
Subcellular Trafficking of Mammalian Lysosomal Proteins: An Extended View

Abstract
:1. Introduction
2. Subcellular Trafficking of Lysosomal Transmembrane Proteins
2.1. Classical Sorting Pathways
2.2. Targeting of Lysosomal Transmembrane Proteins to the Lysosome by Non-Conventional Mechanisms
2.2.1. Atypical Sorting Motifs Identified in Lysosomal Transmembrane Proteins
2.2.2. Sorting Determinants Located in the Luminal Domain of Lysosomal Transmembrane Proteins
2.2.3. Impact of Post-Translational Lipid-Modifications on Lysosomal Membrane Protein Trafficking
2.2.4. Transmembrane Domain(S)-Dependent Sorting of Lysosomal Membrane Proteins
2.2.5. Alternatives to the Clathrin-Coated Carriers
3. Subcellular Trafficking of Lysosomal Hydrolases
3.1. Mannose 6-Phosphate-Dependent Trafficking
3.2. Mannose 6-Phosphate-Independent Sorting Receptors
4. Acquisition of Resident Lysosomal Proteins from the Cytosol
5. New Directions
5.1. Is There a Biosynthetic Trafficking Route to the Endolysosomes That Does Not Require Passage through the Golgi Apparatus?
5.2. Acquisition of the Lysosomal Proteome of Another Cell
6. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- De Duve, C.; Pressman, B.C.; Gianetto, R.; Wattiaux, R.; Appelmans, F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J. 1955, 60, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, D.D.; Adesnik, M. Christian de Duve: Explorer of the cell who discovered new organelles by using a centrifuge. Proc. Natl. Acad. Sci. USA 2013, 110, 13234–13235. [Google Scholar] [CrossRef] [PubMed]
- Schröder, B.; Wrocklage, C.; Pan, C.; Jäger, R.; Kösters, B.; Schäfer, H.; Elsässer, H.-P.; Mann, M.; Hasilik, A. Integral and associated lysosomal membrane proteins. Traffic (Cph. Den.) 2007, 8, 1676–1686. [Google Scholar] [CrossRef] [PubMed]
- Callahan, J.W.; Bagshaw, R.D.; Mahuran, D.J. The integral membrane of lysosomes: Its proteins and their roles in disease. J. Proteom. 2009, 72, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Della Valle, M.C.; Sleat, D.E.; Zheng, H.; Moore, D.F.; Jadot, M.; Lobel, P. Classification of subcellular location by comparative proteomic analysis of native and density-shifted lysosomes. Mol. Cell. Proteom. MCP 2011, 10, M110.006403. [Google Scholar] [CrossRef] [PubMed]
- Sleat, D.E.; Sun, P.; Wiseman, J.A.; Huang, L.; El-Banna, M.; Zheng, H.; Moore, D.F.; Lobel, P. Extending the mannose 6-phosphate glycoproteome by high resolution/accuracy mass spectrometry analysis of control and acid phosphatase 5-deficient mice. Mol. Cell. Proteom. MCP 2013, 12, 1806–1817. [Google Scholar] [CrossRef] [PubMed]
- Chapel, A.; Kieffer-Jaquinod, S.; Sagné, C.; Verdon, Q.; Ivaldi, C.; Mellal, M.; Thirion, J.; Jadot, M.; Bruley, C.; Garin, J.; et al. An extended proteome map of the lysosomal membrane reveals novel potential transporters. Mol. Cell. Proteom. MCP 2013, 12, 1572–1588. [Google Scholar] [CrossRef] [PubMed]
- Ballabio, A.; Gieselmann, V. Lysosomal disorders: From storage to cellular damage. Biochim. Biophys. Acta 2009, 1793, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; Boland, B.; van der Spoel, A.C. The cell biology of disease: Lysosomal storage disorders: The cellular impact of lysosomal dysfunction. J. Cell Biol. 2012, 199, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Dahms, N.M.; Kornfeld, S. Mannose 6-phosphate receptors: New twists in the tale. Nat. Rev. Mol. Cell Biol. 2003, 4, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, J.S.; Traub, L.M. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 2003, 72, 395–447. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.S. Adaptable adaptors for coated vesicles. Trends Cell Biol. 2004, 14, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Braulke, T.; Bonifacino, J.S. Sorting of lysosomal proteins. Biochim. Biophys. Acta 2009, 1793, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.S. Forty years of Clathrin-coated vesicles. Traffic (Cph. Den.) 2015, 16, 1210–1238. [Google Scholar] [CrossRef] [PubMed]
- Kollmann, K.; Pohl, S.; Marschner, K.; Encarnação, M.; Sakwa, I.; Tiede, S.; Poorthuis, B.J.; Lübke, T.; Müller-Loennies, S.; Storch, S.; et al. Mannose phosphorylation in health and disease. Eur. J. Cell Biol. 2010, 89, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J.; Irving, C.; Borner, G.H.H. Adaptor protein complexes AP-4 and AP-5: New players in endosomal trafficking and progressive spastic paraplegia. Traffic (Cph. Den.) 2013, 14, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Saftig, P.; Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J.; Edgar, J.R.; Esteves, T.; Darios, F.; Madeo, M.; Chang, J.; Roda, R.H.; Dürr, A.; Anheim, M.; Gellera, C.; et al. Loss of AP-5 results in accumulation of aberrant endolysosomes: Defining a new type of lysosomal storage disease. Hum. Mol. Genet. 2015, 24, 4984–4996. [Google Scholar] [CrossRef] [PubMed]
- Cherqui, S.; Kalatzis, V.; Trugnan, G.; Antignac, C. The targeting of cystinosin to the lysosomal membrane requires a tyrosine-based signal and a novel sorting motif. J. Biol. Chem. 2001, 276, 13314–13321. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewska, Z.; Névo, N.; Thomas, L.; Bailleux, A.; Chauvet, V.; Benmerah, A.; Antignac, C. Lysosomal targeting of cystinosin requires AP-3. Traffic (Cph. Den.) 2015, 16, 712–726. [Google Scholar] [CrossRef] [PubMed]
- Pryor, P.R.; Jackson, L.; Gray, S.R.; Edeling, M.A.; Thompson, A.; Sanderson, C.M.; Evans, P.R.; Owen, D.J.; Luzio, J.P. Molecular basis for the sorting of the SNARE VAMP7 into endocytic clathrin-coated vesicles by the ArfGAP Hrb. Cell 2008, 134, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Kent, H.M.; Evans, P.R.; Schäfer, I.B.; Gray, S.R.; Sanderson, C.M.; Luzio, J.P.; Peden, A.A.; Owen, D.J. Structural basis of the intracellular sorting of the SNARE VAMP7 by the AP3 adaptor complex. Dev. Cell 2012, 22, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Arca, S.; Rudge, R.; Vacca, M.; Raposo, G.; Camonis, J.; Proux-Gillardeaux, V.; Daviet, L.; Formstecher, E.; Hamburger, A.; Filippini, F.; et al. A dual mechanism controlling the localization and function of exocytic v-SNAREs. Proc. Natl. Acad. Sci. USA 2003, 100, 9011–9016. [Google Scholar] [CrossRef] [PubMed]
