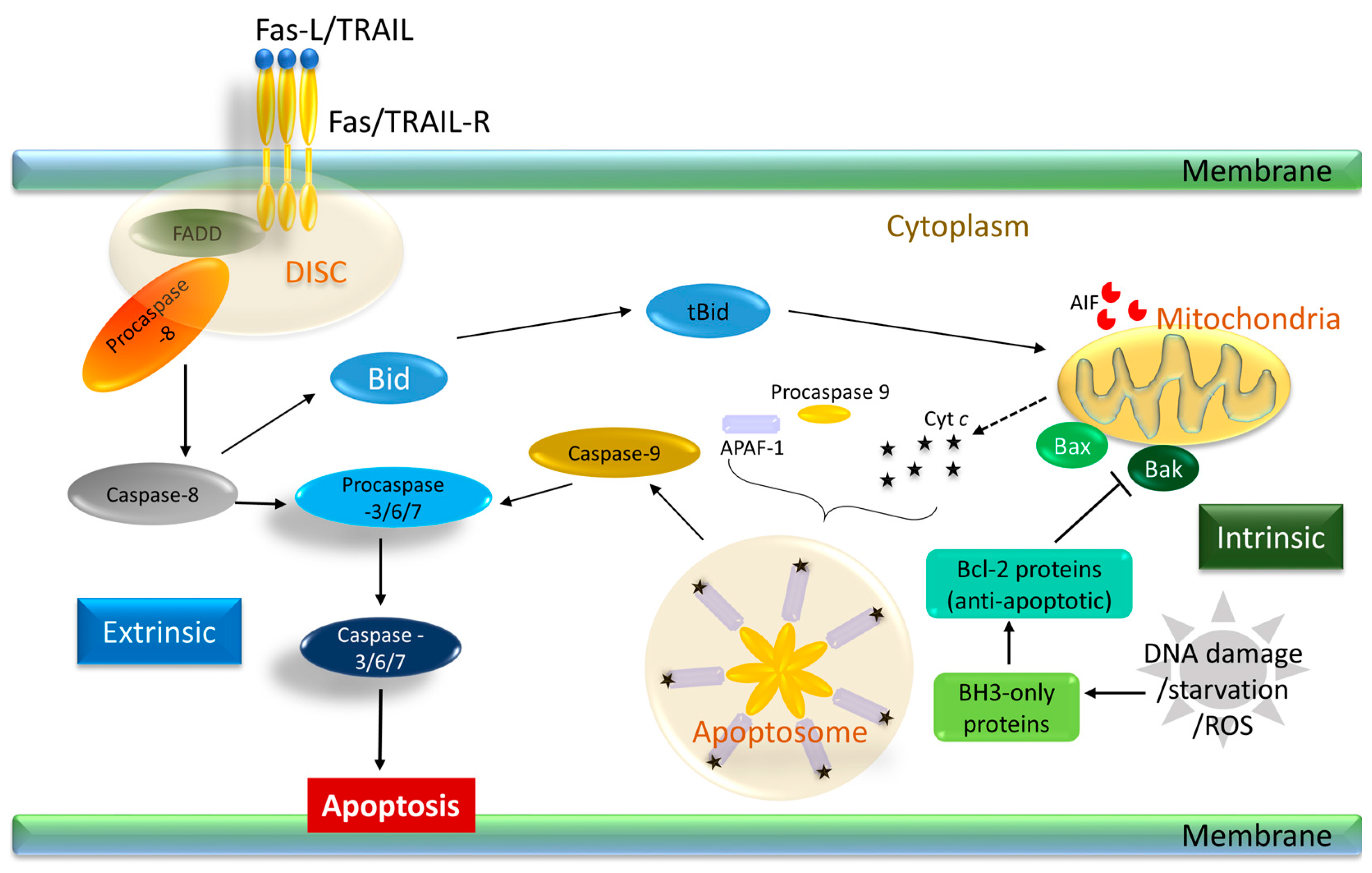
2. Apoptosis
2.1. What Is Apoptosis?
2.2. The Main Players of Apoptosis: Caspases
2.3. Types of Apoptosis
2.4. Apoptosis and Cancer
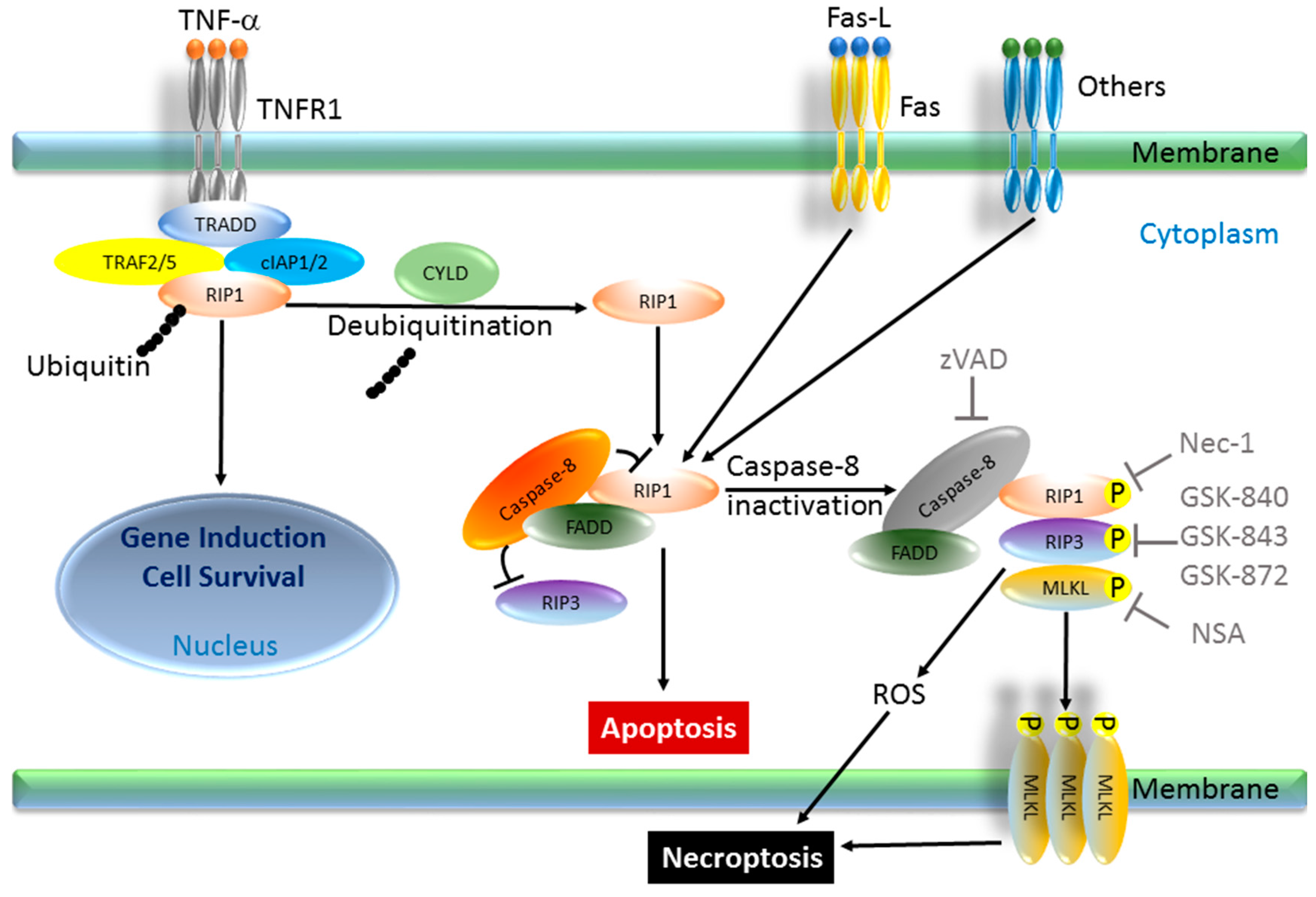
3. Necroptosis
3.1. Genesis of a Novel Concept “Necroptosis”
3.2. Regulation of Necroptosis Machinery
3.3. Downstream and Upstream of Necroptosis Pathway
4. Difference between Apoptosis and Necroptosis
4.1. Morphological Findings
4.2. Functional Findings
5. Exploiting Apoptosis and Necroptosis for Therapeutic Development
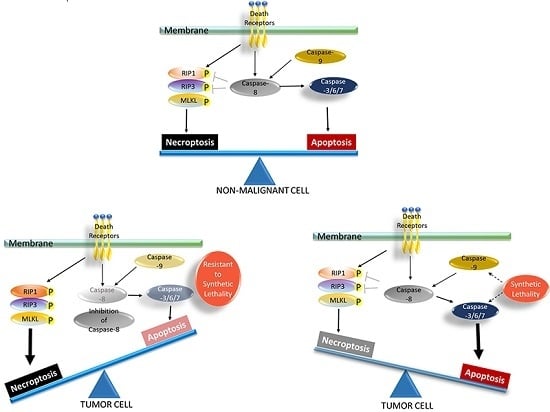
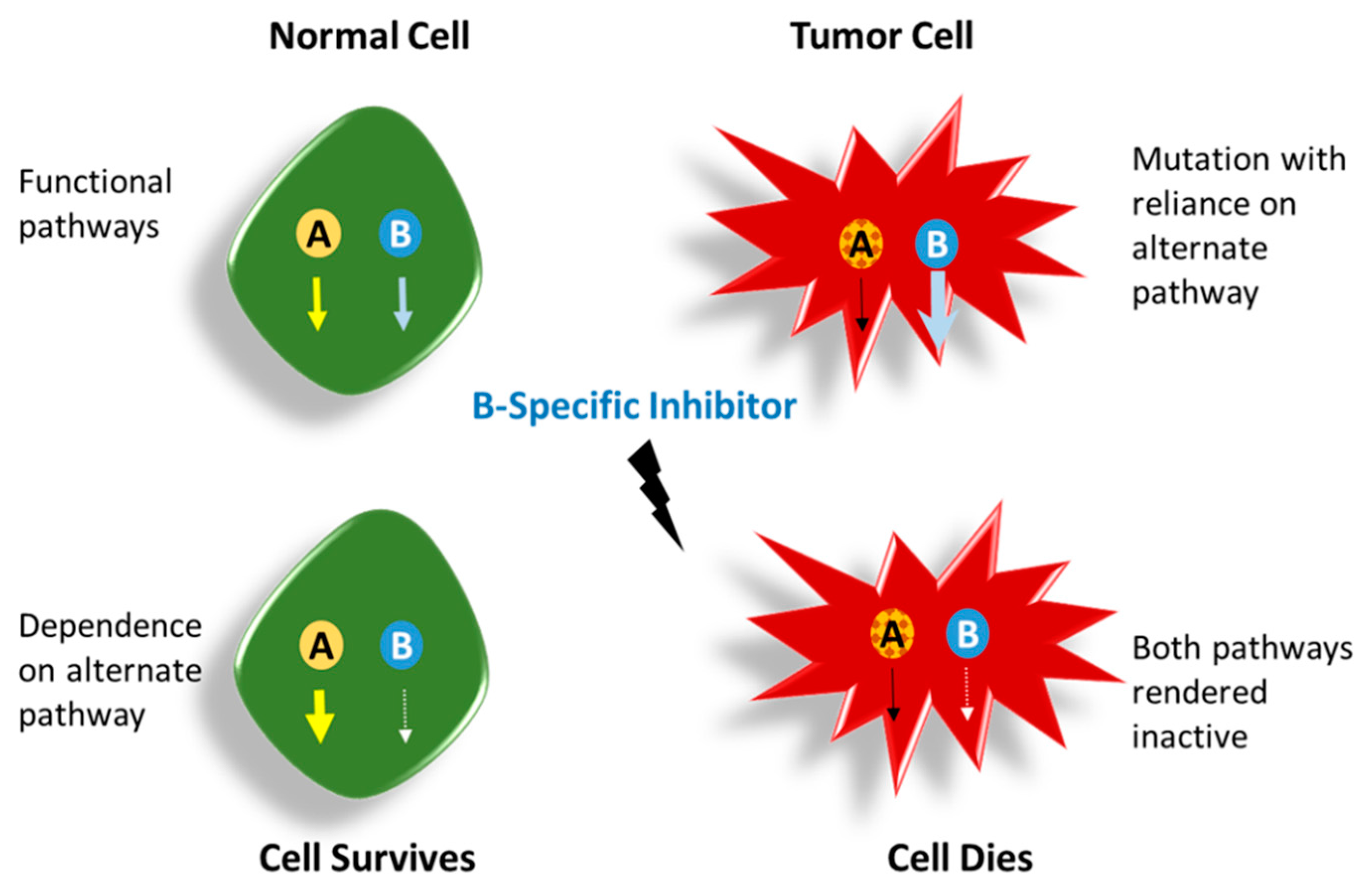
5.1. Synergism and Synthetic Lethality: Inducing Apoptosis in Cancer
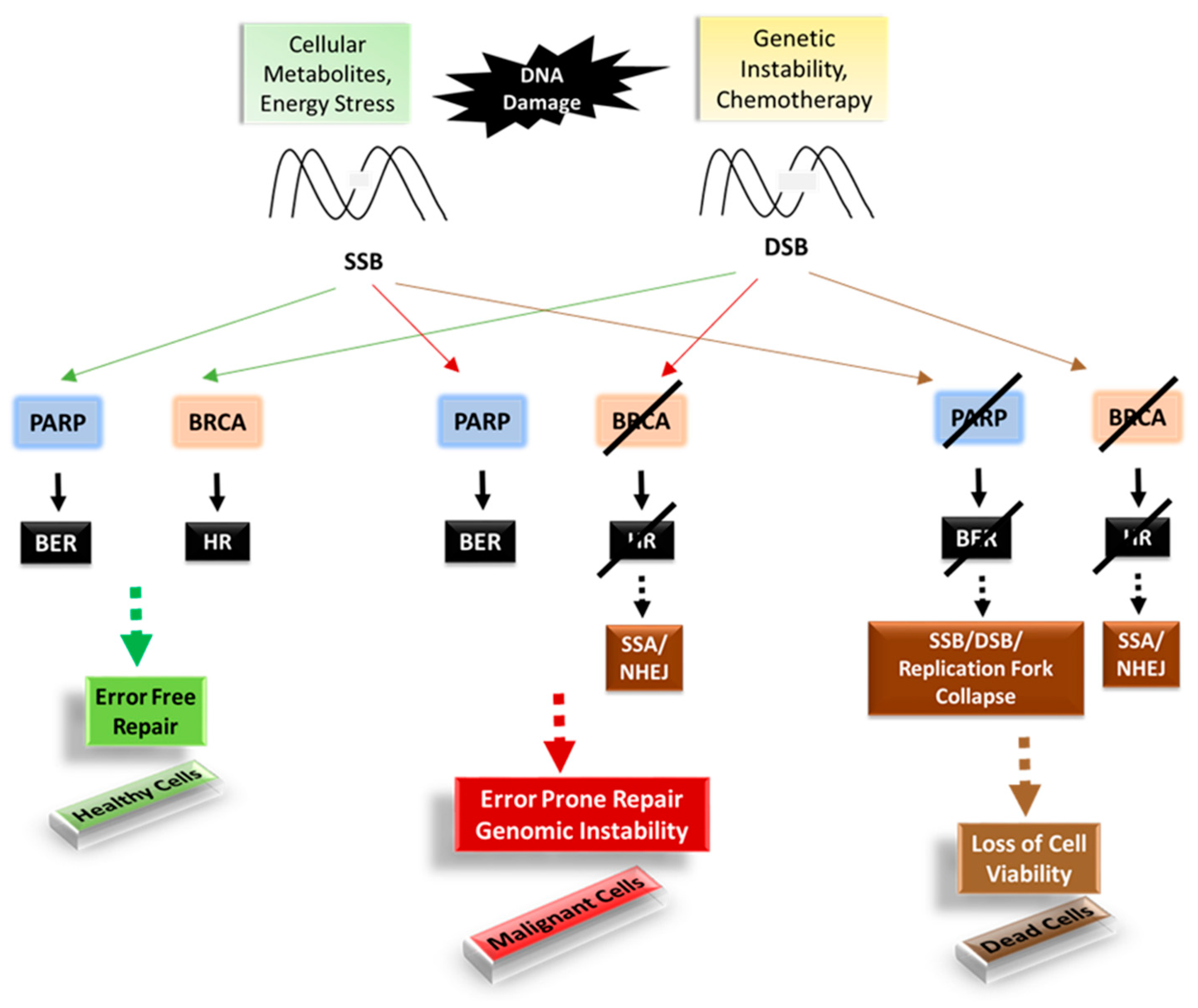
A. Poly (ADP-Ribose) Polymerase-1 (PARP1) and Breast Cancer Susceptibility Gene1/2 (BRCA1/2)
B. RAS/RAF and MEK Pathway
C. MK2, ATM/ATR and Chk1/2 Kinases
D. Avian Myelocytomatosis Viral Oncogene Cellular Homolog (MYC)
E. CREB Binding Protein (CBP) and p300
F. B-Cell Lymphoma 2 (Bcl-2) Small Molecule Inhibitors
G. Tumor Suppressor Gene, p53
5.2. Necroptosis as a Therapeutic Weapon for Apoptosis-Resistant Cancer
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mattson, M.P. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 2000, 1, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Ichim, G.; Tait, S.W. A fate worse than death: Apoptosis as an oncogenic process. Nat. Rev. Cancer 2016, 16, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Yu, J.; Zhang, L. Necroptosis: An alternative cell death program defending against cancer. Biochim. Biophys. Acta 2016, 1865, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Dillon, C.P.; Green, D.R. Molecular cell biology of apoptosis and necroptosis in cancer. Adv. Exp. Med. Biol. 2016, 930, 1–23. [Google Scholar] [PubMed]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Duque-Parra, J.E. Note on the origin and history of the term "apoptosis". Anat. Rec. B New Anat. 2005, 283, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Dexter, R.M.; Wyllie, A.H.; Raff, M.C. The Role of Apoptosis in Development, Tissue Homeostasis and Malignancy; Springer Science & Business Media: Dordecht, The Netherlands, 2012. [Google Scholar]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Hacker, G. The morphology of apoptosis. Cell Tissue Res. 2000, 301, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Savill, J.; Fadok, V. Corpse clearance defines the meaning of cell death. Nature 2000, 407, 784–788. [Google Scholar] [CrossRef] [PubMed]
- Thornberry, N.A.; Lazebnik, Y. Caspases: Enemies within. Science 1998, 281, 1312–1316. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.P.; Black, J.A.; Thomson, J.A.; Kim, E.E.; Griffith, J.P.; Navia, M.A.; Murcko, M.A.; Chambers, S.P.; Aldape, R.A.; Raybuck, S.A.; et al. Structure and mechanism of interleukin-1 β converting enzyme. Nature 1994, 370, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Pop, C.; Salvesen, G.S. Human caspases: Activation, specificity, and regulation. J. Biol. Chem. 2009, 284, 21777–21781. [Google Scholar] [CrossRef] [PubMed]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef] [PubMed]
