APE1/Ref-1 Inhibits Phosphate-Induced Calcification and Osteoblastic Phenotype Changes in Vascular Smooth Muscle Cells


 , and
, and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
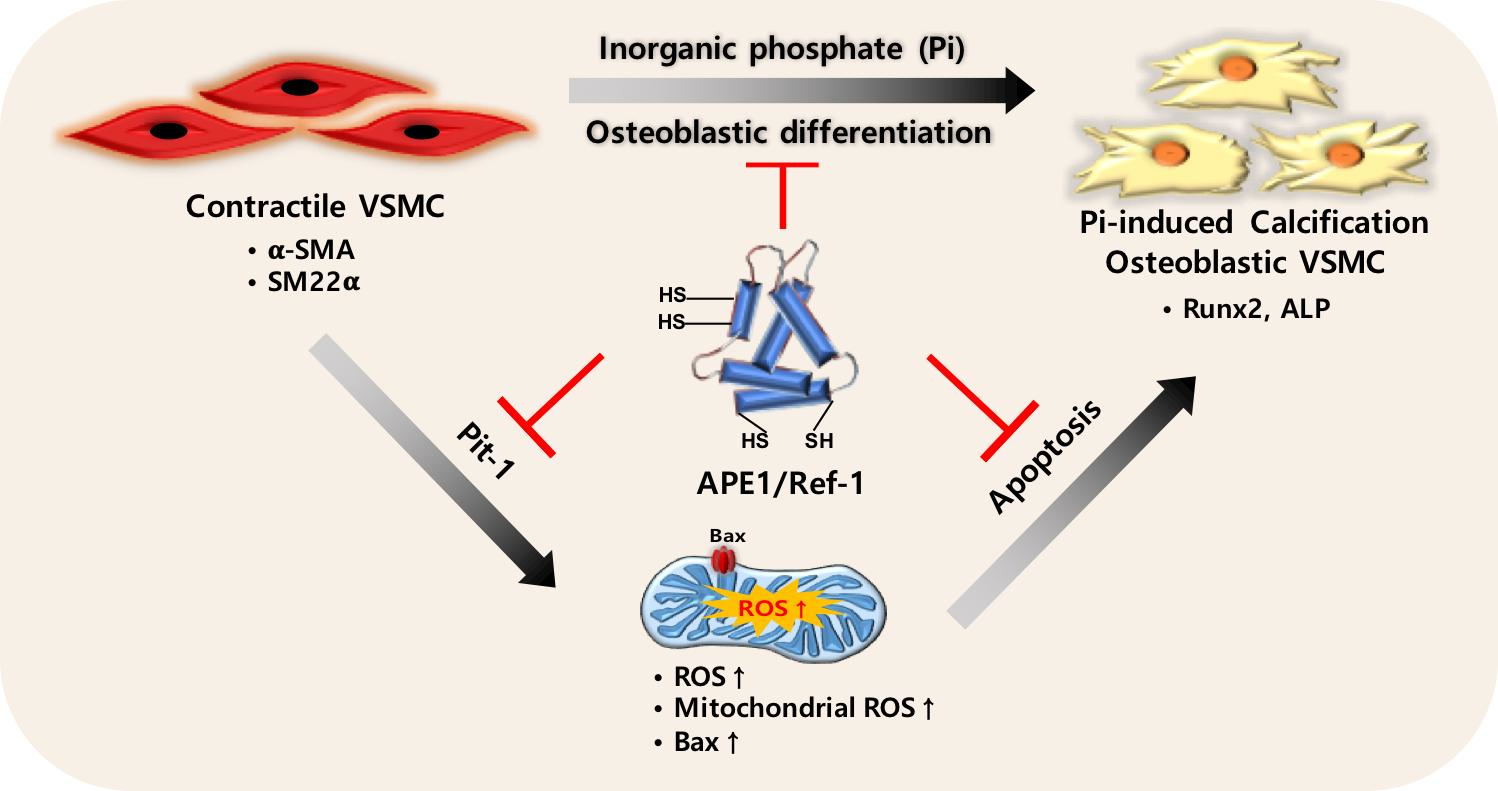
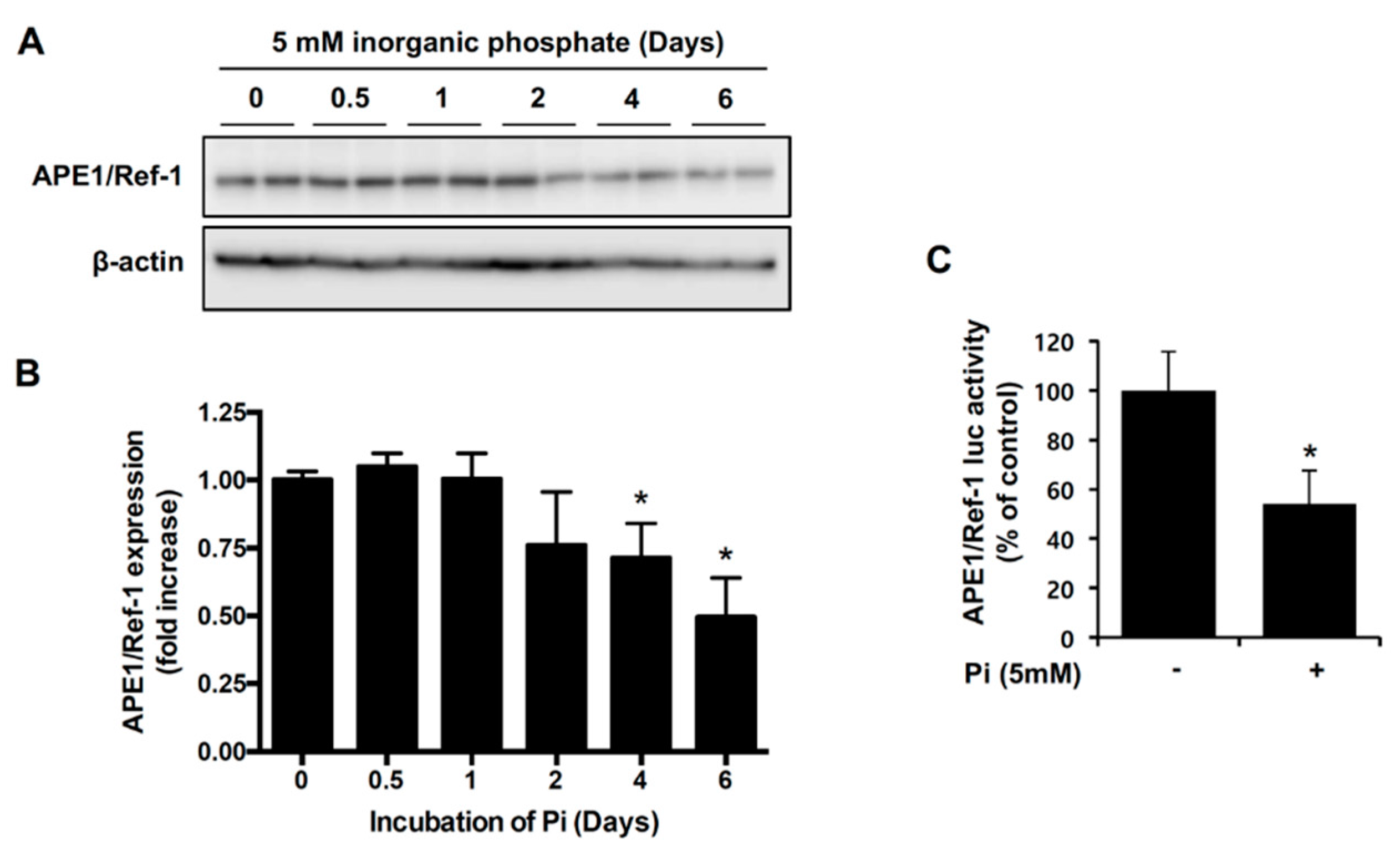
2.1. Pi Inhibits APE1/Ref-1 Transcription and Protein Expression
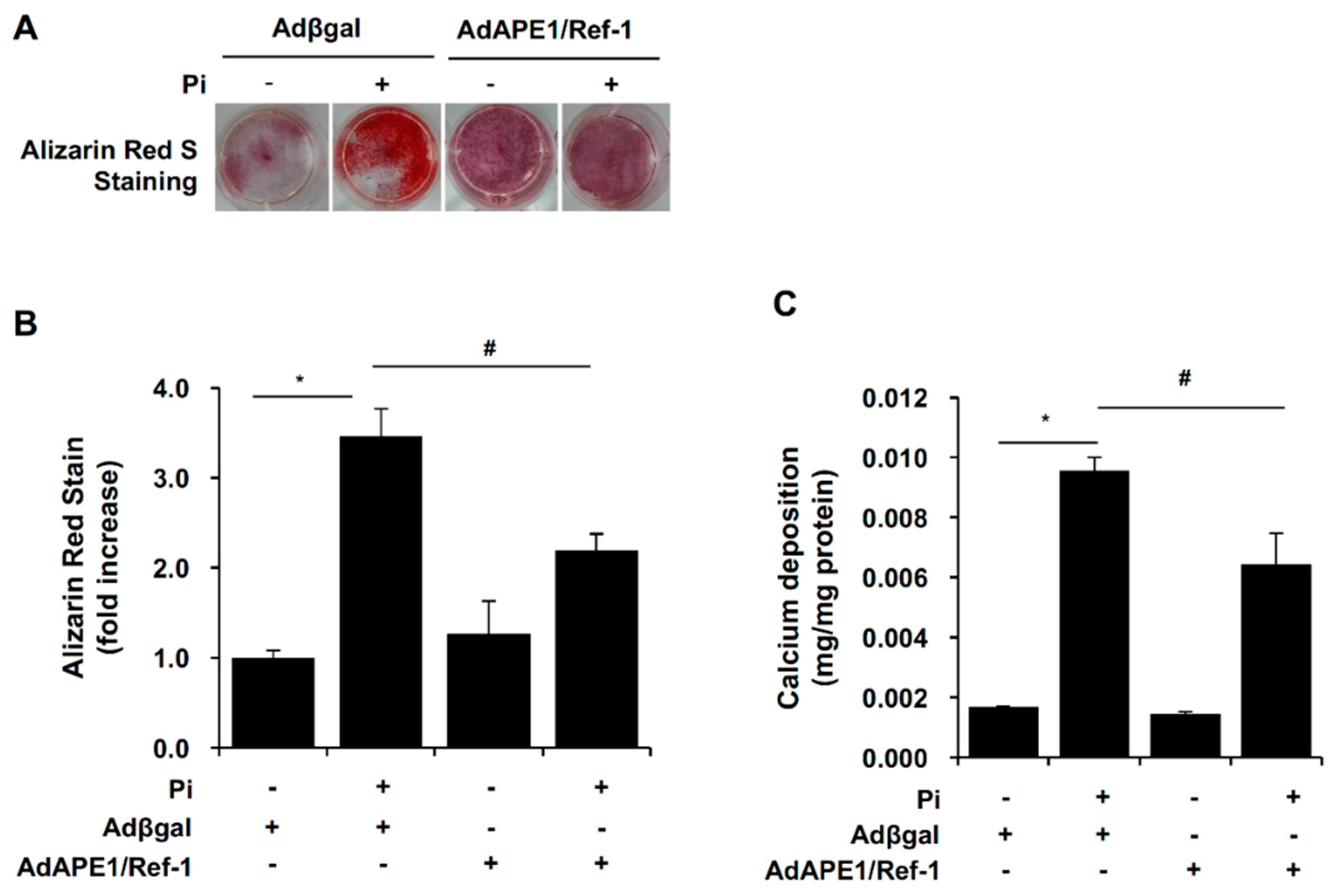
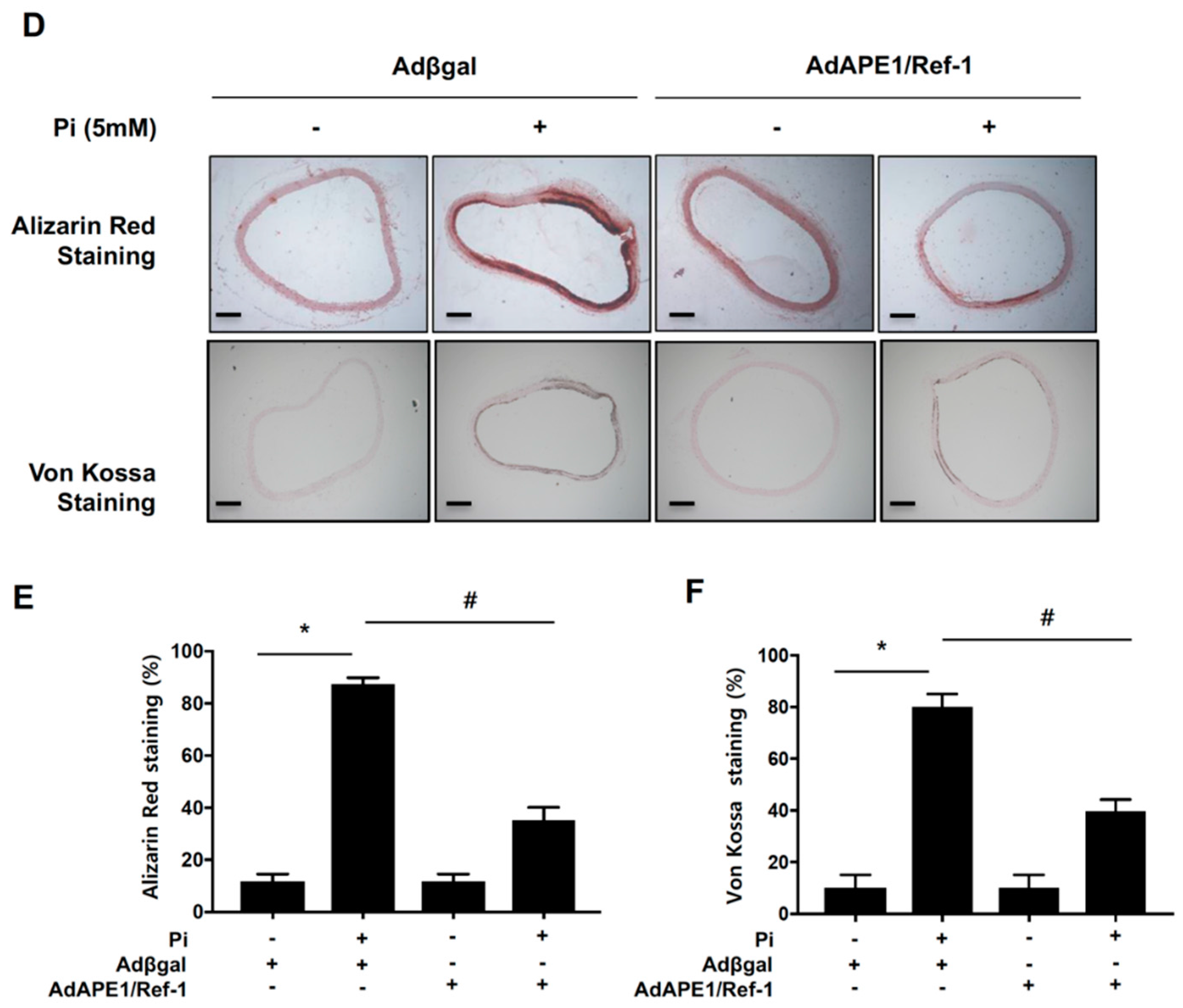
2.2. APE1/Ref-1 Suppresses Pi-Induced Calcification in VSMCs
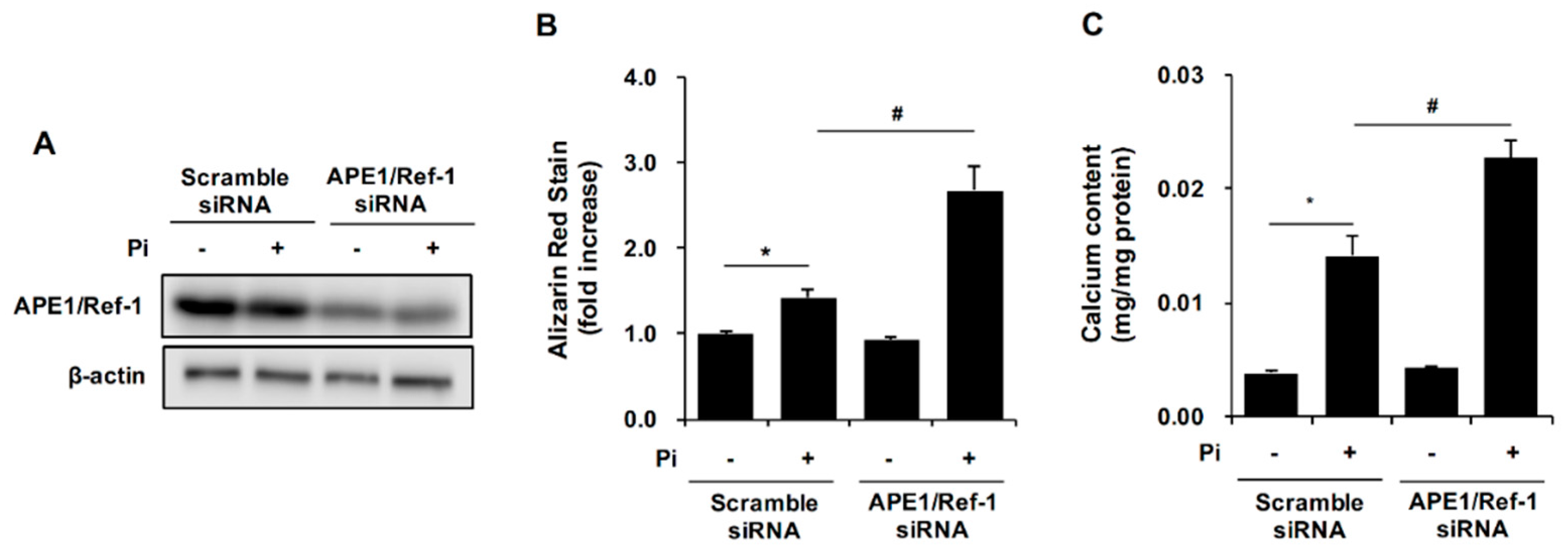
2.3. APE1/Ref-1 Gene Silencing Aggravates Pi-Induced Calcification
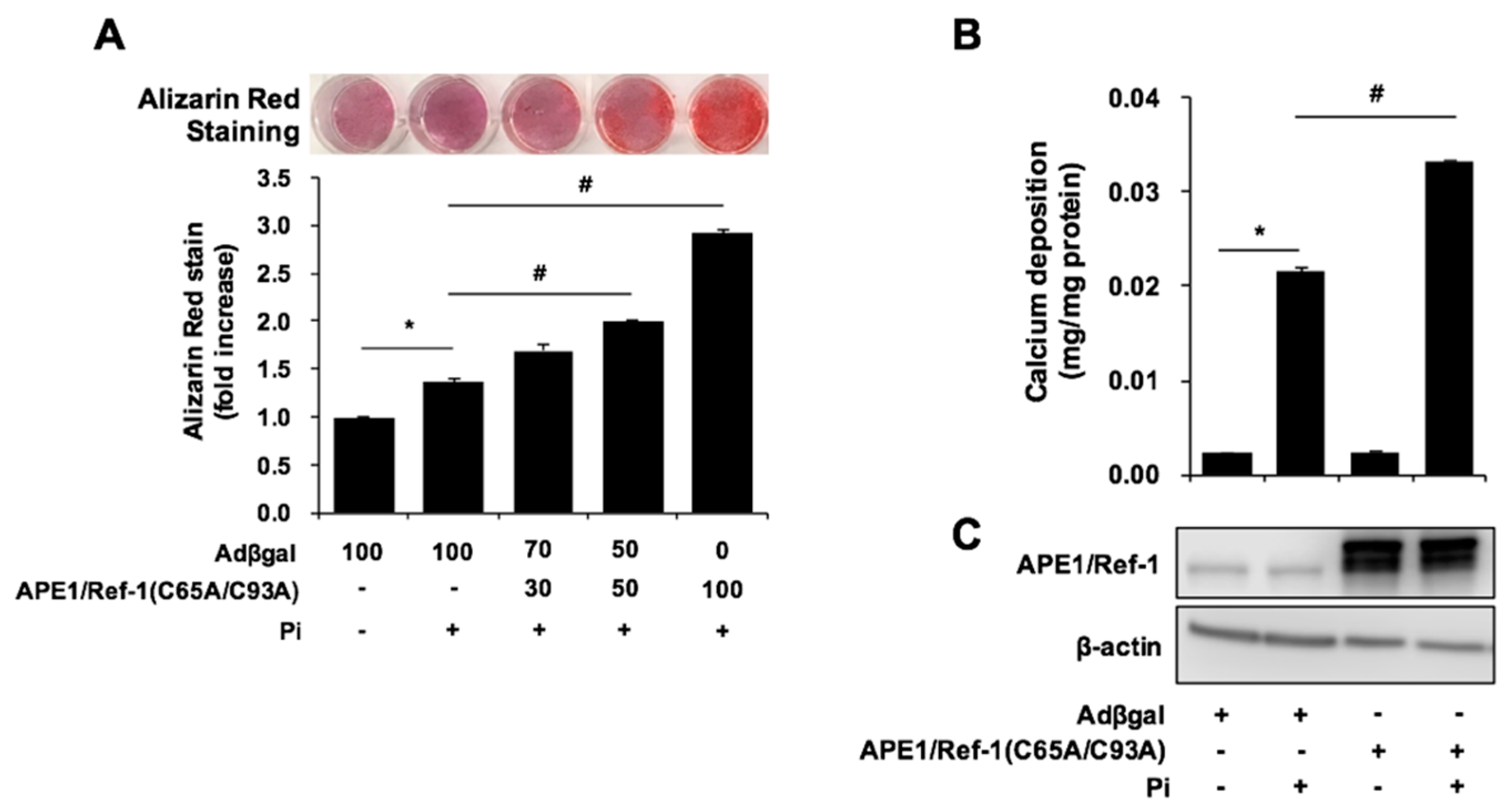
2.4. APE1/Ref-1 Redox Mutants Are Unable to Suppress Pi-Induced VSMC Calcification
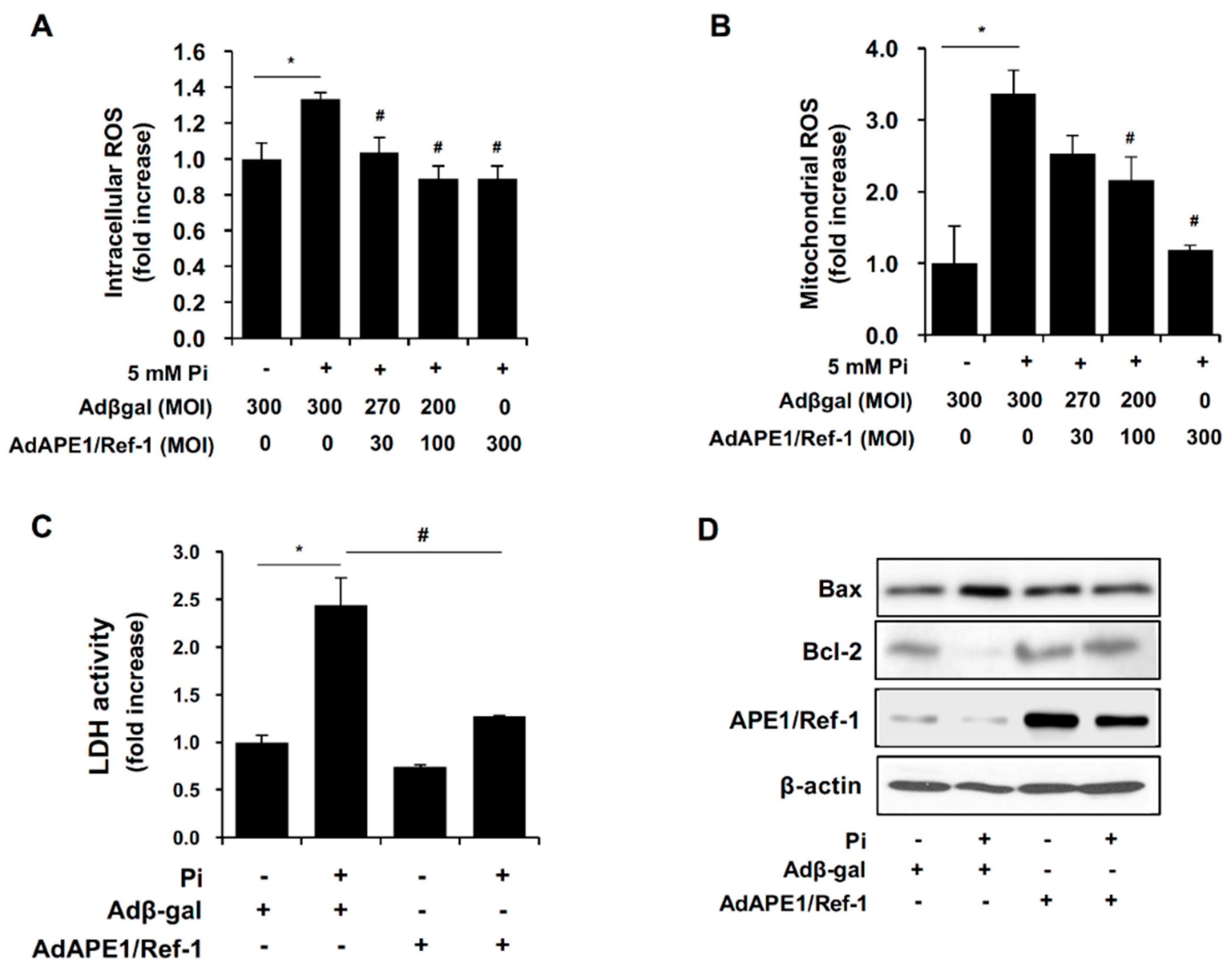
2.5. APE1/Ref-1 Suppresses Pi-Induced ROS Production and Apoptosis
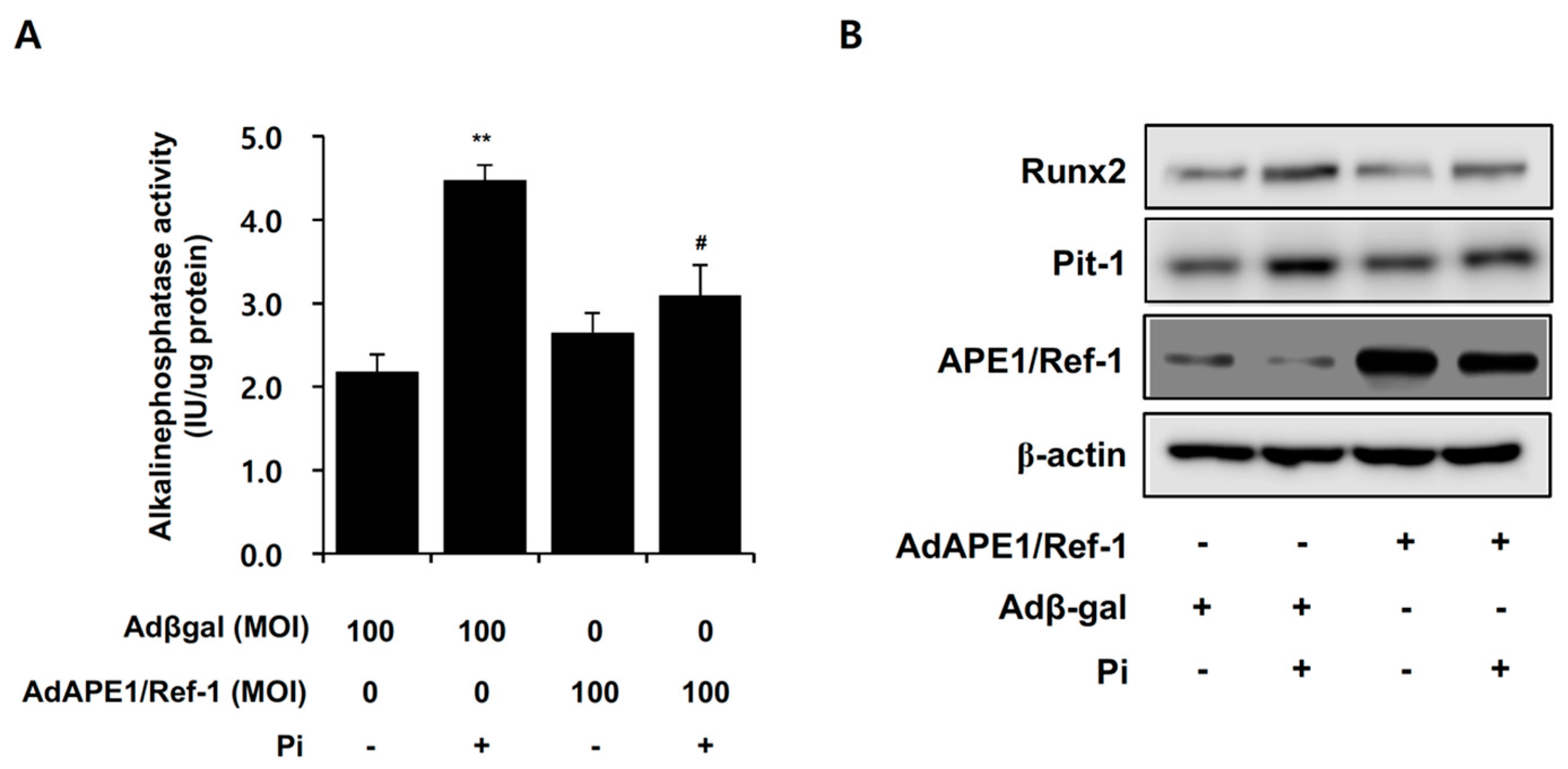
2.6. APE1/Ref-1 Suppresses Pi-Induced Alkaline Phosphatase (ALP) Activity and Osteoblastic Differentiation
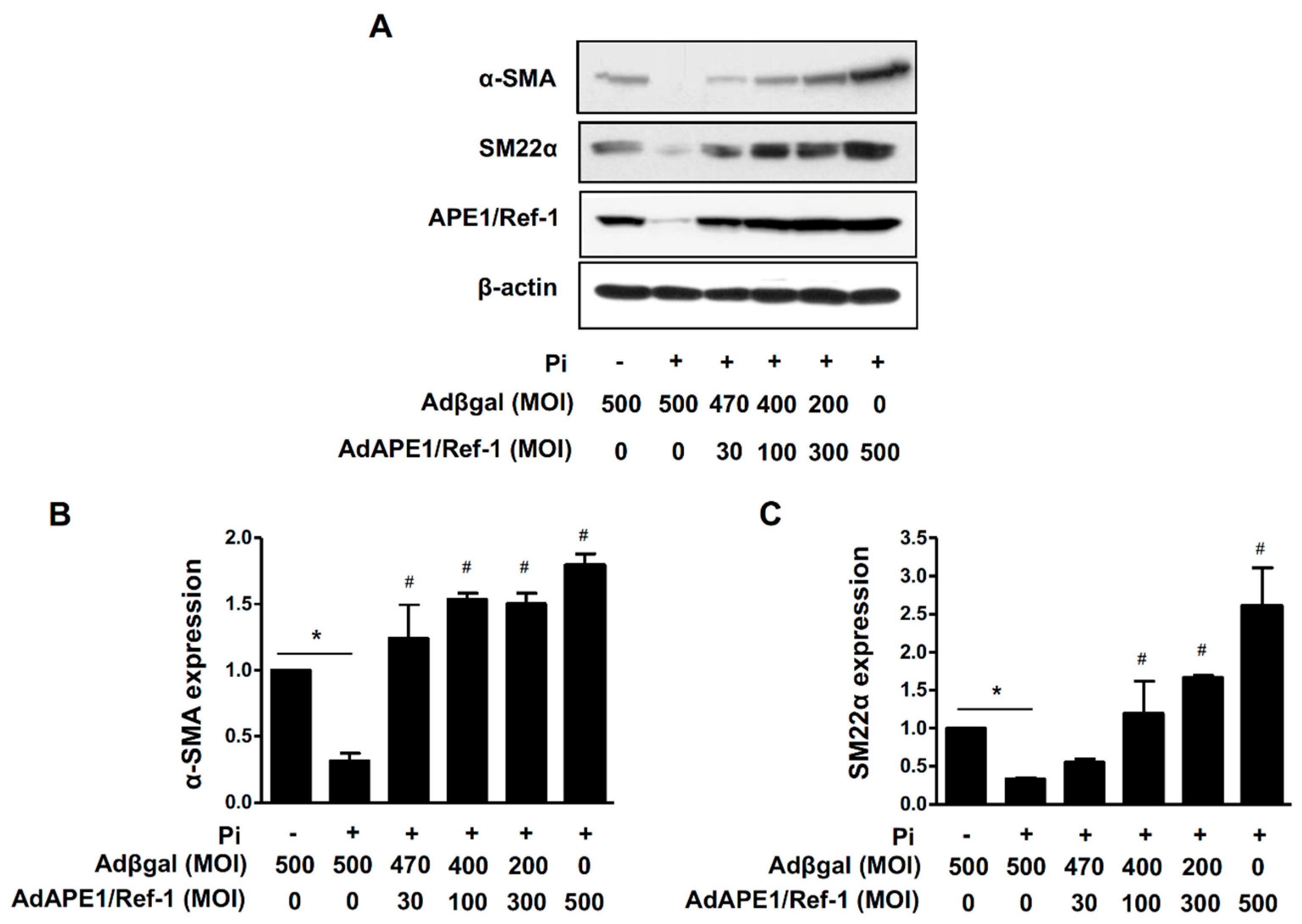
2.7. AP1/Ref-1 Suppresses Pi-Induced Loss of VSMC Phenotype
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Primary VSMC Isolation and Cell Culture
4.3. Induction of VSMC Calcification In Vitro
4.4. Adenoviral Transfections
4.5. APE1/Ref-1 siRNA
4.6. Evaluation of VSMC Calcification
4.7. Western Blot Analysis
4.8. APE1/Ref-1 Luciferase Reporter Assay
4.9. Intracellular and Mitochondrial ROS
4.10. LDH Activity
4.11. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| APE1/Ref-1 | Aprinic/apyrimidinic endonuclease 1/redox factor-1 |
| Pi | Inorganic phosphate |
| VSMC | Vascular smooth muscle cell |
| LDH | Lactate dehydrogenase |
| ALP | Alkaline phosphatase |