- Pak, Y.; Glowacka, W.K.; Bruce, M.C.; Pham, N.; Rotin, D. Transport of LAPTM5 to lysosomes requires association with the ubiquitin ligase Nedd4, but not LAPTM5 ubiquitination. J. Cell Biol. 2006, 175, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Milkereit, R.; Rotin, D. A role for the ubiquitin ligase Nedd4 in membrane sorting of LAPTM4 proteins. PLoS ONE 2011, 6, e27478. [Google Scholar] [CrossRef] [PubMed]
- Busch, J.I.; Unger, T.L.; Jain, N.; Tyler Skrinak, R.; Charan, R.A.; Chen-Plotkin, A.S. Increased expression of the frontotemporal dementia risk factor TMEM106B causes C9orf72-dependent alterations in lysosomes. Hum. Mol. Genet. 2016. [Google Scholar] [CrossRef] [PubMed]
- Storch, S.; Pohl, S.; Braulke, T. A dileucine motif and a cluster of acidic amino acids in the second cytoplasmic domain of the batten disease-related CLN3 protein are required for efficient lysosomal targeting. J. Biol. Chem. 2004, 279, 53625–53634. [Google Scholar] [CrossRef] [PubMed]
- Kyttälä, A.; Ihrke, G.; Vesa, J.; Schell, M.J.; Luzio, J.P. Two motifs target Batten disease protein CLN3 to lysosomes in transfected nonneuronal and neuronal cells. Mol. Biol. Cell 2004, 15, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, A.M.; Rademakers, R. What we know about TMEM106B in neurodegeneration. Acta Neuropathol. 2016, 132, 639–651. [Google Scholar] [CrossRef] [PubMed]
- Cárcel-Trullols, J.; Kovács, A.D.; Pearce, D.A. Cell biology of the NCL proteins: What they do and don’t do. Biochim. Biophys. Acta 2015, 1852, 2242–2255. [Google Scholar] [CrossRef] [PubMed]
- Sleat, D.E.; Gedvilaite, E.; Zhang, Y.; Lobel, P.; Xing, J. Analysis of large-scale whole exome sequencing data to determine the prevalence of genetically-distinct forms of neuronal ceroid lipofuscinosis. Gene 2016, 593, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Kyttälä, A.; Yliannala, K.; Schu, P.; Jalanko, A.; Luzio, J.P. AP-1 and AP-3 facilitate lysosomal targeting of Batten disease protein CLN3 via its dileucine motif. J. Biol. Chem. 2005, 280, 10277–10283. [Google Scholar] [CrossRef] [PubMed]
- Piccirillo, R.; Palmisano, I.; Innamorati, G.; Bagnato, P.; Altimare, D.; Schiaffino, M.V. An unconventional dileucine-based motif and a novel cytosolic motif are required for the lysosomal and melanosomal targeting of OA1. J. Cell Sci. 2006, 119, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Liapis, A.; Chen, F.W.; Davies, J.P.; Wang, R.; Ioannou, Y.A. MLN64 transport to the late endosome is regulated by binding to 14-3-3 via a non-canonical binding site. PLoS ONE 2012, 7, e34424. [Google Scholar] [CrossRef] [PubMed]
- Muslin, A.J.; Xing, H. 14-3-3 proteins: Regulation of subcellular localization by molecular interference. Cell Signal. 2000, 12, 703–709. [Google Scholar] [CrossRef]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.J.; Daut, J.; Schwappach, B. Membrane proteins as 14-3-3 clients in functional regulation and intracellular transport. Physiology (Bethesda) 2011, 26, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Efendiev, R.; Chen, Z.; Krmar, R.T.; Uhles, S.; Katz, A.I.; Pedemonte, C.H.; Bertorello, A.M. The 14-3-3 protein translates the NA+,K+-ATPase α1-subunit phosphorylation signal into binding and activation of phosphoinositide 3-kinase during endocytosis. J. Biol. Chem. 2005, 280, 16272–16277. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liu, P.; Dwyer, N.K.; Christenson, L.K.; Fujimoto, T.; Martinez, F.; Comly, M.; Hanover, J.A.; Blanchette-Mackie, E.J.; Strauss, J.F. MLN64 mediates mobilization of lysosomal cholesterol to steroidogenic mitochondria. J. Biol. Chem. 2002, 277, 33300–33310. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.M.; Fellerer, K.; Schwenk, B.M.; Kuhn, P.-H.; Kremmer, E.; Edbauer, D.; Capell, A.; Haass, C. Membrane orientation and subcellular localization of transmembrane protein 106B (TMEM106B), a major risk factor for frontotemporal lobar degeneration. J. Biol. Chem. 2012, 287, 19355–19365. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, J.R.; Yamazaki, Y.; Palmer, R.H. Tumour-associated mutations of PA-TM-RING ubiquitin ligases RNF167/RNF13 identify the PA domain as a determinant for endosomal localization. Biochem. J. 2014, 459, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Hofmann, K. The protease-associated domain: A homology domain associated with multiple classes of proteases. Trends Biochem. Sci. 2001, 26, 147–148. [Google Scholar] [CrossRef]
- Luo, F.; Fong, Y.H.; Zeng, Y.; Shen, J.; Jiang, L.; Wong, K.-B. How vacuolar sorting receptor proteins interact with their cargo proteins: Crystal structures of apo and cargo-bound forms of the protease-associated domain from an Arabidopsis vacuolar sorting receptor. Plant Cell 2014, 26, 3693–3708. [Google Scholar] [CrossRef] [PubMed]
- Storch, S.; Pohl, S.; Quitsch, A.; Falley, K.; Braulke, T. C-terminal prenylation of the CLN3 membrane glycoprotein is required for efficient endosomal sorting to lysosomes. Traffic (Cph. Den.) 2007, 8, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Vergarajauregui, S.; Puertollano, R. Two di-leucine motifs regulate trafficking of mucolipin-1 to lysosomes. Traffic (Cph. Den.) 2006, 7, 337–353. [Google Scholar] [CrossRef] [PubMed]
- Miedel, M.T.; Weixel, K.M.; Bruns, J.R.; Traub, L.M.; Weisz, O.A. Posttranslational cleavage and adaptor protein complex-dependent trafficking of mucolipin-1. J. Biol. Chem. 2006, 281, 12751–12759. [Google Scholar] [CrossRef] [PubMed]
- Flannery, A.R.; Czibener, C.; Andrews, N.W. Palmitoylation-dependent association with CD63 targets the Ca2+ sensor synaptotagmin VII to lysosomes. J. Cell Biol. 2010, 191, 599–613. [Google Scholar] [CrossRef] [PubMed]
- Cosson, P.; Perrin, J.; Bonifacino, J.S. Anchors aweigh: Protein localization and transport mediated by transmembrane domains. Trends Cell Biol. 2013, 23, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Kiss, K.; Kucsma, N.; Brozik, A.; Tusnady, G.E.; Bergam, P.; van Niel, G.; Szakacs, G. Role of the N-terminal transmembrane domain in the endo-lysosomal targeting and function of the human ABCB6 protein. Biochem. J. 2015, 467, 127–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demirel, O.; Bangert, I.; Tampé, R.; Abele, R. Tuning the cellular trafficking of the lysosomal peptide transporter TAPL by its N-terminal domain. Traffic (Cph. Den.) 2010, 11, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Tseng, L.T.-L.; Lin, C.-L.; Tzen, K.-Y.; Chang, S.C.; Chang, M.-F. LMBD1 protein serves as a specific adaptor for insulin receptor internalization. J. Biol. Chem. 2013, 288, 32424–32432. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, K.; Okamoto, T.; Morita, M.; Imanaka, T. Translocation of the ABC transporter ABCD4 from the endoplasmic reticulum to lysosomes requires the escort protein LMBD1. Sci. Rep. 2016, 6, 30183. [Google Scholar] [CrossRef] [PubMed]
