
Muscle Carnitine Palmitoyltransferase II Deficiency: A Review of Enzymatic Controversy and Clinical Features

Abstract
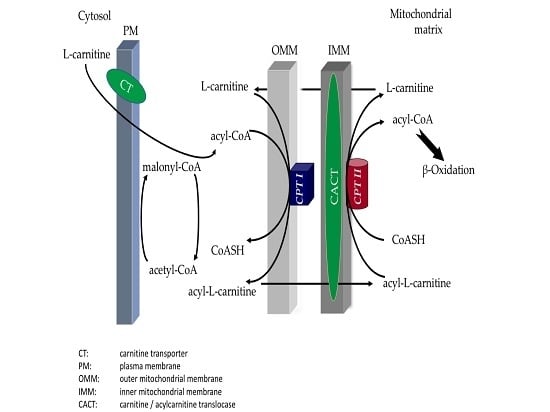
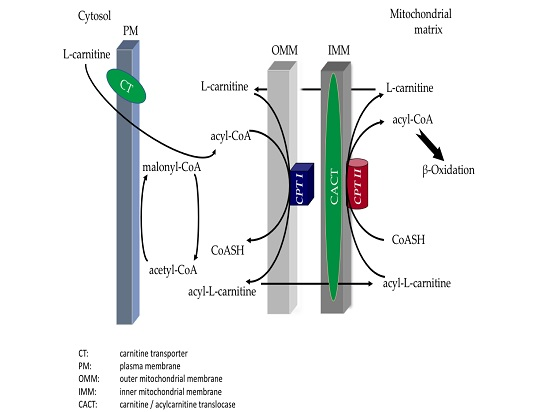
:
{kind=link}
{kind=link}
{kind=link}
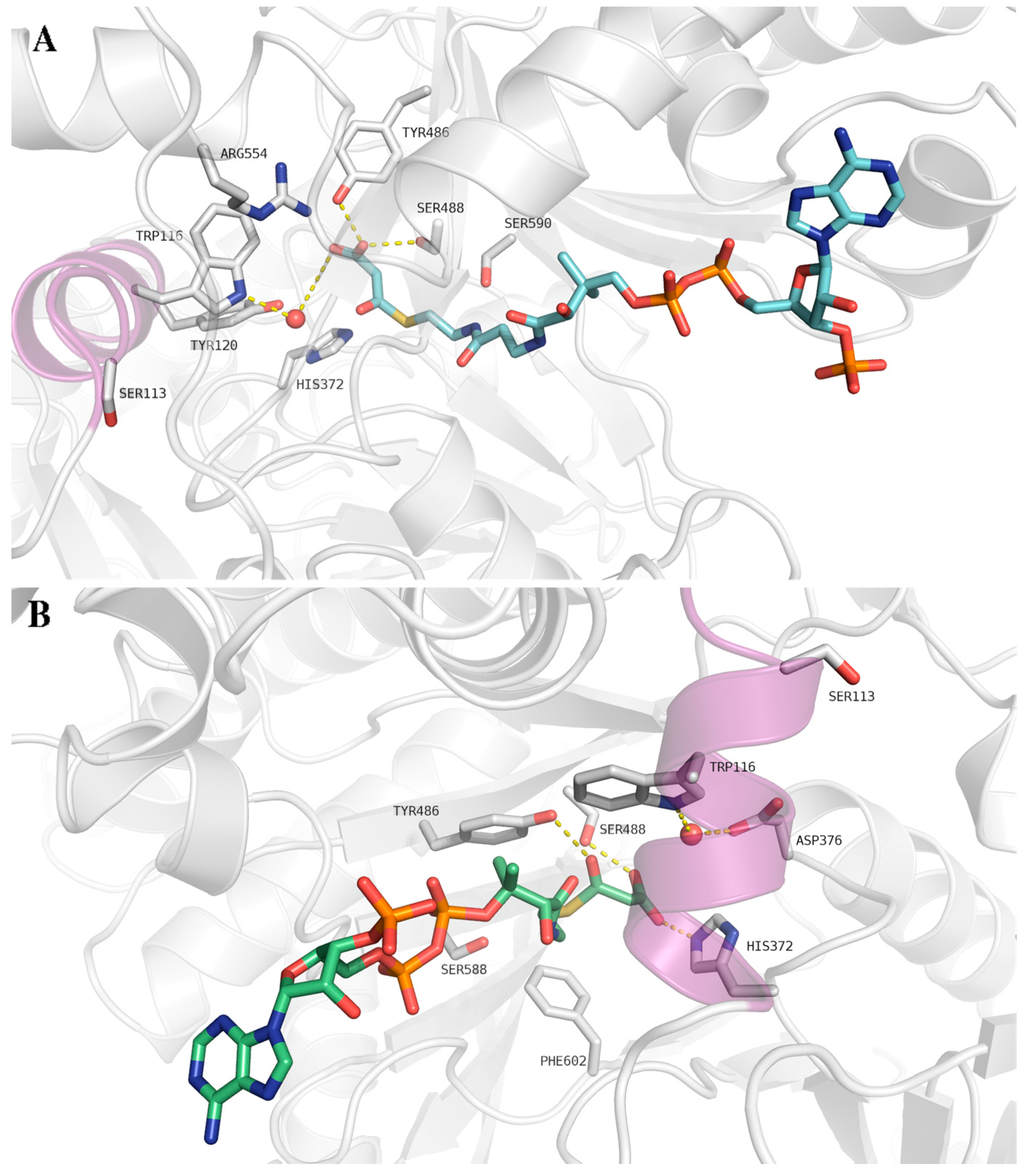{kind=link}
1. Introduction
2. Clinical Presentation of Patients with Muscle CPT II
3. Biochemical Studies in Patients with CPT II Muscle Deficiency
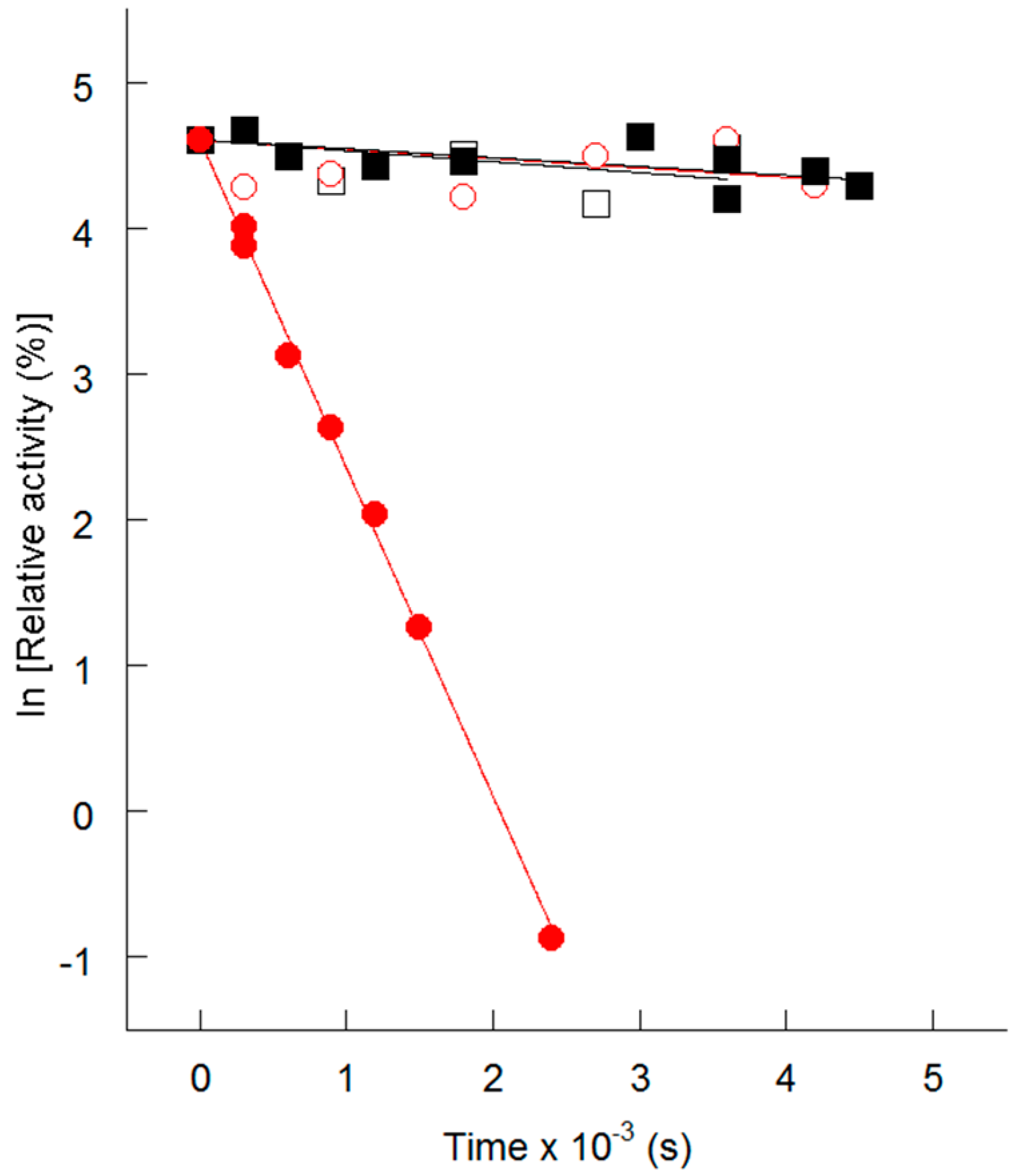
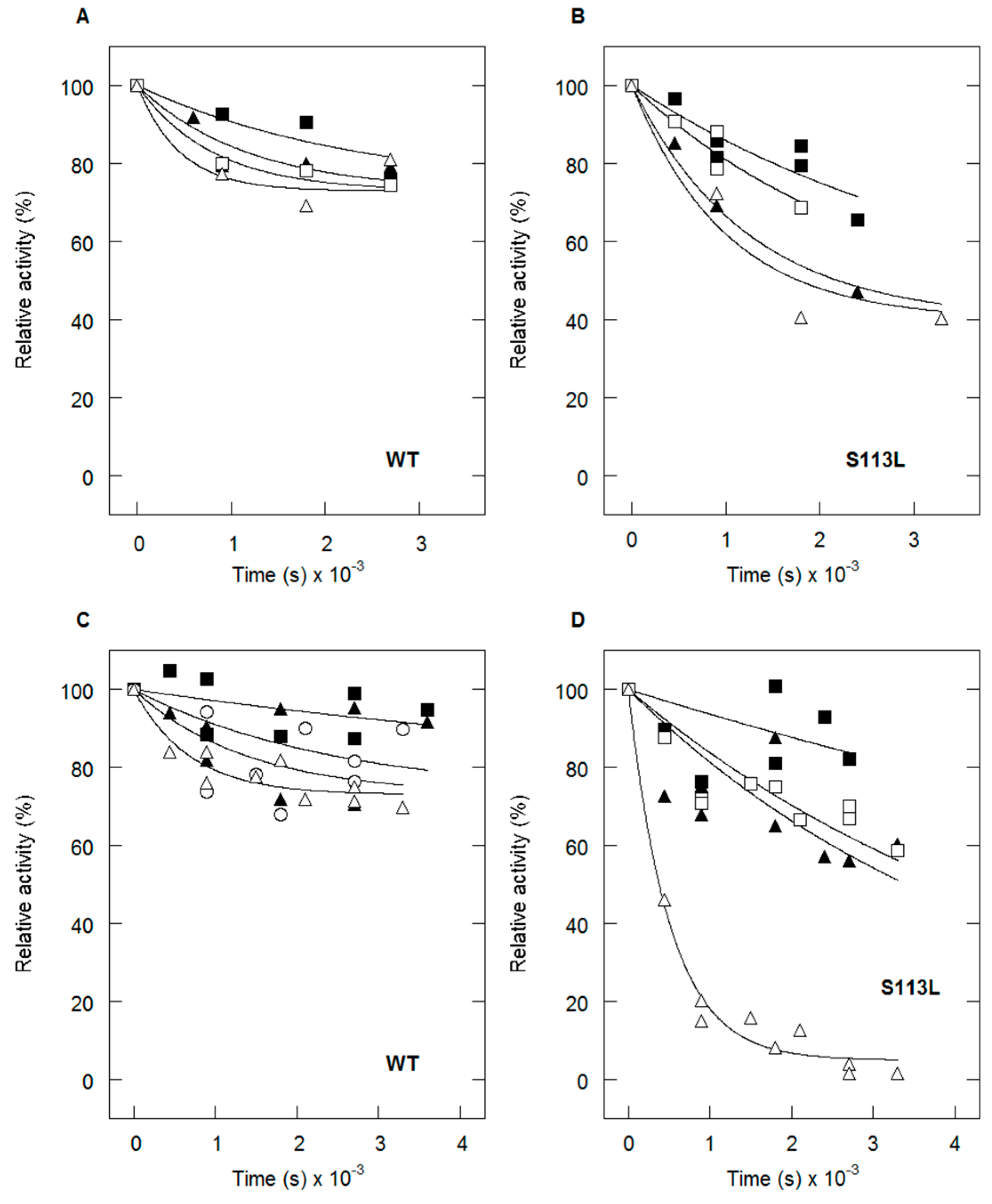
4. Thermolability of the S113L Variant
5. Protective Effect of Natural Substrates
6. Inhibitory Effect of Malonyl-CoA on CPT II
7. Summary and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bonnefont, J.P.; Djouadi, F.; Prip-Buus, C.; Gobin, S.; Munnich, A.; Bastin, J. Carnitine palmitoyltransferases 1 and 2: Biochemical, molecular and medical aspects. Mol. Aspects Med. 2004, 25, 495–520. [Google Scholar] [CrossRef] [PubMed]
- Deschauer, M.; Wieser, T.; Zierz, S. Muscle carnitine palmitoyltransferase II deficiency: Clinical and molecular genetic features and diagnostic aspects. Arch. Neurol. 2005, 62, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Bonnefont, J.P.; Demaugre, F.; Prip-Buus, C.; Saudubray, J.M.; Brivet, M.; Abadi, N.; Thuillier, L. Carnitine palmitoyltransferase deficiencies. Mol. Genet. Metab. 1999, 68, 424–440. [Google Scholar] [CrossRef] [PubMed]
- Isackson, P.J.; Bennett, M.J.; Vladutiu, G.D. Identification of 16 new disease-causing mutations in the CPT2 gene resulting in carnitine palmitoyltransferase II deficiency. Mol. Genet. Metab. 2006, 89, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.R.; Deschauer, M.; Zierz, S. Carnitine palmitoyltransferase II (CPT II) deficiency: Genotype-phenotype analysis of 50 patients. J. Neurol. Sci. 2014, 338, 107–111. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; di Mauro, P.M. Muscle carnitine palmityltransferase deficiency and myoglobinuria. Science 1973, 182, 929–931. [Google Scholar] [CrossRef] [PubMed]