- Dickens, L.S.; Powley, I.R.; Hughes, M.A.; MacFarlane, M. The "complexities" of life and death: Death receptor signalling platforms. Exp. Cell Res. 2012, 318, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Jost, P.J.; Grabow, S.; Gray, D.; McKenzie, M.D.; Nachbur, U.; Huang, D.C.; Bouillet, P.; Thomas, H.E.; Borner, C.; Silke, J.; et al. XIAP discriminates between type I and type II Fas-induced apoptosis. Nature 2009, 460, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Spencer, S.L.; Gaudet, S.; Albeck, J.G.; Burke, J.M.; Sorger, P.K. Non-genetic origins of cell-to-cell variability in trail-induced apoptosis. Nature 2009, 459, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Dean, E.J.; Ranson, M.; Blackhall, F.; Dive, C. X-linked inhibitor of apoptosis protein as a therapeutic target. Expert Opin. Ther. Targets 2007, 11, 1459–1471. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Fisher, J.C.; Dillon, C.P.; Kriwacki, R.W.; Kuwana, T.; Green, D.R. Mechanism of apoptosis induction by inhibition of the anti-apoptotic Bcl-2 proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 20327–20332. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Chipuk, J.E. Apoptosis: Stabbed in the Bax. Nature 2008, 455, 1047–1049. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Green, D.R. How do Bcl-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008, 18, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.; Green, D.R. Cell survival in tough times: The mitochondrial recovery plan. Cell Cycle 2010, 9, 4254–4255. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Cain, K.; Bratton, S.B.; Cohen, G.M. The apaf-1 apoptosome: A large caspase-activating complex. Biochimie 2002, 84, 203–214. [Google Scholar] [CrossRef]
- Degterev, A.; Boyce, M.; Yuan, J. A decade of caspases. Oncogene 2003, 22, 8543–8567. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.L.; MacFarlane, M. Targeting cell death signalling in cancer: Minimising "collateral damage". Br. J. Cancer 2016, 115, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, C.; Fulda, S.; Srinivasan, A.; Friesen, C.; Li, F.; Tomaselli, K.J.; Debatin, K.M.; Krammer, P.H.; Peter, M.E. Two CD95 (Apo-1/Fas) signaling pathways. EMBO J. 1998, 17, 1675–1687. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Budihardjo, I.; Zou, H.; Slaughter, C.; Wang, X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998, 94, 481–490. [Google Scholar] [CrossRef]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [PubMed]