- Milkereit, R.; Persaud, A.; Vanoaica, L.; Guetg, A.; Verrey, F.; Rotin, D. LAPTM4b recruits the LAT1–4F2hc Leu transporter to lysosomes and promotes mTORC1 activation. Nat. Commun. 2015, 6, 7250. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.A.; Fukuda, M. Accumulation of membrane glycoproteins in lysosomes requires a tyrosine residue at a particular position in the cytoplasmic tail. J. Cell Biol. 1990, 111, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Höning, S.; Griffith, J.; Geuze, H.J.; Hunziker, W. The tyrosine-based lysosomal targeting signal in lamp-1 mediates sorting into Golgi-derived clathrin-coated vesicles. EMBO J. 1996, 15, 5230–5239. [Google Scholar] [PubMed]
- Karlsson, K.; Carlsson, S.R. Sorting of lysosomal membrane glycoproteins lamp-1 and lamp-2 into vesicles distinct from mannose 6-phosphate receptor/gamma-adaptin vesicles at the trans-Golgi network. J. Biol. Chem. 1998, 273, 18966–18973. [Google Scholar] [CrossRef] [PubMed]
- Pols, M.S.; van Meel, E.; Oorschot, V.; ten Brink, C.; Fukuda, M.; Swetha, M.G.; Mayor, S.; Klumperman, J. hVps41 and VAMP7 function in direct TGN to late endosome transport of lysosomal membrane proteins. Nat. Commun. 2013, 4, 1361. [Google Scholar] [CrossRef] [PubMed]
- Pols, M.S.; Klumperman, J. Trafficking and function of the tetraspanin CD63. Exp. Cell Res. 2009, 315, 1584–1592. [Google Scholar] [CrossRef] [PubMed]
- Varki, A.; Kornfeld, S. P-type lectins. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009. [Google Scholar]
- Kim, J.-J.P.; Olson, L.J.; Dahms, N.M. Carbohydrate recognition by the mannose-6-phosphate receptors. Curr. Opin. Struct. Biol. 2009, 19, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Pohl, S.; Marschner, K.; Storch, S.; Braulke, T. Glycosylation- and phosphorylation-dependent intracellular transport of lysosomal hydrolases. Biol. Chem. 2009, 390, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Kornfeld, S.; Sly, W.S. I-cell disease and pseudo-Hurler polydystrophy: Disorders of lysosomal enzyme phosphorylation and localization. In The Metabolic and Molecular Bases of Inherited Disease; Schriver, C.R., Baudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New-York, NY, USA, 2000. [Google Scholar]
- Leroy, J.G.; Cathey, S.; Friez, M.J. Mucolipidosis II. In GeneReviews®; Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J., Bird, T.D., Ledbetter, N., Mefford, H.C., Smith, R.J., et al., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Waheed, A.; Pohlmann, R.; Hasilik, A.; von Figura, K.; van Elsen, A.; Leroy, J.G. Deficiency of UDP-N-acetylglucosamine: Lysosomal enzyme N-acetylglucosamine-1-phosphotransferase in organs of I-cell patients. Biochem. Biophys. Res. Commun. 1982, 105, 1052–1058. [Google Scholar] [CrossRef]
- Owada, M.; Neufeld, E.F. Is there a mechanism for introducing acid hydrolases into liver lysosomes that is independent of mannose 6-phosphate recognition? Evidence from I-cell disease. Biochem. Biophys. Res. Commun. 1982, 105, 814–820. [Google Scholar] [CrossRef]
- Little, L.; Alcouloumre, M.; Drotar, A.M.; Herman, S.; Robertson, R.; Yeh, R.Y.; Miller, A.L. Properties of N-acetylglucosamine 1-phosphotransferase from human lymphoblasts. Biochem. J. 1987, 248, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Glickman, J.N.; Kornfeld, S. Mannose 6-phosphate-independent targeting of lysosomal enzymes in I-cell disease B lymphoblasts. J. Cell Biol. 1993, 123, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Boonen, M.; van Meel, E.; Oorschot, V.; Klumperman, J.; Kornfeld, S. Vacuolization of mucolipidosis type II mouse exocrine gland cells represents accumulation of autolysosomes. Mol. Biol. Cell 2011, 22, 1135–1147. [Google Scholar] [CrossRef] [PubMed]
- van Meel, E.; Boonen, M.; Zhao, H.; Oorschot, V.; Ross, F.P.; Kornfeld, S.; Klumperman, J. Disruption of the Man-6-P targeting pathway in mice impairs osteoclast secretory lysosome biogenesis. Traffic (Cph. Den.) 2011, 12, 912–924. [Google Scholar] [CrossRef] [PubMed]
- Kollmann, K.; Pestka, J.M.; Kühn, S.C.; Schöne, E.; Schweizer, M.; Karkmann, K.; Otomo, T.; Catala-Lehnen, P.; Failla, A.V.; Marshall, R.P.; et al. Decreased bone formation and increased osteoclastogenesis cause bone loss in mucolipidosis II. EMBO Mol. Med. 2013, 5, 1871–1886. [Google Scholar] [CrossRef] [PubMed]
- Kollmann, K.; Damme, M.; Markmann, S.; Morelle, W.; Schweizer, M.; Hermans-Borgmeyer, I.; Röchert, A.K.; Pohl, S.; Lübke, T.; Michalski, J.-C.; et al. Lysosomal dysfunction causes neurodegeneration in mucolipidosis II “knock-in” mice. Brain J. Neurol. 2012, 135, 2661–2675. [Google Scholar] [CrossRef] [PubMed]
- Idol, R.A.; Wozniak, D.F.; Fujiwara, H.; Yuede, C.M.; Ory, D.S.; Kornfeld, S.; Vogel, P. Neurologic abnormalities in mouse models of the lysosomal storage disorders mucolipidosis II and mucolipidosis III γ. PLoS ONE 2014, 9, e109768. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, F.; Ulbrich, E.J.; Hafner, A.; Schmahl, W.; Meister, T.; Pohlmann, R.; von Figura, K. Alternative mechanisms for trafficking of lysosomal enzymes in mannose 6-phosphate receptor-deficient mice are cell type-specific. J. Cell Sci. 1999, 112 Pt 10, 1591–1597. [Google Scholar] [PubMed]
- Hogue, D.L.; Nash, C.; Ling, V.; Hobman, T.C. Lysosome-associated protein transmembrane 4 α (LAPTM4 α) requires two tandemly arranged tyrosine-based signals for sorting to lysosomes. Biochem. J. 2002, 365, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Dell’Angelica, E.C.; Shotelersuk, V.; Aguilar, R.C.; Gahl, W.A.; Bonifacino, J.S. Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the β 3A subunit of the AP-3 adaptor. Mol. Cell 1999, 3, 11–21. [Google Scholar] [CrossRef]
- Rous, B.A.; Reaves, B.J.; Ihrke, G.; Briggs, J.A.G.; Gray, S.R.; Stephens, D.J.; Banting, G.; Luzio, J.P. Role of adaptor complex AP-3 in targeting wild-type and mutated CD63 to lysosomes. Mol. Biol. Cell 2002, 13, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J.; Bright, N.A.; Rous, B.; Robinson, M.S. Characterization of a fourth adaptor-related protein complex. Mol. Biol. Cell 1999, 10, 2787–2802. [Google Scholar] [CrossRef] [PubMed]