- Bank, W.J.; DiMauro, S.; Bonilla, E.; Capuzzi, D.M.; Rowland, L.P. A disorder of muscle lipid metabolism and myoglobinuria. Absence of carnitine palmityl transferase. N. Engl. J. Med. 1975, 292, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Reza, M.J.; Kar, N.C.; Pearson, C.M.; Kark, R.A. Recurrent myoglobinuria due to muscle carnitine palmityl transferase deficiency. Ann. Intern. Med. 1978, 88, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Patten, B.M.; Wood, J.M.; Harati, Y.; Hefferan, P.; Howell, R.R. Familial recurrent rhabdomyolysis due to carnitine palmityl transferase deficiency. Am. J. Med. 1979, 67, 167–171. [Google Scholar] [CrossRef]
- Angelini, C.; Freddo, L.; Battistella, P.; Bresolin, N.; Pierobon-Bormioli, S.; Armani, M.; Vergani, L. Carnitine palmityl transferase deficiency: Clinical variability, carrier detection, and autosomal-recessive inheritance. Neurology 1981, 31, 883–886. [Google Scholar] [CrossRef] [PubMed]
- Taroni, F.; Verderio, E.; Fiorucci, S.; Cavadini, P.; Finocchiaro, G.; Uziel, G.; Lamantea, E.; Gellera, C.; DiDonato, S. Molecular characterization of inherited carnitine palmitoyltransferase II deficiency. Proc. Natl. Acad. Sci. USA 1992, 89, 8429–8433. [Google Scholar] [CrossRef] [PubMed]
- Hostetler, K.Y.; Hoppel, C.L.; Romine, J.S.; Sipe, J.C.; Gross, S.R.; Higginbottom, P.A. Partial deficiency of muscle carnitine palmitoyltransferase with normal ketone production. N. Engl. J. Med. 1978, 298, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Ionasescu, V.; Hug, G.; Hoppel, C. Combined