- Joza, N.; Susin, S.A.; Daugas, E.; Stanford, W.L.; Cho, S.K.; Li, C.Y.; Sasaki, T.; Elia, A.J.; Cheng, H.Y.; Ravagnan, L.; et al. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature 2001, 410, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Norberg, E.; Orrenius, S.; Zhivotovsky, B. Mitochondrial regulation of cell death: Processing of apoptosis-inducing factor (AIF). Biochem. Biophys. Res. Commun. 2010, 396, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Castedo, M.; Perfettini, J.L.; Roumier, T.; Andreau, K.; Medema, R.; Kroemer, G. Cell death by mitotic catastrophe: A molecular definition. Oncogene 2004, 23, 2825–2837. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Letai, A.G. Diagnosing and exploiting cancer's addiction to blocks in apoptosis. Nat. Rev. Cancer 2008, 8, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y.; Cossman, J.; Jaffe, E.; Croce, C.M. Involvement of the Bcl-2 gene in human follicular lymphoma. Science 1985, 228, 1440–1443. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y.; Jaffe, E.; Cossman, J.; Gorham, J.; Nowell, P.C.; Croce, C.M. Clustering of breakpoints on chromosome 11 in human B-cell neoplasms with the t(11;14) chromosome translocation. Nature 1985, 315, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W.; Lin, A.W. Apoptosis in cancer. Carcinogenesis 2000, 21, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Yip, K.W.; Reed, J.C. Bcl-2 family proteins and cancer. Oncogene 2008, 27, 6398–6406. [Google Scholar] [CrossRef] [PubMed]
- Rampino, N.; Yamamoto, H.; Ionov, Y.; Li, Y.; Sawai, H.; Reed, J.C.; Perucho, M. Somatic frameshift mutations in the Bax gene in colon cancers of the microsatellite mutator phenotype. Science 1997, 275, 967–969. [Google Scholar] [CrossRef] [PubMed]
- Meijerink, J.P.; Mensink, E.J.; Wang, K.; Sedlak, T.W.; Sloetjes, A.W.; de Witte, T.; Waksman, G.; Korsmeyer, S.J. Hematopoietic malignancies demonstrate loss-of-function mutations of Bax. Blood 1998, 91, 2991–2997. [Google Scholar] [PubMed]
- Pierce, R.H.; Vail, M.E.; Ralph, L.; Campbell, J.S.; Fausto, N. Bcl-2 expression inhibits liver carcinogenesis and delays the development of proliferating foci. Am. J. Pathol. 2002, 160, 1555–1560. [Google Scholar] [CrossRef]
- Bai, L.; Ni, H.M.; Chen, X.; DiFrancesca, D.; Yin, X.M. Deletion of Bid impedes cell proliferation and hepatic carcinogenesis. Am. J. Pathol. 2005, 166, 1523–1532. [Google Scholar] [CrossRef]
- Labi, V.; Erlacher, M. How cell death shapes cancer. Cell Death Dis. 2015, 6, e1675. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Monack, D.M. Inflammasome adaptors and sensors: Intracellular regulators of infection and inflammation. Nat. Rev. Immunol. 2007, 7, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Peter, M.E. Programmed cell death: Apoptosis meets necrosis. Nature 2011, 471, 310–312. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kroemer, G. Necroptosis: A specialized pathway of programmed necrosis. Cell 2008, 135, 1161–1163. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Declercq, W.; Vanden Berghe, T.; Vandenabeele, P. RIP kinases at the crossroads of cell death and survival. Cell 2009, 138, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Oberst, A.; Dillon, C.P.; Weinlich, R.; Salvesen, G.S. RIPK-dependent necrosis and its regulation by caspases: A mystery in five acts. Mol. Cell 2011, 44, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Safa, A.R. Roles of c-FLIP in apoptosis, necroptosis, and autophagy. J. Carcinog. Mutagen. 2013, 6. [Google Scholar] [CrossRef] [PubMed]
