Primary Cilium-Dependent Signaling Mechanisms


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
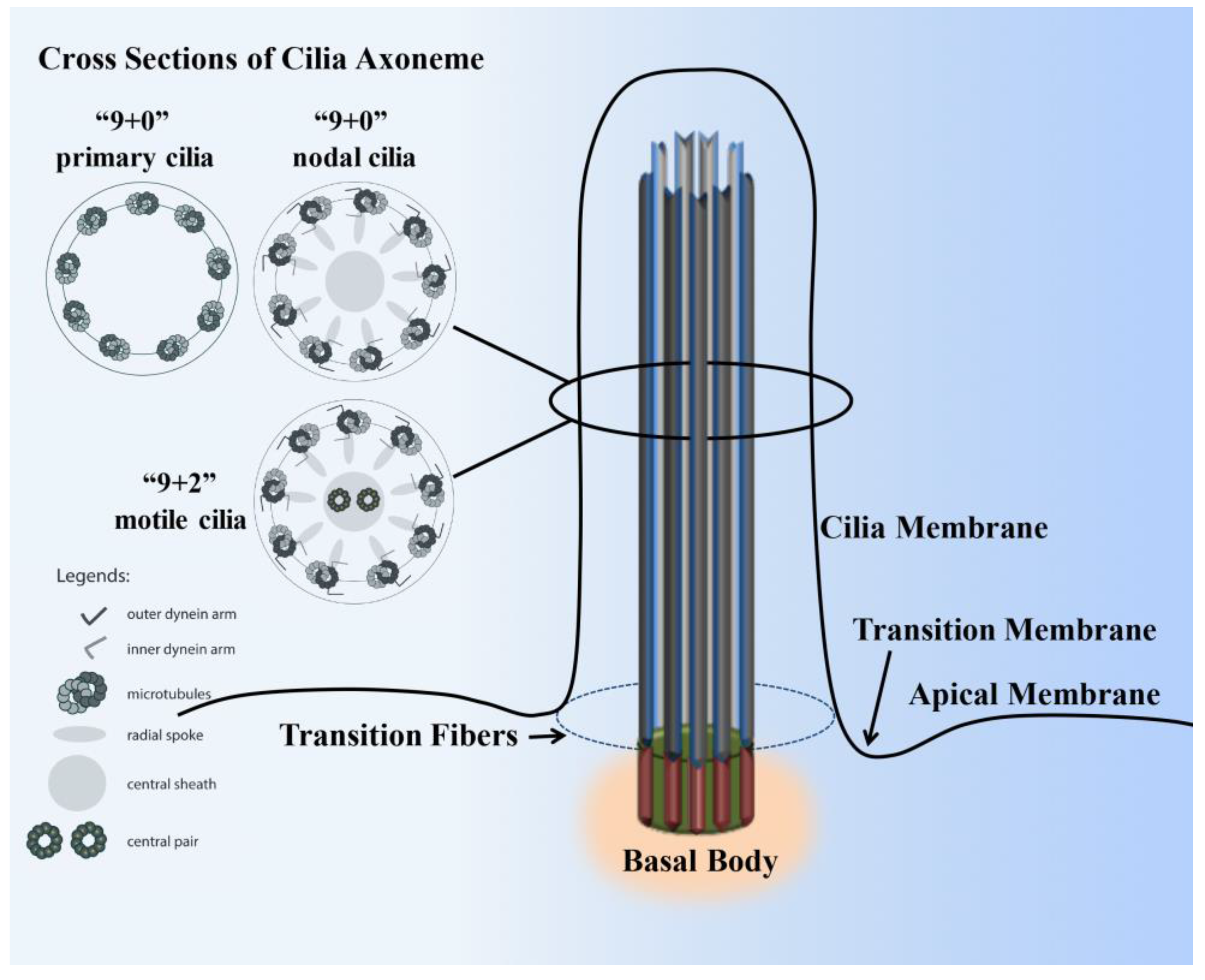