| Runx2 | Runt-related transcription factor 2 |
| Pit-1 | Pituitary-specific positive transcription factor 1 |
| α-SMA | α-Smooth muscle actin |
| SM22α | Smooth muscle protein 22-α |
| ROS | Reactive oxygen species |
| NF-κB | Nuclear factor-kappa B |
| FBS | Fetal bovine serum |
| MOI | Multiplicity of infection |
| DMEM | Dulbecco’s modified Eagle medium |
| H2DCFDA | 2′,7′-dichlorofluorescin diacetate |
| siRNA | Small-interfering RNA |
References
- London, G.M.; Guerin, A.P.; Marchais, S.J.; Metivier, F.; Pannier, B.; Adda, H. Arterial media calcification in end-stage renal disease: Impact on all-cause and cardiovascular mortality. Nephrol. Dial. Transplant. 2003, 18, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Agharazii, M.; St-Louis, R.; Gautier-Bastien, A.; Ung, R.V.; Mokas, S.; Lariviere, R.; Richard, D.E. Inflammatory cytokines and reactive oxygen species as mediators of chronic kidney disease-related vascular calcification. Am. J. Hypertens. 2015, 28, 746–755. [Google Scholar] [CrossRef] [PubMed]
- Angkeow, P.; Deshpande, S.S.; Qi, B.; Liu, Y.X.; Park, Y.C.; Jeon, B.H.; Ozaki, M.; Irani, K. Redox factor-1: An extra-nuclear role in the regulation of endothelial oxidative stress and apoptosis. Cell Death Differ. 2002, 9, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, M.; Suzuki, S.; Irani, K. Redox factor-1/APE suppresses oxidative stress by inhibiting the rac1 GTPase. FASEB J. 2002, 16, 889–890. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Chen, J.; Zhao, T.; Fan, Z. Granzyme K degrades the redox/DNA repair enzyme Ape1 to trigger oxidative stress of target cells leading to cytotoxicity. Mol. Immunol. 2008, 45, 2225–2235. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.S.; Son, S.J.; Kim, E.K.; Kim, S.N.; Yoo, D.G.; Kim, H.S.; Ryoo, S.W.; Lee, S.D.; Irani, K.; Jeon, B.H. Apurinic/apyrimidinic endonuclease1/redox factor-1 inhibits monocyte adhesion in endothelial cells. Cardiovasc. Res. 2006, 69, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.L.; Wang, X.; Van, A.; Zhao, Y.; Gibbons, G.H. Overexpression of Ref-1 inhibits hypoxia and tumor necrosis factor-induced endothelial cell apoptosis through nuclear factor-kappab-independent and -dependent pathways. Circ. Res. 2001, 88, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Yuk, J.M.; Yang, C.S.; Shin, D.M.; Kim, K.K.; Lee, S.K.; Song, Y.J.; Lee, H.M.; Cho, C.H.; Jeon, B.H.; Jo, E.K. A dual regulatory role of apurinic/apyrimidinic endonuclease 1/redox factor-1 in HMGB1-induced inflammatory responses. Antioxid. Redox Signal. 2009, 11, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Iyemere, V.P.; Proudfoot, D.; Weissberg, P.L.; Shanahan, C.M. Vascular smooth muscle cell phenotypic plasticity and the regulation of vascular calcification. J. Intern. Med. 2006, 260, 192–210. [Google Scholar] [CrossRef] [PubMed]
- Byon, C.H.; Javed, A.; Dai, Q.; Kappes, J.C.; Clemens, T.L.; Darley-Usmar, V.M.; McDonald, J.M.; Chen, Y. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J. Biol. Chem. 2008, 283, 15319–15327. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Joo, H.K.; Jeon, B.H. Dynamic Regulation of APE1/Ref-1 as a Therapeutic Target Protein. Chonnam Med. J. 2016, 52, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.Y.; Jing, K.; Song, K.S.; Seo, K.S.; Park, J.H.; Kim, J.S.; Jung, Y.J.; Hur, G.M.; Jo, D.Y.; Kweon, G.R.; et al. Downregulation of APE1/Ref-1 is involved in the senescence of mesenchymal stem cells. Stem Cells 2009, 27, 1455–1462. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Liu, Z.; Sun, W.; Li, J.; Liang, Y.; Yang, X.; Xu, Y.; Yu, M.; Tian, W.; Chen, G.; et al. Inhibition of Ape1 Redox Activity Promotes Odonto/osteogenic Differentiation of Dental Papilla Cells. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Ciceri, P.; Volpi, E.; Brenna, I.; Arnaboldi, L.; Neri, L.; Brancaccio, D.; Cozzolino, M. Combined effects of ascorbic acid and phosphate on rat VSMC osteoblastic differentiation. Nephrol. Dial. Transplant. 2012, 27, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Akiyoshi, T.; Ota, H.; Iijima, K.; Son, B.K.; Kahyo, T.; Setou, M.; Ogawa, S.; Ouchi, Y.; Akishita, M. A novel organ culture model of aorta for vascular calcification. Atherosclerosis 2016, 244, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Park, M.S.; Kim, C.S.; Joo, H.K.; Lee, Y.R.; Kang, G.; Kim, S.J.; Choi, S.; Lee, S.D.; Park, J.B.; Jeon, B.H. Cytoplasmic localization and redox cysteine residue of APE1/Ref-1 are associated with its anti-inflammatory activity in cultured endothelial cells. Mol. Cells 2013, 36, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Xanthoudakis, S.; Miao, G.G.; Curran, T. The redox and DNA-repair activities of Ref-1 are encoded by nonoverlapping domains. Proc. Natl. Acad. Sci. USA 1994, 91, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Orimo, H. The mechanism of mineralization and the role of alkaline phosphatase in health and disease. J. Nippon Med. Sch. 2010, 77, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Lau, W.L.; Pai, A.; Moe, S.M.; Giachelli, C.M. Direct effects