- Vandenabeele, P.; Declercq, W.; Van Herreweghe, F.; Vanden Berghe, T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci. Signal. 2010, 3, re4. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.G.; Liu, Z.G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch'en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the caspase-8-FLIP(l) complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, W.J.; Upton, J.W.; Long, A.B.; Livingston-Rosanoff, D.; Daley-Bauer, L.P.; Hakem, R.; Caspary, T.; Mocarski, E.S. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 2011, 471, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Jouan-Lanhouet, S.; Riquet, F.; Duprez, L.; Vanden Berghe, T.; Takahashi, N.; Vandenabeele, P. Necroptosis, in vivo detection in experimental disease models. Semin. Cell Dev. Biol. 2014, 35, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Yang, Z.; Xie, L.; DeWitt, J.P.; Chen, Y. Cancer therapy in the necroptosis era. Cell Death Differ. 2016, 23, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Tabata, K.; Hamano, A.; Akihisa, T.; Suzuki, T. Kuguaglycoside c, a constituent of momordica charantia, induces caspase-independent cell death of neuroblastoma cells. Cancer Sci. 2012, 103, 2153–2158. [Google Scholar] [CrossRef] [PubMed]
- Nomura, M.; Ueno, A.; Saga, K.; Fukuzawa, M.; Kaneda, Y. Accumulation of cytosolic calcium induces necroptotic cell death in human neuroblastoma. Cancer Res. 2014, 74, 1056–1066. [Google Scholar] [CrossRef] [PubMed]
- Delavallee, L.; Cabon, L.; Galan-Malo, P.; Lorenzo, H.K.; Susin, S.A. AIF-mediated caspase-independent necroptosis: A new chance for targeted therapeutics. IUBMB Life 2011, 63, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhang, Y.; Cui, M.; Jin, L.; Wang, Y.; Lv, F.; Liu, Y.; Zheng, W.; Shang, H.; Zhang, J.; et al. Camkii is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat. Med. 2016, 22, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Iglehart, J.D.; Silver, D.P. Synthetic lethality—A new direction in cancer-drug development. N. Engl. J. Med. 2009, 361, 189–191. [Google Scholar] [CrossRef] [PubMed]
- Whitehurst, A.W.; Bodemann, B.O.; Cardenas, J.; Ferguson, D.; Girard, L.; Peyton, M.; Minna, J.D.; Michnoff, C.; Hao, W.; Roth, M.G.; et al. Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature 2007, 446, 815–819. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Lord, C.J.; Iorns, E.; Brough, R.; Swift, S.; Elliott, R.; Rayter, S.; Tutt, A.N.; Ashworth, A. A synthetic lethal sirna screen identifying genes mediating sensitivity to a PARP inhibitor. EMBO J. 2008, 27, 1368–1377. [Google Scholar] [CrossRef] [PubMed]