2. Cilia Structure
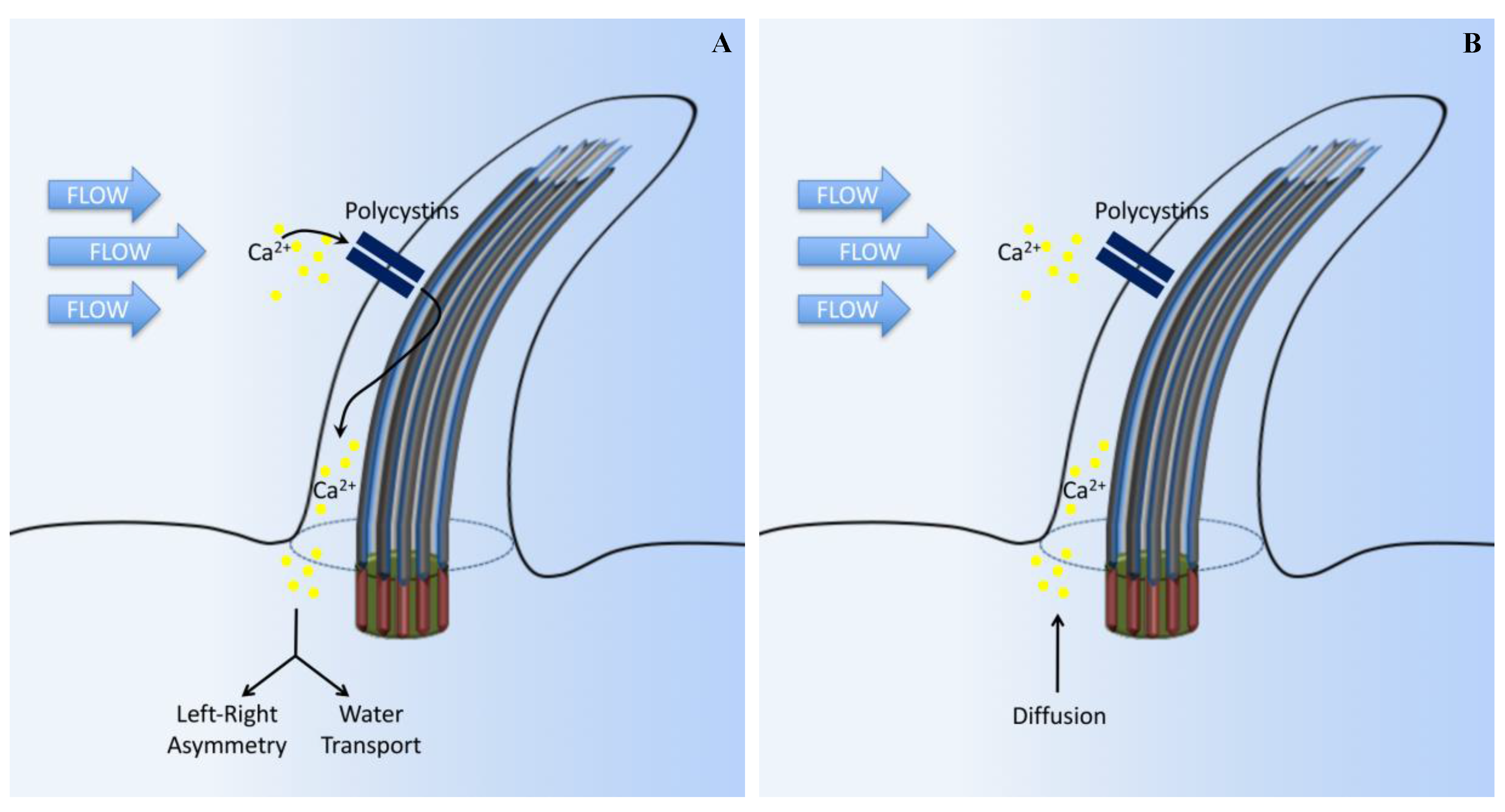
3. Calcium Signaling