- Janvier, K.; Bonifacino, J.S. Role of the endocytic machinery in the sorting of lysosome-associated membrane proteins. Mol. Biol. Cell 2005, 16, 4231–4242. [Google Scholar] [CrossRef] [PubMed]
- Harter, C.; Mellman, I. Transport of the lysosomal membrane glycoprotein lgp120 (lgp-A) to lysosomes does not require appearance on the plasma membrane. J. Cell Biol. 1992, 117, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Rohrer, J.; Schweizer, A.; Russell, D.; Kornfeld, S. The targeting of Lamp1 to lysosomes is dependent on the spacing of its cytoplasmic tail tyrosine sorting motif relative to the membrane. J. Cell Biol. 1996, 132, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Obermüller, S.; Kiecke, C.; von Figura, K.; Höning, S. The tyrosine motifs of Lamp 1 and LAP determine their direct and indirect targetting to lysosomes. J. Cell Sci. 2002, 115, 185–194. [Google Scholar] [PubMed]
- Peden, A.A.; Oorschot, V.; Hesser, B.A.; Austin, C.D.; Scheller, R.H.; Klumperman, J. Localization of the AP-3 adaptor complex defines a novel endosomal exit site for lysosomal membrane proteins. J. Cell Biol. 2004, 164, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; Tikkanen, R.; Mühlhausen, C.; Wenzel, D.; von Figura, K.; Höning, S. AP-1 and AP-3 mediate sorting of melanosomal and lysosomal membrane proteins into distinct post-Golgi trafficking pathways. Traffic (Cph. Den.) 2008, 9, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, R.C.; Boehm, M.; Gorshkova, I.; Crouch, R.J.; Tomita, K.; Saito, T.; Ohno, H.; Bonifacino, J.S. Signal-binding specificity of the mu4 subunit of the adaptor protein complex AP-4. J. Biol. Chem. 2001, 276, 13145–13152. [Google Scholar] [CrossRef] [PubMed]
- Lefrancois, S.; Zeng, J.; Hassan, A.J.; Canuel, M.; Morales, C.R. The lysosomal trafficking of sphingolipid activator proteins (SAPs) is mediated by sortilin. EMBO J. 2003, 22, 6430–6437. [Google Scholar] [CrossRef] [PubMed]
- Canuel, M.; Lefrancois, S.; Zeng, J.; Morales, C.R. AP-1 and retromer play opposite roles in the trafficking of sortilin between the Golgi apparatus and the lysosomes. Biochem. Biophys. Res. Commun. 2008, 366, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.S.; Madsen, P.; Christensen, E.I.; Nykjaer, A.; Gliemann, J.; Kasper, D.; Pohlmann, R.; Petersen, C.M. The sortilin cytoplasmic tail conveys Golgi-endosome transport and binds the VHS domain of the GGA2 sorting protein. EMBO J. 2001, 20, 2180–2190. [Google Scholar] [CrossRef] [PubMed]
- Takatsu, H.; Katoh, Y.; Shiba, Y.; Nakayama, K. Golgi-localizing, gamma-adaptin ear homology domain, ADP-ribosylation factor-binding (GGA) proteins interact with acidic dileucine sequences within the cytoplasmic domains of sorting receptors through their Vps27p/Hrs/STAM (VHS) domains. J. Biol. Chem. 2001, 276, 28541–28545. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.J.; Zeng, J.; Ni, X.; Morales, C.R. The trafficking of prosaposin (SGP-1) and GM2AP to the lysosomes of TM4 Sertoli cells is mediated by sortilin and monomeric adaptor proteins. Mol. Reprod. Dev. 2004, 68, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Le Borgne, R.; Alconada, A.; Bauer, U.; Hoflack, B. The mammalian AP-3 adaptor-like complex mediates the intracellular transport of lysosomal membrane glycoproteins. J. Biol. Chem. 1998, 273, 29451–29461. [Google Scholar] [CrossRef] [PubMed]
- Höning, S.; Sandoval, I.V.; von Figura, K. A di-leucine-based motif in the cytoplasmic tail of LIMP-II and tyrosinase mediates selective binding of AP-3. EMBO J. 1998, 17, 1304–1314. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Saeki, M.; Yasunaga, K.; Ueda, T.; Imoto, T.; Himeno, M. In vitro binding study of adaptor protein complex (AP-1) to lysosomal targeting motif (LI-motif). Biochem. Biophys. Res. Commun. 1999, 255, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Tabuchi, N.; Akasaki, K.; Tsuji, H. Two acidic amino acid residues, Asp(470) and Glu(471), contained in the carboxyl cytoplasmic tail of a major lysosomal membrane protein, LGP85/LIMP II, are important for its accumulation in secondary lysosomes. Biochem. Biophys. Res. Commun. 2000, 270, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Tabuchi, N.; Akasaki, K.; Tsuji, H. Ile (476), a constituent of di-leucine-based motif of a major lysosomal membrane protein, LGP85/LIMP II, is important for its proper distribution in late endosomes and lysosomes. Biochem. Biophys. Res. Commun. 2002, 295, 149–156. [Google Scholar] [CrossRef]
- Janvier, K.; Kato, Y.; Boehm, M.; Rose, J.R.; Martina, J.A.; Kim, B.-Y.; Venkatesan, S.; Bonifacino, J.S. Recognition of dileucine-based sorting signals from HIV-1 Nef and LIMP-II by the AP-1 γ-sigma1 and AP-3 δ/σ3 hemicomplexes. J. Cell Biol. 2003, 163, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.J.; Goldstein, J.L.; Brown, M.S. NPXY, a sequence often found in cytoplasmic tails, is required for coated pit-mediated internalization of the low density lipoprotein receptor. J. Biol. Chem. 1990, 265, 3116–3123. [Google Scholar] [PubMed]
- Garcia, C.K.; Wilund, K.; Arca, M.; Zuliani, G.; Fellin, R.; Maioli, M.; Calandra, S.; Bertolini, S.; Cossu, F.; Grishin, N.; et al. Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science 2001, 292, 1394–1398. [Google Scholar] [CrossRef] [PubMed]
- Eden, E.R.; Patel, D.D.; Sun, X.-M.; Burden, J.J.; Themis, M.; Edwards, M.; Lee, P.; Neuwirth, C.; Naoumova, R.P.; Soutar, A.K. Restoration of LDL receptor function in cells from patients with autosomal recessive hypercholesterolemia by retroviral expression of ARH1. J. Clin. Investig. 2002, 110, 1695–1702. [Google Scholar] [CrossRef] [PubMed]
- Boll, W.; Rapoport, I.; Brunner, C.; Modis, Y.; Prehn, S.; Kirchhausen, T. The mu2 subunit of the clathrin adaptor AP-2 binds to FDNPVY and YppØ sorting signals at distinct sites. Traffic (Cph. Den.) 2002, 3, 590–600. [Google Scholar] [CrossRef]
- Li, Y.; Marzolo, M.P.; van Kerkhof, P.; Strous, G.J.; Bu, G. The YXXL motif, but not the two NPXY motifs, serves as the dominant endocytosis signal for low density lipoprotein receptor-related protein. J. Biol. Chem. 2000, 275, 17187–17194. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Betts, G.N.; Barnes, H.; Ghassemian, M.; van der Geer, P.; Komives, E.A. Interactions of the NPXY microdomains of the low density lipoprotein receptor-related protein 1. Proteomics 2009, 9, 5016–5028. [Google Scholar] [CrossRef] [PubMed]