partial deficiency of muscle carnitine palmitoyltransferase and carnitine with autosomal dominant inheritance. J. Neurol. Neurosurg. Psychiatry 1980, 43, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Layzer, R.B.; Havel, R.J.; McIlroy, M.B. Partial deficiency of carnitine palmityltransferase: Physiologic and biochemical consequences. Neurology 1980, 30, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Zierz, S.; Engel, A.G. Regulatory properties of a mutant carnitine palmitoyltransferase in human skeletal muscle. Eur. J. Biochem. 1985, 149, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Vladutiu, G.D.; Saponara, I.; Conroy, J.M.; Grier, R.E.; Brady, L.; Brady, P. Immunoquantitation of carnitine palmitoyl transferase in skeletal muscle of 31 patients. Neuromuscul. Disord. 1992, 2, 249–259. [Google Scholar] [CrossRef]
- Trevisan, C.P.; Angelini, C.; Freddo, L.; Isaya, G.; Martinuzzi, A. Myoglobinuria and carnitine palmityltransferase (CPT) deficiency: Studies with malonyl-CoA suggest absence of only CPT-II. Neurology 1984, 34, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Zierz, S.; Neumann-Schmidt, S.; Jerusalem, F. Inhibition of carnitine palmitoyltransferase in normal human skeletal muscle and in muscle of patients with carnitine palmitoyltransferase deficiency by long- and short-chain acylcarnitine and acyl-coenzyme A. Clin. Investig. 1993, 71, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Zierz, S.; Mundegar, R.R.; Jerusalem, F. Biochemical evidence for heterozygosity in muscular carnitine palmitoyltransferase deficiency. Clin. Investig. 1993, 72, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Zierz, S. Limited trypsin proteolysis renders carnitine palmitoyltransferase insensitive to inhibition by malonyl-CoA in patients with muscle carnitine palmitoyltransferase deficiency. Clin. Investig. 