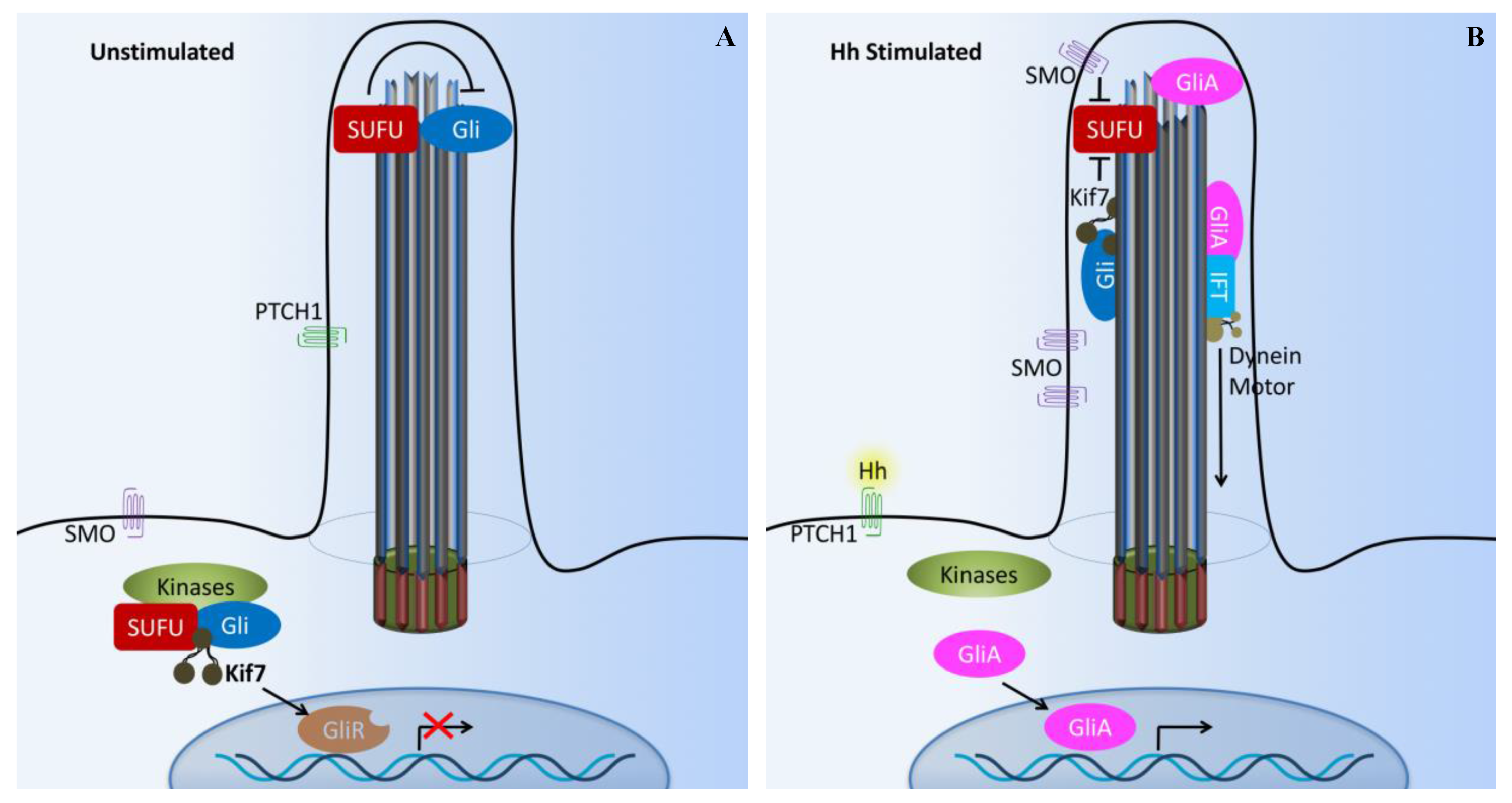
4. Hedgehog Signaling