- Oleinikov, A.V.; Zhao, J.; Makker, S.P. Cytosolic adaptor protein Dab2 is an intracellular ligand of endocytic receptor gp600/megalin. Biochem. J. 2000, 347 Pt 3, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Yamazaki, H.; Farquhar, M.G. Identification of an apical sorting determinant in the cytoplasmic tail of megalin. Am. J. Physiol. Cell Physiol. 2003, 284, C1105–C1113. [Google Scholar] [CrossRef] [PubMed]
- Nagai, M.; Meerloo, T.; Takeda, T.; Farquhar, M.G. The adaptor protein ARH escorts megalin to and through endosomes. Mol. Biol. Cell 2003, 14, 4984–4996. [Google Scholar] [CrossRef] [PubMed]
- Boonen, M.; Staudt, C.; Gilis, F.; Oorschot, V.; Klumperman, J.; Jadot, M. Cathepsin D and its newly identified transport receptor SEZ6L2 can modulate neurite outgrowth. J. Cell Sci. 2016, 129, 557–568. [Google Scholar] [CrossRef] [PubMed]
- East, L.; Isacke, C.M. The mannose receptor family. Biochim. Biophys. Acta 2002, 1572, 364–386. [Google Scholar] [CrossRef]
- Kruskal, B.A.; Sastry, K.; Warner, A.B.; Mathieu, C.E.; Ezekowitz, R.A. Phagocytic chimeric receptors require both transmembrane and cytoplasmic domains from the mannose receptor. J. Exp. Med. 1992, 176, 1673–1680. [Google Scholar] [CrossRef] [PubMed]
- Beaujouin, M.; Prébois, C.; Derocq, D.; Laurent-Matha, V.; Masson, O.; Pattingre, S.; Coopman, P.; Bettache, N.; Grossfield, J.; Hollingsworth, R.E.; et al. Pro-cathepsin D interacts with the extracellular domain of the β chain of LRP1 and promotes LRP1-dependent fibroblast outgrowth. J. Cell Sci. 2010, 123, 3336–3346. [Google Scholar] [CrossRef] [PubMed]
- Zunke, F.; Andresen, L.; Wesseler, S.; Groth, J.; Arnold, P.; Rothaug, M.; Mazzulli, J.R.; Krainc, D.; Blanz, J.; Saftig, P.; et al. Characterization of the complex formed by β-glucocerebrosidase and the lysosomal integral membrane protein type-2. Proc. Natl. Acad. Sci. USA 2016, 113, 3791–3796. [Google Scholar] [CrossRef] [PubMed]
- Westergaard, U.B.; Sørensen, E.S.; Hermey, G.; Nielsen, M.S.; Nykjaer, A.; Kirkegaard, K.; Jacobsen, C.; Gliemann, J.; Madsen, P.; Petersen, C.M. Functional organization of the sortilin Vps10p domain. J. Biol. Chem. 2004, 279, 50221–50229. [Google Scholar] [CrossRef] [PubMed]
- van Meel, E.; Klumperman, J. Imaging and imagination: Understanding the endo-lysosomal system. Histochem. Cell Biol. 2008, 129, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, M.F.; Prata, M.J.; Alves, S. Mannose-6-phosphate pathway: A review on its role in lysosomal function and dysfunction. Mol. Genet. Metab. 2012, 105, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Reczek, D.; Schwake, M.; Schröder, J.; Hughes, H.; Blanz, J.; Jin, X.; Brondyk, W.; van Patten, S.; Edmunds, T.; Saftig, P. LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of β-glucocerebrosidase. Cell 2007, 131, 770–783. [Google Scholar] [CrossRef] [PubMed]
- Kuronita, T.; Eskelinen, E.-L.; Fujita, H.; Saftig, P.; Himeno, M.; Tanaka, Y. A role for the lysosomal membrane protein LGP85 in the biogenesis and maintenance of endosomal and lysosomal morphology. J. Cell Sci. 2002, 115, 4117–4131. [Google Scholar] [CrossRef] [PubMed]
- Rothaug, M.; Zunke, F.; Mazzulli, J.R.; Schweizer, M.; Altmeppen, H.; Lüllmann-Rauch, R.; Kallemeijn, W.W.; Gaspar, P.; Aerts, J.M.; Glatzel, M.; et al. LIMP-2 expression is critical for β-glucocerebrosidase activity and α-synuclein clearance. Proc. Natl. Acad. Sci. USA 2014, 111, 15573–15578. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Morales, C.R. The lysosomal trafficking of acid sphingomyelinase is mediated by sortilin and mannose 6-phosphate receptor. Traffic (Cph. Den.) 2006, 7, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Canuel, M.; Korkidakis, A.; Konnyu, K.; Morales, C.R. Sortilin mediates the lysosomal targeting of cathepsins D and H. Biochem. Biophys. Res. Commun. 2008, 373, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Racicott, J.; Morales, C.R. The inactivation of the sortilin gene leads to a partial disruption of prosaposin trafficking to the lysosomes. Exp. Cell Res. 2009, 315, 3112–3124. [Google Scholar] [CrossRef] [PubMed]
- Canuel, M.; Libin, Y.; Morales, C.R. The interactomics of sortilin: An ancient lysosomal receptor evolving new functions. Histol. Histopathol. 2009, 24, 481–492. [Google Scholar] [PubMed]
- Wähe, A.; Kasmapour, B.; Schmaderer, C.; Liebl, D.; Sandhoff, K.; Nykjaer, A.; Griffiths, G.; Gutierrez, M.G. Golgi-to-phagosome transport of acid sphingomyelinase and prosaposin is mediated by sortilin. J. Cell Sci. 2010, 123, 2502–2511. [Google Scholar] [CrossRef] [PubMed]
- Markmann, S.; Thelen, M.; Cornils, K.; Schweizer, M.; Brocke-Ahmadinejad, N.; Willnow, T.; Heeren, J.; Gieselmann, V.; Braulke, T.; Kollmann, K. Lrp1/LDL receptor play critical roles in mannose 6-phosphate-independent lysosomal enzyme targeting. Traffic (Cph. Den.) 2015, 16, 743–759. [Google Scholar] [CrossRef] [PubMed]
- Christensen, E.I.; Zhou, Q.; Sørensen, S.S.; Rasmussen, A.K.; Jacobsen, C.; Feldt-Rasmussen, U.; Nielsen, R. Distribution of α-galactosidase A in normal human kidney and renal accumulation and distribution of recombinant alpha-galactosidase A in Fabry mice. J. Am. Soc. Nephrol. JASN 2007, 18, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Prabakaran, T.; Nielsen, R.; Larsen, J.V.; Sørensen, S.S.; Feldt-Rasmussen, U.; Saleem, M.A.; Petersen, C.M.; Verroust, P.J.; Christensen, E.I. Receptor-mediated endocytosis of α-galactosidase A in human podocytes in Fabry disease. PLoS ONE 2011, 6, e25065. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.; Courtoy, P.J.; Jacobsen, C.; Dom, G.; Lima, W.R.; Jadot, M.; Willnow, T.E.; Devuyst, O.; Christensen, E.I. Endocytosis provides a major alternative pathway for lysosomal biogenesis in kidney proximal tubular cells. Proc. Natl. Acad. Sci. USA 2007, 104, 5407–5412. [Google Scholar] [CrossRef] [PubMed]
- Derocq, D.; Prébois, C.; Beaujouin, M.; Laurent-Matha, V.; Pattingre, S.; Smith, G.K.; Liaudet-Coopman, E. Cathepsin D is partly endocytosed by the LRP1 receptor and inhibits LRP1-regulated intramembrane proteolysis. Oncogene 2012, 31, 3202–3212. [Google Scholar] [CrossRef] [PubMed]