1994, 72, 957–960. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Shepherd, I.M.; Derrick, J.P.; Ramsay, R.R.; Sherratt, H.S.; Turnbull, D.M. A case of carnitine palmitoyltransferase ii deficiency in human skeletal muscle. FEBS Lett. 1988, 241, 126–130. [Google Scholar] [CrossRef]
- Taroni, F.; Verderio, E.; Dworzak, F.; Willems, P.J.; Cavadini, P.; DiDonato, S. Identification of a common mutation in the carnitine palmitoyltransferase II gene in familial recurrent myoglobinuria patients. Nat. Genet. 1993, 4, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Olpin, S.E.; Afifi, A.; Clark, S.; Manning, N.J.; Bonham, J.R.; Dalton, A.; Leonard, J.V.; Land, J.M.; Andresen, B.S.; Morris, A.A.; et al. Mutation and biochemical analysis in carnitine palmitoyltransferase type II (CPT II) deficiency. J. Inherit. Metab. Dis. 2003, 26, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.R.; Deschauer, M.; Zierz, S. Clinically symptomatic heterozygous carnitine palmitoyltransferase II (CPT II) deficiency. Wien. Klinische Wochenschr. 2012, 124, 851–854. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, D.; Zierz, S. Normal protein content but abnormally inhibited enzyme activity in muscle carnitine palmitoyltransferase II deficiency. J. Neurol. Sci. 2014, 339, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.R.; Young, P.; Deschauer, M.; Zierz, S. Expanding mutation spectrum in CPT II gene: Identification of four novel mutations. J. Neurol. 2013, 260, 1412–1414. [Google Scholar] [CrossRef] [PubMed]