- Jerby-Arnon, L.; Pfetzer, N.; Waldman, Y.Y.; McGarry, L.; James, D.; Shanks, E.; Seashore-Ludlow, B.; Weinstock, A.; Geiger, T.; Clemons, P.A.; et al. Predicting cancer-specific vulnerability via data-driven detection of synthetic lethality. Cell 2014, 158, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in brca mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Venkitaraman, A.R. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 2002, 108, 171–182. [Google Scholar] [CrossRef]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of "brcaness" in sporadic cancers. Nat. Rev. Cancer 2004, 4, 814–819. [Google Scholar] [CrossRef] [PubMed]
- O′Donovan, P.J.; Livingston, D.M. BRCA1 and BRCA2: Breast/ovarian cancer susceptibility gene products and participants in DNA double-strand break repair. Carcinogenesis 2010, 31, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Wooster, R.; Weber, B.L. Breast and ovarian cancer. N. Engl. J. Med. 2003, 348, 2339–2347. [Google Scholar] [PubMed]
- Brown, J.S.; Kaye, S.B.; Yap, T.A. Parp inhibitors: The race is on. Br. J. Cancer 2016, 114, 713–715. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Ison, G.; McKee, A.E.; Zhang, H.; Tang, S.; Gwise, T.; Sridhara, R.; Lee, E.; Tzou, A.; Philip, R.; et al. FDA approval summary: Olaparib monotherapy in patients with deleterious germline BRCA-mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin. Cancer Res. 2015, 21, 4257–4261. [Google Scholar] [CrossRef] [PubMed]
- Ricks, T.K.; Chiu, H.J.; Ison, G.; Kim, G.; McKee, A.E.; Kluetz, P.; Pazdur, R. Successes and challenges of PARP inhibitors in cancer therapy. Front. Oncol. 2015, 5, 222. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [PubMed]
- She, Q.B.; Halilovic, E.; Ye, Q.; Zhen, W.; Shirasawa, S.; Sasazuki, T.; Solit, D.B.; Rosen, N. 4E-BP1 is a key effector of the oncogenic activation of the AKT and ERK signaling pathways that integrates their function in tumors. Cancer Cell 2010, 18, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O′Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated braf in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Janne, P.A.; Shaw, A.T.; Pereira, J.R.; Jeannin, G.; Vansteenkiste, J.; Barrios, C.; Franke, F.A.; Grinsted, L.; Zazulina, V.; Smith, P.; et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: A randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013, 14, 38–47. [Google Scholar] [CrossRef]
- Lin, L.; Asthana, S.; Chan, E.; Bandyopadhyay, S.; Martins, M.M.; Olivas, V.; Yan, J.J.; Pham, L.; Wang, M.M.; Bollag, G.; et al. Mapping the molecular determinants of BRAF oncogene dependence in human lung cancer. Proc. Natl. Acad. Sci. USA 2014, 111, E748–E757. [Google Scholar] [CrossRef] [PubMed]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF (v600e) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; Della Pelle, P.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of braf mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Bivona, T.G. The hippo effector YAP regulates the response of cancer cells to MAPK pathway inhibitors. Mol. Cell. Oncol. 2016, 3, e1021441. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Ren, X.; Wang, L.; Zhang, L.; Wu, X. Lung-cancer chemoprevention by induction of synthetic lethality in mutant KRAS premalignant cells in vitro and in vivo. Cancer Prev. Res. 2011, 4, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Lamba, S.; Russo, M.; Sun, C.; Lazzari, L.; Cancelliere, C.; Grernrum, W.; Lieftink, C.; Bernards, R.; di Nicolantonio, F.; Bardelli, A. RAF suppression synergizes with MEK inhibition in KRAS mutant cancer cells. Cell Rep. 2014, 8, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; Cheng, K.A.; Hata, A.N.; Faber, A.C.; Ebi, H.; Coffee, E.M.; Greninger, P.; Brown, R.D.; Godfrey, J.T.; Cohoon, T.J.; et al. Synthetic lethal interaction of combined Bcl-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell 2013, 23, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003, 3, 421–429. [Google Scholar] [CrossRef]
- Shiloh, Y. ATM and related protein kinases: Safeguarding genome integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [PubMed]
- Medema, R.H.; Macurek, L. Checkpoint control and cancer. Oncogene 2012, 31, 2601–2613. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lu, X. Live or let die: The cell's response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, H.C.; Aslanian, A.S.; Lees, J.A.; Yaffe, M.B. P53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 2007, 11, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Dietlein, F.; Kalb, B.; Jokic, M.; Noll, E.M.; Strong, A.; Tharun, L.; Ozretic, L.; Kunstlinger, H.; Kambartel, K.; Randerath, W.J.; et al. A synergistic interaction between Chk1- and MK2 inhibitors in KRAS-mutant cancer. Cell 2015, 162, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Engels, I.H.; Knee, D.A.; Nasoff, M.; Deveraux, Q.L.; Quon, K.C. Synthetic lethal targeting of MYC by activation of the DR5 death receptor pathway. Cancer Cell 2004, 5, 501–512. [Google Scholar] [CrossRef]
- Goga, A.; Yang, D.; Tward, A.D.; Morgan, D.O.; Bishop, J.M. Inhibition of CDK1 as a potential therapy for tumors over-expressing MYC. Nat. Med. 2007, 13, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, D.; Kusdra, L.; Huskey, N.E.; Chandriani, S.; Lenburg, M.E.; Gonzalez-Angulo, A.M.; Creasman, K.J.; Bazarov, A.V.; Smyth, J.W.; Davis, S.E.; et al. MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J. Exp. Med. 2012, 209, 679–696. [Google Scholar] [CrossRef] [PubMed]
- Pourdehnad, M.; Truitt, M.L.; Siddiqi, I.N.; Ducker, G.S.; Shokat, K.M.; Ruggero, D. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in MYC-driven cancers. Proc. Natl. Acad. Sci. USA 2013, 110, 11988–11993. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Liu, H.; Goga, A.; Kim, S.; Yuneva, M.; Bishop, J.M. Therapeutic potential of a synthetic lethal interaction between the MYC proto-oncogene and inhibition of aurora-B kinase. Proc. Natl. Acad. Sci. USA 2010, 107, 13836–13841. [Google Scholar] [CrossRef] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-MYC. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Murga, M.; Campaner, S.; Lopez-Contreras, A.J.; Toledo, L.I.; Soria, R.; Montana, M.F.; D′Artista, L.; Schleker, T.; Guerra, C.; Garcia, E.; et al. Exploiting oncogene-induced replicative stress for the selective killing of MYC-driven tumors. Nat. Struct. Mol. Biol. 2011, 18, 1331–1335. [Google Scholar] [CrossRef] [PubMed]