- Liaudet-Coopman, E.; Beaujouin, M.; Derocq, D.; Garcia, M.; Glondu-Lassis, M.; Laurent-Matha, V.; Prébois, C.; Rochefort, H.; Vignon, F. Cathepsin D: Newly discovered functions of a long-standing aspartic protease in cancer and apoptosis. Cancer Lett. 2006, 237, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Laurent-Matha, V.; Farnoud, M.R.; Lucas, A.; Rougeot, C.; Garcia, M.; Rochefort, H. Endocytosis of pro-cathepsin D into breast cancer cells is mostly independent of mannose-6-phosphate receptors. J. Cell Sci. 1998, 111 Pt 17, 2539–2549. [Google Scholar] [PubMed]
- Stützer, I.; Selevsek, N.; Esterházy, D.; Schmidt, A.; Aebersold, R.; Stoffel, M. Systematic proteomic analysis identifies β-site amyloid precursor protein cleaving enzyme 2 and 1 (BACE2 and BACE1) substrates in pancreatic β-cells. J. Biol. Chem. 2013, 288, 10536–10547. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.-H.; Koroniak, K.; Hogl, S.; Colombo, A.; Zeitschel, U.; Willem, M.; Volbracht, C.; Schepers, U.; Imhof, A.; Hoffmeister, A.; et al. Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J. 2012, 31, 3157–3168. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Hashimoto, K.; Uda, A.; Sakagami, H.; Nakamura, Y.; Saito, S.; Nishi, M.; Kume, H.; Tohgo, A.; Kaneko, I.; et al. Disturbance of cerebellar synaptic maturation in mutant mice lacking BSRPs, a novel brain-specific receptor-like protein family. FEBS Lett. 2006, 580, 4057–4064. [Google Scholar] [CrossRef] [PubMed]
- Gunnersen, J.M.; Kim, M.H.; Fuller, S.J.; de Silva, M.; Britto, J.M.; Hammond, V.E.; Davies, P.J.; Petrou, S.; Faber, E.S.L.; Sah, P.; et al. Sez-6 proteins affect dendritic arborization patterns and excitability of cortical pyramidal neurons. Neuron 2007, 56, 621–639. [Google Scholar] [CrossRef] [PubMed]
- Achord, D.T.; Brot, F.E.; Bell, C.E.; Sly, W.S. Human beta-glucuronidase: In vivo clearance and in vitro uptake by a glycoprotein recognition system on reticuloendothelial cells. Cell 1978, 15, 269–278. [Google Scholar] [CrossRef]
- Hubbard, A.L.; Wilson, G.; Ashwell, G.; Stukenbrok, H. An electron microscope autoradiographic study of the carbohydrate recognition systems in rat liver. I. Distribution of 125I-ligands among the liver cell types. J. Cell Biol. 1979, 83, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Maynard, Y.; Baenziger, J.U. Oligosaccharide specific endocytosis by isolated rat hepatic reticuloendothelial cells. J. Biol. Chem. 1981, 256, 8063–8068. [Google Scholar] [PubMed]
- Lennartz, M.R.; Wileman, T.E.; Stahl, P.D. Isolation and characterization of a mannose-specific endocytosis receptor from rabbit alveolar macrophages. Biochem. J. 1987, 245, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.E.; Conary, J.T.; Lennartz, M.R.; Stahl, P.D.; Drickamer, K. Primary structure of the mannose receptor contains multiple motifs resembling carbohydrate-recognition domains. J. Biol. Chem. 1990, 265, 12156–12162. [Google Scholar] [PubMed]
- Lee, S.J.; Evers, S.; Roeder, D.; Parlow, A.F.; Risteli, J.; Risteli, L.; Lee, Y.C.; Feizi, T.; Langen, H.; Nussenzweig, M.C. Mannose receptor-mediated regulation of serum glycoprotein homeostasis. Science 2002, 295, 1898–1901. [Google Scholar] [CrossRef] [PubMed]
- Elvevold, K.; Simon-Santamaria, J.; Hasvold, H.; McCourt, P.; Smedsrød, B.; Sørensen, K.K. Liver sinusoidal endothelial cells depend on mannose receptor-mediated recruitment of lysosomal enzymes for normal degradation capacity. Hepatology 2008, 48, 2007–2015. [Google Scholar] [CrossRef] [PubMed]
- Sleat, D.E.; Della Valle, M.C.; Zheng, H.; Moore, D.F.; Lobel, P. The mannose 6-phosphate glycoprotein proteome. J. Proteome Res. 2008, 7, 3010–3021. [Google Scholar] [CrossRef] [PubMed]
- Puissant, E.; Gilis, F.; Dogné, S.; Flamion, B.; Jadot, M.; Boonen, M. Subcellular trafficking and activity of Hyal-1 and its processed forms in murine macrophages. Traffic (Cph. Den.) 2014, 15, 500–515. [Google Scholar] [CrossRef] [PubMed]
- Boonen, M.; Puissant, E.; Gilis, F.; Flamion, B.; Jadot, M. Mouse liver lysosomes contain enzymatically active processed forms of Hyal-1. Biochem. Biophys. Res. Commun. 2014, 446, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Furbish, F.S.; Steer, C.J.; Krett, N.L.; Barranger, J.A. Uptake and distribution of placental glucocerebrosidase in rat hepatic cells and effects of sequential deglycosylation. Biochim. Biophys. Acta 1981, 673, 425–434. [Google Scholar] [CrossRef]
- Brady, R.O.; Murray, G.J.; Barton, N.W. Modifying exogenous glucocerebrosidase for effective replacement therapy in Gaucher disease. J. Inherit. Metab. Dis. 1994, 17, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Puissant, E.; Boonen, M. Monocytes/macrophages upregulate the hyaluronidase HYAL1 and adapt its subcellular trafficking to promote extracellular residency upon differentiation into osteoclasts. PLoS ONE 2016, 11, e0165004. [Google Scholar] [CrossRef] [PubMed]
- Kowalewski, B.; Lübke, T.; Kollmann, K.; Braulke, T.; Reinheckel, T.; Dierks, T.; Damme, M. Molecular characterization of arylsulfatase G: Expression, processing, glycosylation, transport, and activity. J. Biol. Chem. 2014, 289, 27992–28005. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, A.; Omura, K.; Suzuki, Y. Intracellular transport of acid α-glucosidase in human fibroblasts: Evidence for involvement of phosphomannosyl receptor-independent system. J. Biochem. (Tokyo) 1988, 104, 276–278. [Google Scholar] [PubMed]
- Díaz, E.; Pfeffer, S.R. TIP47: A cargo selection device for mannose 6-phosphate receptor trafficking. Cell 1998, 93, 433–443. [Google Scholar] [CrossRef]
- Carroll, K.S.; Hanna, J.; Simon, I.; Krise, J.; Barbero, P.; Pfeffer, S.R. Role of Rab9 GTPase in facilitating receptor recruitment by TIP47. Science 2001, 292, 1373–1376. [Google Scholar] [CrossRef] [PubMed]
- Puertollano, R. mTOR and lysosome regulation. F1000prime Rep. 2014, 6, 52. [Google Scholar] [CrossRef] [PubMed]
- Efeyan, A.; Zoncu, R.; Sabatini, D.M. Amino acids and mTORC1: From lysosomes to disease. Trends Mol. Med. 2012, 18, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Stransky, L.A.; Forgac, M. Amino acid availability modulates vacuolar H+-ATPase assembly. J. Biol. Chem. 2015, 290, 27360–27369. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J.; Borner, G.H.H.; Edgar, J.; Hein, M.Y.; Mann, M.; Buchholz, F.; Antrobus, R.; Robinson, M.S. Interaction between AP-5 and the hereditary spastic paraplegia proteins SPG11 and SPG15. Mol. Biol. Cell 2013, 24, 2558–2569. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Lee, S.; Blackstone, C. Spastic paraplegia proteins spastizin and spatacsin mediate autophagic lysosome reformation. J. Clin. Investig. 2014, 124, 5249–5262. [Google Scholar] [CrossRef] [PubMed]