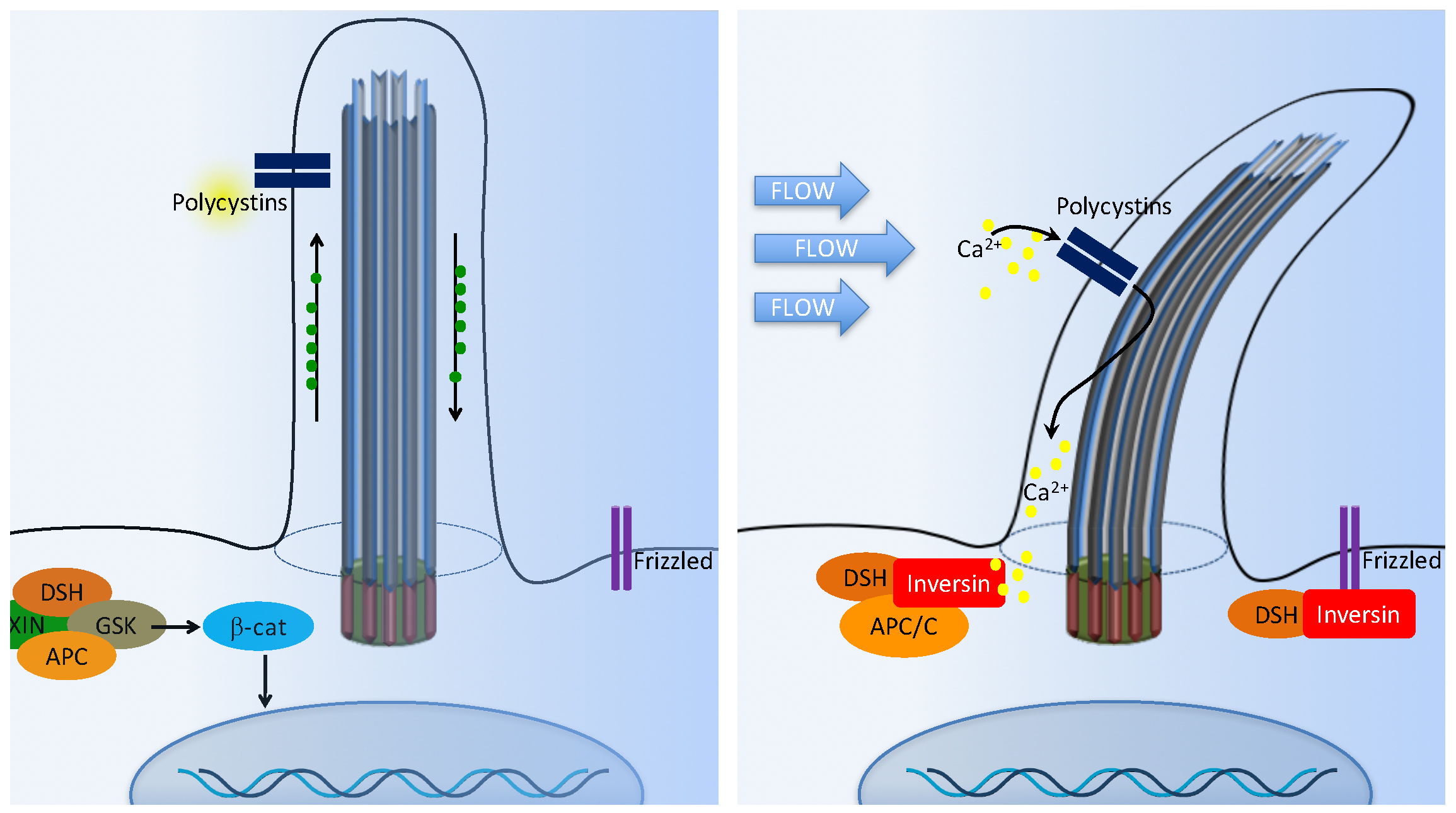
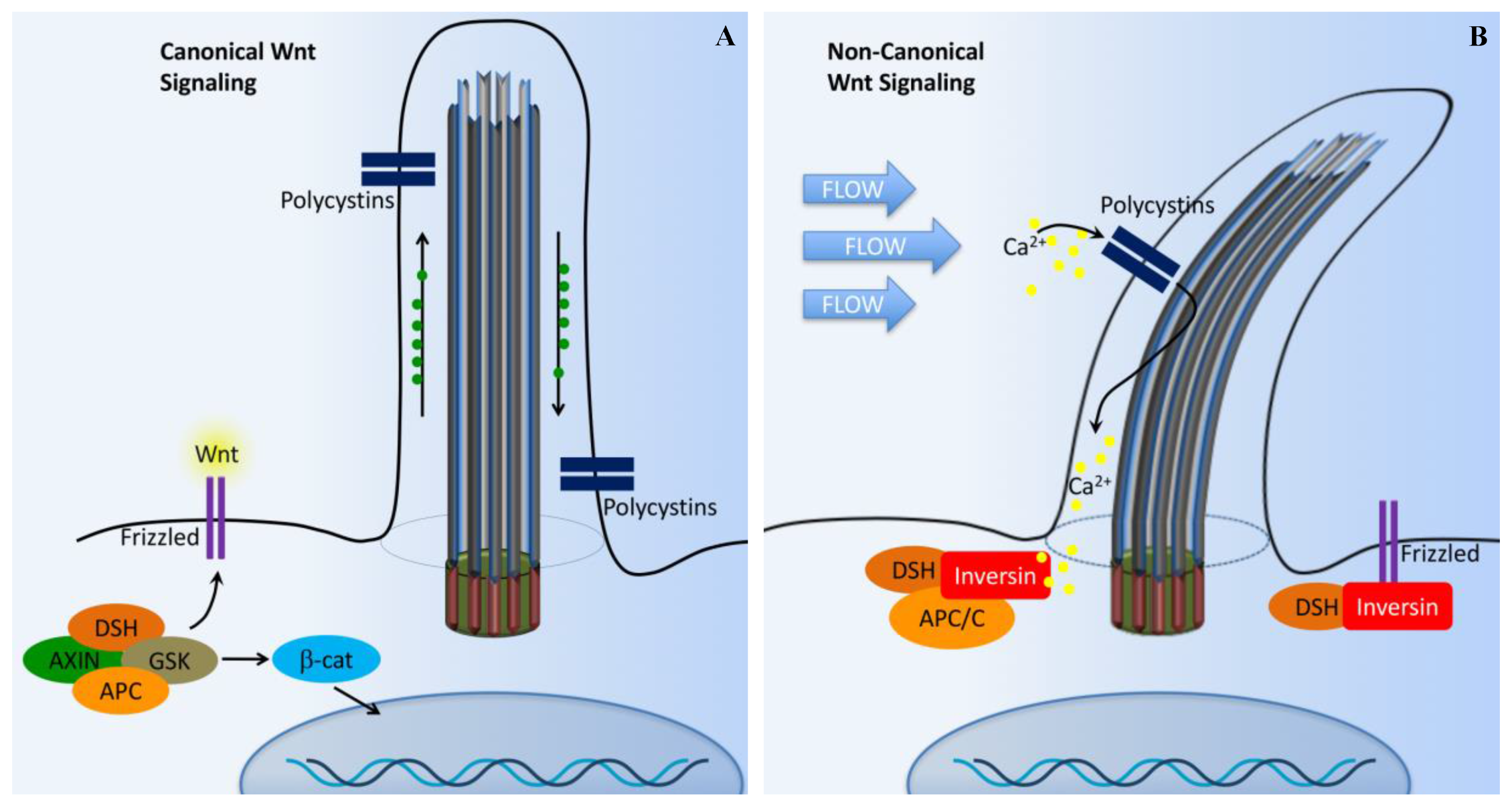