- Ferrao, P.T.; Bukczynska, E.P.; Johnstone, R.W.; McArthur, G.A. Efficacy of Chk inhibitors as single agents in MYC-driven lymphoma cells. Oncogene 2012, 31, 1661–1672. [Google Scholar] [CrossRef] [PubMed]
- Kelly, G.L.; Grabow, S.; Glaser, S.P.; Fitzsimmons, L.; Aubrey, B.J.; Okamoto, T.; Valente, L.J.; Robati, M.; Tai, L.; Fairlie, W.D.; et al. Targeting of MCL-1 kills MYC-driven mouse and human lymphomas even when they bear mutations in p53. Genes Dev. 2014, 28, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.S.; Redkar, S.; Bearss, D.; Wierda, W.G.; Gandhi, V. Pim kinase inhibitor, SGI-1776, induces apoptosis in chronic lymphocytic leukemia cells. Blood 2009, 114, 4150–4157. [Google Scholar] [CrossRef] [PubMed]
- Asano, J.; Nakano, A.; Oda, A.; Amou, H.; Hiasa, M.; Takeuchi, K.; Miki, H.; Nakamura, S.; Harada, T.; Fujii, S.; et al. The serine/threonine kinase Pim-2 is a novel anti-apoptotic mediator in myeloma cells. Leukemia 2011, 25, 1182–1188. [Google Scholar] [CrossRef] [PubMed]
- Oike, T.; Ogiwara, H.; Nakano, T.; Kohno, T. Proposal for a synthetic lethality therapy using the paralog dependence of cancer cells-response. Cancer Res. 2014, 74, 4948–4949. [Google Scholar] [CrossRef] [PubMed]
- Ogiwara, H.; Sasaki, M.; Mitachi, T.; Oike, T.; Higuchi, S.; Tominaga, Y.; Kohno, T. Targeting p300 addiction in CBP-deficient cancers causes synthetic lethality by apoptotic cell death due to abrogation of MYC expression. Cancer Discov. 2016, 6, 430–445. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, R.; Giri, A.; Goustin, A.S. Small-molecule inhibitors of Bcl-2 family proteins as therapeutic agents in cancer. Recent Pat. Anticancer Drug Discov. 2008, 3, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.M.; Thomas, D.; Corces-Zimmerman, M.R.; Xavy, S.; Rastogi, S.; Hong, W.J.; Zhao, F.; Medeiros, B.C.; Tyvoll, D.A.; Majeti, R. Isocitrate dehydrogenase 1 and 2 mutations induce Bcl-2 dependence in acute myeloid leukemia. Nat. Med. 2015, 21, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Fridman, J.S.; Lowe, S.W. Control of apoptosis by p53. Oncogene 2003, 22, 9030–9040. [Google Scholar] [CrossRef] [PubMed]
- Leszczynska, K.B.; Foskolou, I.P.; Abraham, A.G.; Anbalagan, S.; Tellier, C.; Haider, S.; Span, P.N.; O′Neill, E.E.; Buffa, F.M.; Hammond, E.M. Hypoxia-induced p53 modulates both apoptosis and radiosensitivity via AKT. J. Clin. Investig. 2015, 125, 2385–2398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Fan, S.; Eastman, A.; Worland, P.J.; Sausville, E.A.; O'Connor, P.M. UCN-01: A potent abrogator of G2 checkpoint function in cancer cells with disrupted p53. J. Natl. Cancer Inst. 1996, 88, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Sur, S.; Pagliarini, R.; Bunz, F.; Rago, C.; Diaz, L.A., Jr.; Kinzler, K.W.; Vogelstein, B.; Papadopoulos, N. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc. Natl. Acad. Sci. USA 2009, 106, 3964–3969. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, J.; Booher, R.N.; Kraker, A.; Lawrence, T.; Leopold, W.R.; Sun, Y. Radiosensitization of p53 mutant cells by PD0166285, a novel G2 checkpoint abrogator. Cancer Res. 2001, 61, 8211–8217. [Google Scholar] [PubMed]