- Renvoisé, B.; Chang, J.; Singh, R.; Yonekawa, S.; FitzGibbon, E.J.; Mankodi, A.; Vanderver, A.; Schindler, A.; Toro, C.; Gahl, W.A.; et al. Lysosomal abnormalities in hereditary spastic paraplegia types SPG15 and SPG11. Ann. Clin. Transl. Neurol. 2014, 1, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, F.; Grieve, A.; Rabouille, C. Unconventional secretion: A stress on GRASP. Curr. Opin. Cell Biol. 2011, 23, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Nickel, W.; Rabouille, C. Mechanisms of regulated unconventional protein secretion. Nat. Rev. Mol. Cell Biol. 2009, 10, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Jiang, S.; Dupont, N. Autophagy intersections with conventional and unconventional secretion in tissue development, remodeling and inflammation. Trends Cell Biol. 2012, 22, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Rabouille, C.; Linstedt, A.D. GRASP: A multitasking tether. Front. Cell Dev. Biol. 2016, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Young, A.R.J.; Chan, E.Y.W.; Hu, X.W.; Köchl, R.; Crawshaw, S.G.; High, S.; Hailey, D.W.; Lippincott-Schwartz, J.; Tooze, S.A. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci. 2006, 119, 3888–3900. [Google Scholar] [CrossRef] [PubMed]
- Staudt, C.; Gilis, F.; Boonen, M.; Jadot, M. Molecular determinants that mediate the sorting of human ATG9A from the endoplasmic reticulum. Biochim. Biophys. Acta 2016, 1863, 2299–2310. [Google Scholar] [CrossRef] [PubMed]
- Tveit, H.; Akslen, L.K.A.; Fagereng, G.L.; Tranulis, M.A.; Prydz, K. A secretory Golgi bypass route to the apical surface domain of epithelial MDCK cells. Traffic (Cph. Den.) 2009, 10, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Syres, K.; Harrison, F.; Tadlock, M.; Jester, J.V.; Simpson, J.; Roy, S.; Salomon, D.R.; Cherqui, S. Successful treatment of the murine model of cystinosis using bone marrow cell transplantation. Blood 2009, 114, 2542–2552. [Google Scholar] [CrossRef] [PubMed]
- Rocca, C.J.; Kreymerman, A.; Ur, S.N.; Frizzi, K.E.; Naphade, S.; Lau, A.; Tran, T.; Calcutt, N.A.; Goldberg, J.L.; Cherqui, S. Treatment of inherited eye defects by systemic hematopoietic stem cell transplantation. Investig. Ophthalmol. Vis. Sci. 2015, 56, 7214–7223. [Google Scholar] [CrossRef] [PubMed]
- Gaide Chevronnay, H.P.; Janssens, V.; van der Smissen, P.; Rocca, C.J.; Liao, X.H.; Refetoff, S.; Pierreux, C.E.; Cherqui, S.; Courtoy, P.J. Hematopoietic stem cells transplantation can normalize thyroid function in a cystinosis mouse model. Endocrinology 2016, 157, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Naphade, S.; Sharma, J.; Gaide Chevronnay, H.P.; Shook, M.A.; Yeagy, B.A.; Rocca, C.J.; Ur, S.N.; Lau, A.J.; Courtoy, P.J.; Cherqui, S. Brief reports: Lysosomal cross-correction by hematopoietic stem cell-derived macrophages via tunneling nanotubes. Stem Cells 2015, 33, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.-H. Nanotubular highways for intercellular organelle transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef] [PubMed]
- Rustom, A. The missing link: Does tunnelling nanotube-based supercellularity provide a new understanding of chronic and lifestyle diseases? Open Biol. 2016, 6, 160057. [Google Scholar] [CrossRef] [PubMed]
- Cocucci, E.; Meldolesi, J. Ectosomes and exosomes: Shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015, 25, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, D.M.; El-Kares, R.; Taranta, A.; Bellomo, F.; Emma, F.; Besouw, M.; Levtchenko, E.; Toelen, J.; van den Heuvel, L.; Chu, L.; et al. Stem cell microvesicles transfer cystinosin to human cystinotic cells and reduce cystine accumulation in vitro. PLoS ONE 2012, 7, e42840. [Google Scholar] [CrossRef] [PubMed]
- Keerthikumar, S.; Chisanga, D.; Ariyaratne, D.; Al Saffar, H.; Anand, S.; Zhao, K.; Samuel, M.; Pathan, M.; Jois, M.; Chilamkurti, N.; et al. ExoCarta: A web-based compendium of exosomal cargo. J. Mol. Biol. 2016, 428, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Sleat, D.E.; Zheng, H.; Moore, D.; Lobel, P. Proteomics analysis of serum from mutant mice reveals lysosomal proteins selectively transported by each of the two mannose 6-phosphate receptors. Mol. Cell. Proteom. MCP 2008, 7, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Matzner, U.; von Figura, K.; Pohlmann, R. Expression of the two mannose 6-phosphate receptors is spatially and temporally different during mouse embryogenesis. Development (Camb.) 1992, 114, 965–972. [Google Scholar]
- Waguri, S.; Kohmura, M.; Kanamori, S.; Watanabe, T.; Ohsawa, Y.; Koike, M.; Tomiyama, Y.; Wakasugi, M.; Kominami, E.; Uchiyama, Y. Different distribution patterns of the two mannose 6-phosphate receptors in rat liver. J. Histochem. Cytochem. 2001, 49, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Konishi, Y.; Fushimi, S.; Shirabe, T. Immunohistochemical distribution of cation-dependent mannose 6-phosphate receptors in the mouse central nervous system: Comparison with that of cation-independent mannose 6-phophate receptors. Neurosci. Lett. 2005, 378, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]



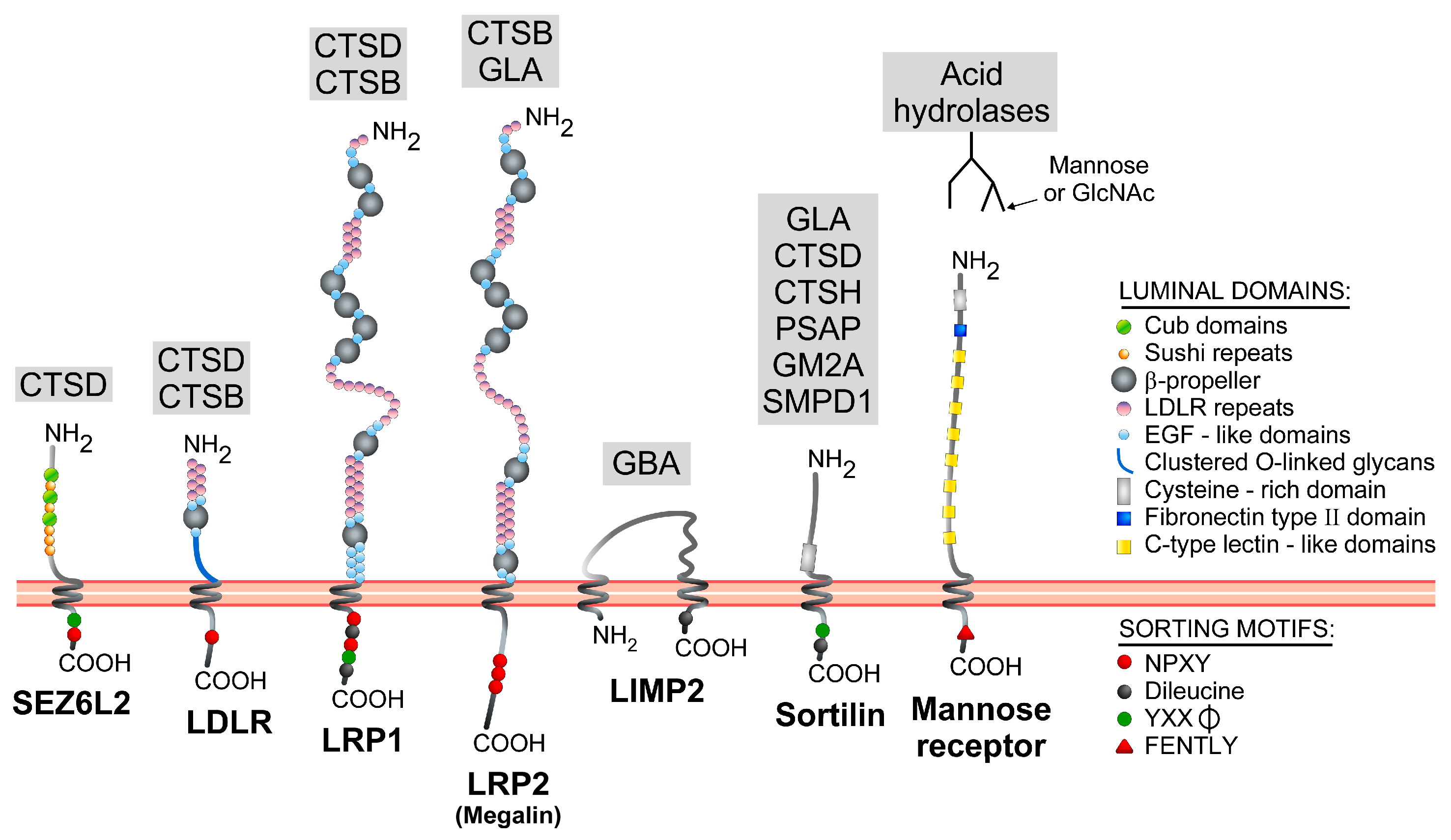{kind=link}
{kind=link}
| Gene Symbol | Protein Name | Conventional Sorting Determinant(s) | Atypical Sorting Determinant(s) | Trafficking Mechanism(s) | References |
|---|---|---|---|---|---|
| CTNS | Cystinosin | GYDQL in C-ter tail | YFPQA in 3rd cytoplasmic loop | AP-3 -dependent intracellular/direct trafficking. | [19,20] |
| VAMP7 | Vesicle-associated membrane protein 7 | N-ter longin domain (critical residues: Leu43/Tyr45) | cis-SNARE complex transported by AP-3-dependent direct trafficking, and by Hrb-dependent endocytosis. | [21,22,23] | |
| LAPTM5 | Lysosomal-associated transmembrane protein 5 | PY motifs (L/PPxY) + ubiquitin-interacting motif (LKVALPSYEE) | PY motifs recruits GGA3, which binds to the ubiquitin-interacting motif of LAPTM5 and mediates transport to endolysosomes. | [24] | |