of phosphate on vascular cell function. Adv. Chronic Kidney Dis. 2011, 18, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Miano, J.M.; Cserjesi, P.; Olson, E.N. SM22 alpha, a marker of adult smooth muscle, is expressed in multiple myogenic lineages during embryogenesis. Circ. Res. 1996, 78, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Bhakat, K.K.; Mantha, A.K.; Mitra, S. Transcriptional regulatory functions of mammalian AP-endonuclease (APE1/Ref-1), an essential multifunctional protein. Antioxid. Redox Signal. 2009, 11, 621–638. [Google Scholar] [CrossRef] [PubMed]
- Fritz, G.; Grosch, S.; Tomicic, M.; Kaina, B. APE/Ref-1 and the mammalian response to genotoxic stress. Toxicology 2003, 193, 67–78. [Google Scholar] [CrossRef]
- Edwards, M.; Kent, T.A.; Rea, H.C.; Wei, J.; Quast, M.; Izumi, T.; Mitra, S.; Perez-Polo, J.R. APE/Ref-1 responses to ischemia in rat brain. Neuroreport 1998, 9, 4015–4018. [Google Scholar] [CrossRef] [PubMed]
- Fridman, J.S.; Lowe, S.W. Control of apoptosis by p53. Oncogene 2003, 22, 9030–9040. [Google Scholar] [CrossRef] [PubMed]
- Zaky, A.; Busso, C.; Izumi, T.; Chattopadhyay, R.; Bassiouny, A.; Mitra, S.; Bhakat, K.K. Regulation of the human AP-endonuclease (APE1/Ref-1) expression by the tumor suppressor p53 in response to DNA damage. Nucleic Acids Res. 2008, 36, 1555–1566. [Google Scholar] [CrossRef] [PubMed]
- Park, M.S.; Choi, S.; Lee, Y.R.; Joo, H.K.; Kang, G.; Kim, C.S.; Kim, S.J.; Lee, S.D.; Jeon, B.H. Secreted APE1/Ref-1 inhibits TNF-alpha-stimulated endothelial inflammation via thiol-disulfide exchange in TNF receptor. Sci. Rep. 2016, 6, 23015. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Luo, M.; Kelley, M.R. Human apurinic endonuclease 1 (APE1) expression and prognostic significance in osteosarcoma: Enhanced sensitivity of osteosarcoma to DNA damaging agents using silencing RNA APE1 expression inhibition. Mol. Cancer Ther. 2004, 3, 679–686. [Google Scholar] [PubMed]
- Mansfield, K.; Pucci, B.; Adams, C.S.; Shapiro, I.M. Induction of apoptosis in skeletal tissues: Phosphate-mediated chick chondrocyte apoptosis is calcium dependent. Calcif. Tissue Int. 2003, 73, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.M.; Xu, M.J.; Cai, Y.; Zhao, G.; Guan, Y.; Kong, W.; Tang, C.; Wang, X. Mitochondrial reactive oxygen species promote p65 nuclear translocation mediating high-phosphate-induced vascular calcification in vitro and in vivo. Kidney Int. 2011, 79, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Alesutan, I.; Voelkl, J.; Feger, M.; Kratschmar, D.V.; Castor, T.; Mia, S.; Sacherer, M.; Viereck, R.; Borst, O.; Leibrock, C.; et al. Involvement of Vascular Aldosterone Synthase in Phosphate-Induced Osteogenic Transformation of Vascular Smooth Muscle Cells. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.M.; Campbell, G.R.; Campbell, J.H. Replication of smooth muscle cells in vascular disease. Circ. Res. 1986, 58, 427–444. [Google Scholar] [CrossRef] [PubMed]
- Kendrick, J.; Chonchol, M. The role of phosphorus in the development and progression of vascular calcification. Am. J. Kidney Dis. 2011, 58, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Bennett, M.R.; Shanahan, C.M.; Weissberg, P.L. Apoptosis regulates human vascular calcification in vitro: Evidence for initiation of vascular calcification by apoptotic bodies. Circ. Res. 2000, 87, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Son, B.K.; Kozaki, K.; Iijima, K.; Eto, M.; Kojima, T.; Ota, H.; Senda, Y.; Maemura, K.; Nakano, T.; Akishita, M.; et al. Statins protect human aortic smooth muscle cells from inorganic phosphate-induced calcification by restoring Gas6-Axl survival pathway. Circ. Res. 2006, 98, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
- Shavelle, D.M.; Takasu, J.; Budoff, M.J.; Mao, S.; Zhao, X.Q.; O’Brien, K.D. HMG CoA reductase inhibitor (statin) and aortic valve calcium. Lancet 2002, 359, 1125–1126. [Google Scholar] [CrossRef]
- Montes de Oca, A.; Madueno, J.A.; Martinez-Moreno, J.M.; Guerrero, F.; Munoz-Castaneda, J.; Rodriguez-Ortiz, M.E.; Mendoza, F.J.; Almaden, Y.; Lopez, I.; Rodriguez, M.; et al. High-phosphate-induced calcification is related to SM22alpha promoter methylation in vascular smooth muscle cells. J. Bone Miner. Res. 2010, 25, 1996–2005. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Fu, J.; Chen, J.; Xiao, P.; Lan, T.; Le, K.; Cheng, F.; He, L.; Shen, X.; Huang, H.; et al. Development of an optimized protocol for primary culture of smooth muscle cells from rat thoracic aortas. Cytotechnology 2009, 61, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Jeon, B.H.; Gupta, G.; Park, Y.C.; Qi, B.; Haile, A.; Khanday, F.A.; Liu, Y.X.; Kim, J.M.; Ozaki, M.; White, A.R.; et al. Apurinic/apyrmidinic endonuclease 1 regulates endothelial NO production and vascular tone. Circ. Res. 2004, 95, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.Y.; Kwun, I.S. Phosphate-induced rat vascular smooth muscle cell calcification and the implication of zinc deficiency in a7r5 cell viability. Prev. Nutr. Food Sci. 2013, 18, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Joo, H.K.; Lee, Y.R.; Kang, G.; Choi, S.; Kim, C.S.; Ryoo, S.; Park, J.B.; Jeon, B.H. The 18-kDa Translocator Protein Inhibits Vascular Cell Adhesion Molecule-1 Expression via Inhibition of Mitochondrial Reactive Oxygen Species. Mol. Cells 2015, 38, 1064–1070. [Google Scholar] [PubMed]








© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, K.M.; Lee, E.O.; Lee, Y.R.; Joo, H.K.; Park, M.S.; Kim, C.-S.; Choi, S.; Jeong, J.-O.; Jeon, B.H. APE1/Ref-1 Inhibits Phosphate-Induced Calcification and Osteoblastic Phenotype Changes in Vascular Smooth Muscle Cells. Int. J. Mol. Sci. 2017, 18, 2053. https://doi.org/10.3390/ijms18102053
Lee KM, Lee EO, Lee YR, Joo HK, Park MS, Kim C-S, Choi S, Jeong J-O, Jeon BH. APE1/Ref-1 Inhibits Phosphate-Induced Calcification and Osteoblastic Phenotype Changes in Vascular Smooth Muscle Cells. International Journal of Molecular Sciences. 2017; 18(10):2053. https://doi.org/10.3390/ijms18102053
Chicago/Turabian StyleLee, Ki Mo, Eun Ok Lee, Yu Ran Lee, Hee Kyoung Joo, Myoung Soo Park, Cuk-Seong Kim, Sunga Choi, Jin-Ok Jeong, and Byeong Hwa Jeon. 2017. "APE1/Ref-1 Inhibits Phosphate-Induced Calcification and Osteoblastic Phenotype Changes in Vascular Smooth Muscle Cells" International Journal of Molecular Sciences 18, no. 10: 2053. https://doi.org/10.3390/ijms18102053
APA StyleLee, K. M., Lee, E. O., Lee, Y. R., Joo, H. K., Park, M. S., Kim, C. -S., Choi, S., Jeong, J. -O., & Jeon, B. H. (2017). APE1/Ref-1 Inhibits Phosphate-Induced Calcification and Osteoblastic Phenotype Changes in Vascular Smooth Muscle Cells. International Journal of Molecular Sciences, 18(10), 2053. https://doi.org/10.3390/ijms18102053