- Rufer, A.C.; Thoma, R.; Benz, J.; Stihle, M.; Gsell, B.; De Roo, E.; Banner, D.W.; Mueller, F.; Chomienne, O.; Hennig, M. The crystal structure of carnitine palmitoyltransferase 2 and implications for diabetes treatment. Structure 2006, 14, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Motlagh, L.; Golbik, R.; Sippl, W.; Zierz, S. Malony-CoA inhibits the S113L variant of carnitine-palmitoyltransferase II. Biochim. Biophys. Acta 2016, 1861, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Motlagh, L.; Golbik, R.; Sippl, W.; Zierz, S. Stabilization of the thermolabile variant S113L of carnitine palmitoyltransferase II. Neurol. Genet. 2016, 2, e53. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Cai, M.; Yao, D.; Xu, X.; Yang, R.; Li, Y.; Zhang, Y.; Kido, H.; Yao, D. Abbreviated half-lives and impaired fuel utilization in carnitine palmitoyltransferase II variant fibroblasts. PLoS ONE 2015, 10, e0119936. [Google Scholar] [CrossRef] [PubMed]
- Abu-Elheiga, L.; Almarza-Ortega, D.B.; Baldini, A.; Wakil, S.J. Human acetyl-CoA carboxylase 2. Molecular cloning, characterization, chromosomal mapping, and evidence for two isoforms. J. Biol. Chem. 1997, 272, 10669–10677. [Google Scholar] [PubMed]
- Saggerson, D. Malonyl-CoA, a key signaling molecule in mammalian cells. Annu. Rev. Nutr. 2008, 28, 253–272. [Google Scholar] [CrossRef] [PubMed]
- McGarry, J.D.; Stark, M.J.; Foster, D.W. Hepatic malonyl-CoA levels of fed, fasted and diabetic rats as measured using a simple radioisotopic assay. J. Biol. Chem. 1978, 253, 8291–8293. [Google Scholar] [PubMed]
- McGarry, J.D.; Mills, S.E.; Long, C.S.; Foster, D.W. Observations on the affinity for carnitine, and malonyl-CoA sensitivity, of carnitine palmitoyltransferase I in animal and human tissues. Demonstration of the presence of malonyl-CoA in non-hepatic tissues of the rat. Biochem. J. 1983, 214, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Corti, S.; Bordoni, A.; Ronchi, D.; Musumeci, O.; Aguennouz, M.; Toscano, A.; Lamperti, C.; Bresolin, N.; Comi, G.P. Clinical features and new molecular findings in carnitine palmitoyltransferase II (CPT II) deficiency. J. Neurol. Sci. 2008, 266, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.A.; Rubio, J.C.; De Bustos, F.; Del Hoyo, P.; Campos, Y.; Garcia, A.; Bornstein, B.; Cabello, A.; Arenas, J. Molecular analysis in spanish patients with muscle carnitine palmitoyltransferase deficiency. Muscle Nerve 1999, 22, 941–943. [Google Scholar] [CrossRef]
- Vladutiu, G.D. Biochemical and molecular correlations in carnitine palmitoyltransferase II deficiency. Muscle Nerve 1999, 22, 949–951. [Google Scholar] [CrossRef]
- Gempel, K.; von Praun, C.; Baumkotter, J.; Lehnert, W.; Ensenauer, R.; Gerbitz, K.D.; Bauer, M.F. “Adult” form of muscular carnitine palmitoyltransferase II deficiency: Manifestation in a 2-year-old child. Eur. J. Pediatr. 2001, 160, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Hurvitz, H.; Klar, A.; Korn-Lubetzki, I.; Wanders, R.J.; Elpeleg, O.N. Muscular carnitine palmitoyltransferase II deficiency in infancy. Pediatr. Neurol. 2000, 22, 148–150. [Google Scholar] [CrossRef]
- Fanin, M.; Anichini, A.; Cassandrini, D.; Fiorillo, C.; Scapolan, S.; Minetti, C.; Cassanello, M.; Donati, M.A.; Siciliano, G.; D’Amico, A.; et al. Allelic and phenotypic heterogeneity in 49 italian patients with the muscle form of CPT-II deficiency. Clin. Genet. 2012, 82, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Anichini, A.; Fanin, M.; Vianey-Saban, C.; Cassandrini, D.; Fiorillo, C.; Bruno, C.; Angelini, C. Genotype-phenotype correlations in a large series of patients with muscle type CPT II deficiency. Neurol. Res. 2011, 33, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Thuillier, L.; Sevin, C.; Demaugre, F.; Brivet, M.; Rabier, D.; Droin, V.; Aupetit, J.; Abadi, N.; Kamoun, P.; Saudubray, J.M.; et al. Genotype/phenotype correlation in carnitine palmitoyl transferase II deficiency: Lessons from a compound heterozygous patient. Neuromuscul. Disord. 2000, 10, 200–205. [Google Scholar] [CrossRef]
- Reuschenbach, C.; Zierz, S. Mutant carnitine palmitoyltransferase associated with myoadenylate deaminase deficiency in skeletal muscle. J. Pediatr. 1988, 112, 600–603. [Google Scholar] [CrossRef]
- Engel, A.G.; Angelini, C. Carnitine deficiency of human skeletal muscle with associated lipid storage myopathy: A new syndrome. Science 1973, 179, 899–902. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lehmann, D.; Motlagh, L.; Robaa, D.; Zierz, S. Muscle Carnitine Palmitoyltransferase II Deficiency: A Review of Enzymatic Controversy and Clinical Features. Int. J. Mol. Sci. 2017, 18, 82. https://doi.org/10.3390/ijms18010082
Lehmann D, Motlagh L, Robaa D, Zierz S. Muscle Carnitine Palmitoyltransferase II Deficiency: A Review of Enzymatic Controversy and Clinical Features. International Journal of Molecular Sciences. 2017; 18(1):82. https://doi.org/10.3390/ijms18010082
Chicago/Turabian StyleLehmann, Diana, Leila Motlagh, Dina Robaa, and Stephan Zierz. 2017. "Muscle Carnitine Palmitoyltransferase II Deficiency: A Review of Enzymatic Controversy and Clinical Features" International Journal of Molecular Sciences 18, no. 1: 82. https://doi.org/10.3390/ijms18010082
APA StyleLehmann, D., Motlagh, L., Robaa, D., & Zierz, S. (2017). Muscle Carnitine Palmitoyltransferase II Deficiency: A Review of Enzymatic Controversy and Clinical Features. International Journal of Molecular Sciences, 18(1), 82. https://doi.org/10.3390/ijms18010082