| LAPTM4A | Lysosomal-associated transmembrane protein 4A | YXXφ motif in C-ter region | PY motifs in C-ter tail | Nedd4-dependent sorting to endolysosomes. | [25,74] |
| LAPTM4B | Lysosomal-associated transmembrane protein 4B | PY motifs in C-ter tail | Nedd4-dependent sorting to endolysosomes. | [25] | |
| CLN3 | Battenin | Atypical dileucine motif (EEEX(8)LI) in a cytoplasmic loop; MX9G in C-ter tail; C-ter CAAX farnesylation motif (C435QLS) | Mostly AP-1 and AP-3-mediated intracellular sorting. Inhibition of farnesylation induces relocalization to the PM and slows transport to endolysosomes. | [27,28,32,44] | |
| TMEM106B | Transmembrane protein 106B | Extended dileucine motif (ENQLVALI) in the N-ter region; 4th and 5th N-glycosylation sites | Mutation of the 4th and 5th N-glycosylation sites results in ER retention and relocalization at the PM, respectively. Mutation of LI in the atypical dileucine signal results in a diffuse cytoplasmic localization. | [26,40] | |
| GRP143/OA1 | G-protein coupled receptor 143 | Unconventional dileucine motif (SLLKGRQGIY) in the 3rd cytosolic loop; WE (tryptophan/Glutamic acid) motif in C-ter tail | [33] | ||
| STARD3 | StAR-related lipid transfer protein 3/MLN64 (metastatic lymph node 64) | 14-3-3 binding motif (K392SASNP) in the START domain (C-ter); unidentified internalization motif in the N-ter cytosolic region or transmembrane domains | Indirect trafficking via PM. Mutation of the 14-3-3 binding site delays transport to endosomes via the cell surface. | [34,39] | |
| RNF13 | E3 ubiquitin-protein ligase RNF13 (Ring finger protein 13) | Luminal protease-associated domain | A114P substitution in the luminal protease-associated domain prevents sorting to endolysosomes. | [41] | |
| RNF167 | E3 ubiquitin-protein ligase RNF167 (Ring finger protein 167) | Luminal protease-associated domain | A104P and V98G substitutions in the luminal protease-associated domain prevent sorting to endolysosomes. | [41] | |
| MCOLN1 | Mucolipin-1 | ETERLL in N-ter domain; EEHSLL in C-ter domain | Cysteines 565–567 | ETERLL-mediated direct transport, likely mediated by AP-1. EEHSLL-mediated internalization, AP-2–mediated. Palmitoylation of cysteines 565–567 promotes internalization, possibly by bringing the C-ter dileucine signal closer to the membrane. | [45,46] |
| CD63 | CD63 antigen/LAMP3 | GYEVM in C-ter region | Direct and indirect transport. C-ter domain binds to AP-2, AP-3, and AP-4. Internalization from the cell surface via caveolae. | [58,75,76,77,78] | |
| SYT7 | Synaptotagmin-7 | Cysteines 35, 38 and 41 close to and in the transmembrane domain | Palmitoylation-dependent piggybacking on CD63. | [47] | |
| ABCB6 | ATP-binding cassette subfamily B member 9 | Extended N-ter domain (TMD0) which contains 5 transmembrane helices | Clathrin-dependent internalization. | [49] | |
| ABCB9 | ATP-binding cassette subfamily B member 9 | TMD0, composed of four transmembrane helices | [50] | ||
| ABCD4 | ATP-binding cassette subfamily D member 4 | Possibly transmembrane domains 2 and 5 | Clathrin-dependent internalization. Piggybacking on LMBD1, which uses a Yxxφ sorting signal. | [51,52] | |
| LAMP1 | Lysosome-associated membrane glycoprotein 1 | C-ter GYQTI | AP-1- and AP-3-dependent direct sorting. AP-2-dependent internalization. Sorted in vesicles positive for hVps41 and VAMP7, negative for CI-MPR, AP-1 and clathrin. | [54,55,56,57,75,78,79,80,81,82,83] | |
| LAMP2 | Lysosome-associated membrane glycoprotein 2 | C-ter YEQF | AP-1-and AP-3-dependent direct sorting. AP-2-dependent internalization. Binds to AP-4. Sorted in vesicles positive for hVps41 and VAMP7, negative for CI-MPR, AP-1 and clathrin. | [56,57,75,78,82,84] | |
| SLC3A2/SLC7A5 | 4F2hc/LAT1 | Piggybacking on LAPTM4b. | [53] |
| Gene Name | Protein Name | Endosomal Sorting Motif(s) | Endosomal Sorting Mechanism(s) | References |
|---|---|---|---|---|
| SORT1 | Sortilin | YXXφ (YSVL) and dileucine (DDSDEDLI) signals in C-ter region | Direct route: GGA-mediated (dileucine); AP-1-mediated (YXXφ). Indirect route: Clathrin-dependent internalization (YXXφ). | [85,86,87,88,89] |
| SCARB2 | LIMP2 | Dileucine signal (DERAPLI) in C-ter region | Direct route: AP-1 and AP-3-mediated via the dileucine motif. Indirect route: minor in some cell types. | [90,91,92,93,94,95] |
| LDLR | LDL (low-density lipoprotein) receptor | NPXY signal (NPVY) in the C-ter tail | Indirect route: NPXY- and ARH-dependent internalization. Binding to AP-2 reported. | [96,97,98,99] |
| LRP1 | LDL receptor-related protein 1 | YXXφ (YATL), NPXY (NPTY and NPVY), dileucine (DDVGGLL and DEKRELL) signals in C-ter region | Indirect route: mediated by YXXφ and dileucine motifs closest to the C-ter end. Minor involvement of the other signals. Binding to AP-2 and clathrin reported. | [100,101] |
| LRP2 | Megalin/low-density lipoprotein receptor-related protein 2 | NPXY signals in the C-ter region | Indirect route: NPXY-dependent internalization. Proximal NPXY binds to ARH; distal NPXY binds to Dab2. | [102,103,104] |
| SEZ6L2 | Seizure 6-like protein 2/Brain Specific Receptor-like Protein A (BSRP-A) | YXXφ (YSPI) and NPXY (NPLY) signals in the C-ter region | Direct route: likely YXXφ -mediated; SEZ6L2 detected in AP-1 positive clathrin-coated vesicles. Indirect route: NPXY- and Dab-2 mediated internalization. | [105] |
| MRC1 | Mannose receptor | FENTLY in the C-ter domain | Indirect route: Transmembrane domain and FENTLY-dependent internalization. | [106,107] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Staudt, C.; Puissant, E.; Boonen, M. Subcellular Trafficking of Mammalian Lysosomal Proteins: An Extended View. Int. J. Mol. Sci. 2017, 18, 47. https://doi.org/10.3390/ijms18010047
Staudt C, Puissant E, Boonen M. Subcellular Trafficking of Mammalian Lysosomal Proteins: An Extended View. International Journal of Molecular Sciences. 2017; 18(1):47. https://doi.org/10.3390/ijms18010047
Chicago/Turabian StyleStaudt, Catherine, Emeline Puissant, and Marielle Boonen. 2017. "Subcellular Trafficking of Mammalian Lysosomal Proteins: An Extended View" International Journal of Molecular Sciences 18, no. 1: 47. https://doi.org/10.3390/ijms18010047
APA StyleStaudt, C., Puissant, E., & Boonen, M. (2017). Subcellular Trafficking of Mammalian Lysosomal Proteins: An Extended View. International Journal of Molecular Sciences, 18(1), 47. https://doi.org/10.3390/ijms18010047