- Trinidad, A.G.; Muller, P.A.; Cuellar, J.; Klejnot, M.; Nobis, M.; Valpuesta, J.M.; Vousden, K.H. Interaction of p53 with the CCT complex promotes protein folding and wild-type p53 activity. Mol. Cell 2013, 50, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Mouratidis, P.X.; Rivens, I.; Ter Haar, G. A study of thermal dose-induced autophagy, apoptosis and necroptosis in colon cancer cells. Int. J. Hyperthermia. 2015, 31, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.V.; Seibert, S.; Walch-Ruckheim, B.; Vicinus, B.; Kamionka, E.M.; Pahne-Zeppenfeld, J.; Solomayer, E.F.; Kim, Y.J.; Bohle, R.M.; Smola, S. RIPK3 expression in cervical cancer cells is required for polyic-induced necroptosis, IL-1α release, and efficient paracrine dendritic cell activation. Oncotarget 2015, 6, 8635–8647. [Google Scholar] [CrossRef] [PubMed]
- Meng, M.B.; Wang, H.H.; Cui, Y.L.; Wu, Z.Q.; Shi, Y.Y.; Zaorsky, N.G.; Deng, L.; Yuan, Z.Y.; Lu, Y.; Wang, P. Necroptosis in tumorigenesis, activation of anti-tumor immunity, and cancer therapy. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features | Apoptosis | Necroptosis |
|---|---|---|
| Membrane blebbing | Yes | No |
| Cytoplasmic shrinkage | Yes | No |
| Apoptotic body | Yes | No |
| Nuclear fragmentation | Yes | No |
| Chromatin condensation | Yes | No |
| Swollen organelle and mitochondria | No | Yes |
| Membrane permeabilization | No | Yes |
| Caspase activation | Yes | No |
| RIP1/RIP3/MLKL activation | No | Yes |
| ROS production | Yes | Yes |
| ATP depletion | No | Yes |
| ATP increase | Yes | No |
| Executioner of cell death | caspase-3, caspase-7 | ROS, homotrimerized MLKL |
| Selective inhibitor of cell death | zVAD (caspase inhibitor) | Nec-1 (RIP1 inhibitor) |
| GSK-840/-843/-872 (RIP3 inhibitor) | ||
| NSA (MLKL inhibitor) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dasgupta, A.; Nomura, M.; Shuck, R.; Yustein, J. Cancer’s Achilles’ Heel: Apoptosis and Necroptosis to the Rescue. Int. J. Mol. Sci. 2017, 18, 23. https://doi.org/10.3390/ijms18010023
Dasgupta A, Nomura M, Shuck R, Yustein J. Cancer’s Achilles’ Heel: Apoptosis and Necroptosis to the Rescue. International Journal of Molecular Sciences. 2017; 18(1):23. https://doi.org/10.3390/ijms18010023
Chicago/Turabian StyleDasgupta, Atreyi, Motonari Nomura, Ryan Shuck, and Jason Yustein. 2017. "Cancer’s Achilles’ Heel: Apoptosis and Necroptosis to the Rescue" International Journal of Molecular Sciences 18, no. 1: 23. https://doi.org/10.3390/ijms18010023
APA StyleDasgupta, A., Nomura, M., Shuck, R., & Yustein, J. (2017). Cancer’s Achilles’ Heel: Apoptosis and Necroptosis to the Rescue. International Journal of Molecular Sciences, 18(1), 23. https://doi.org/10.3390/ijms18010023