5. Wnt Signaling
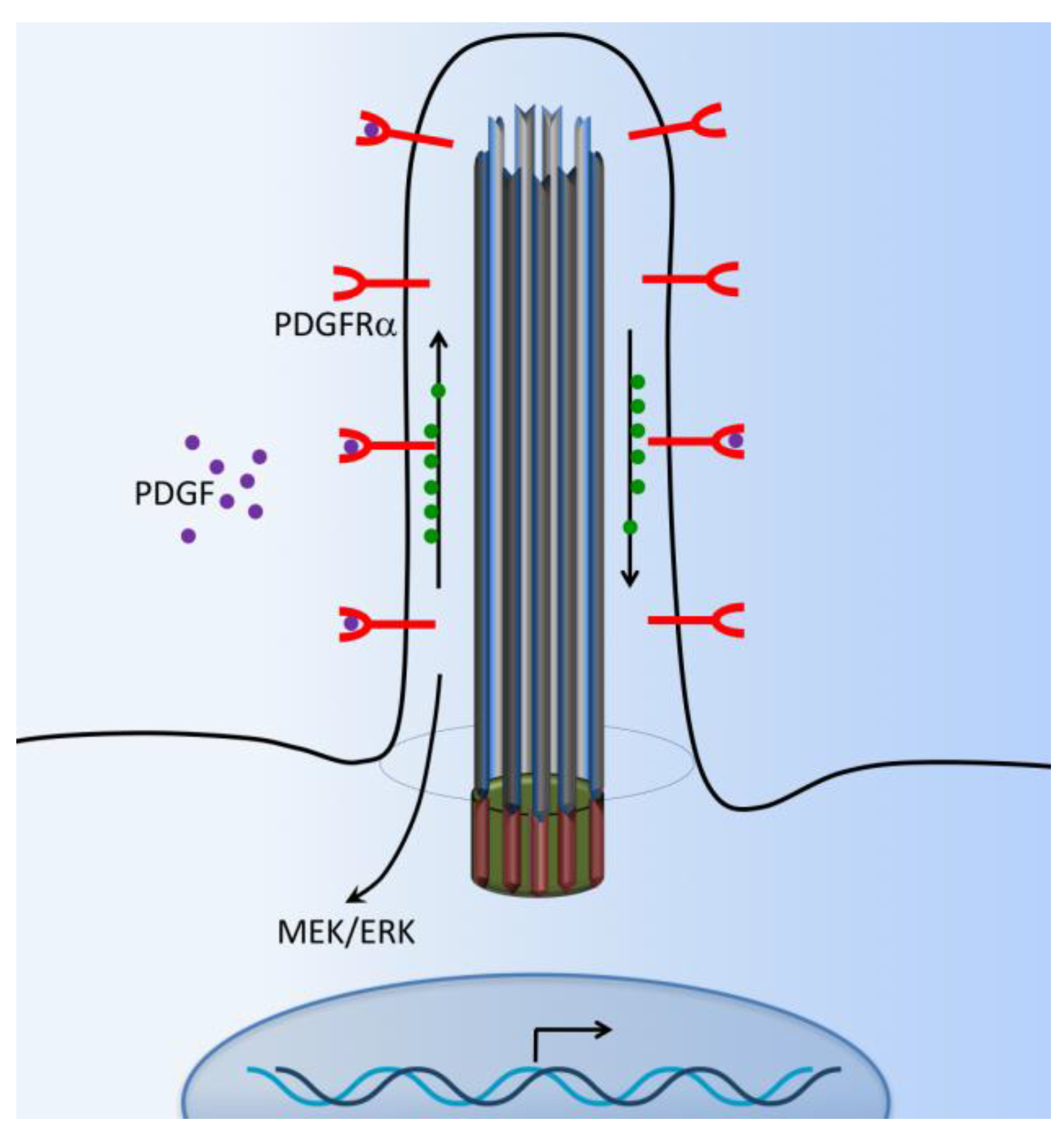
6. PDGFR Signaling
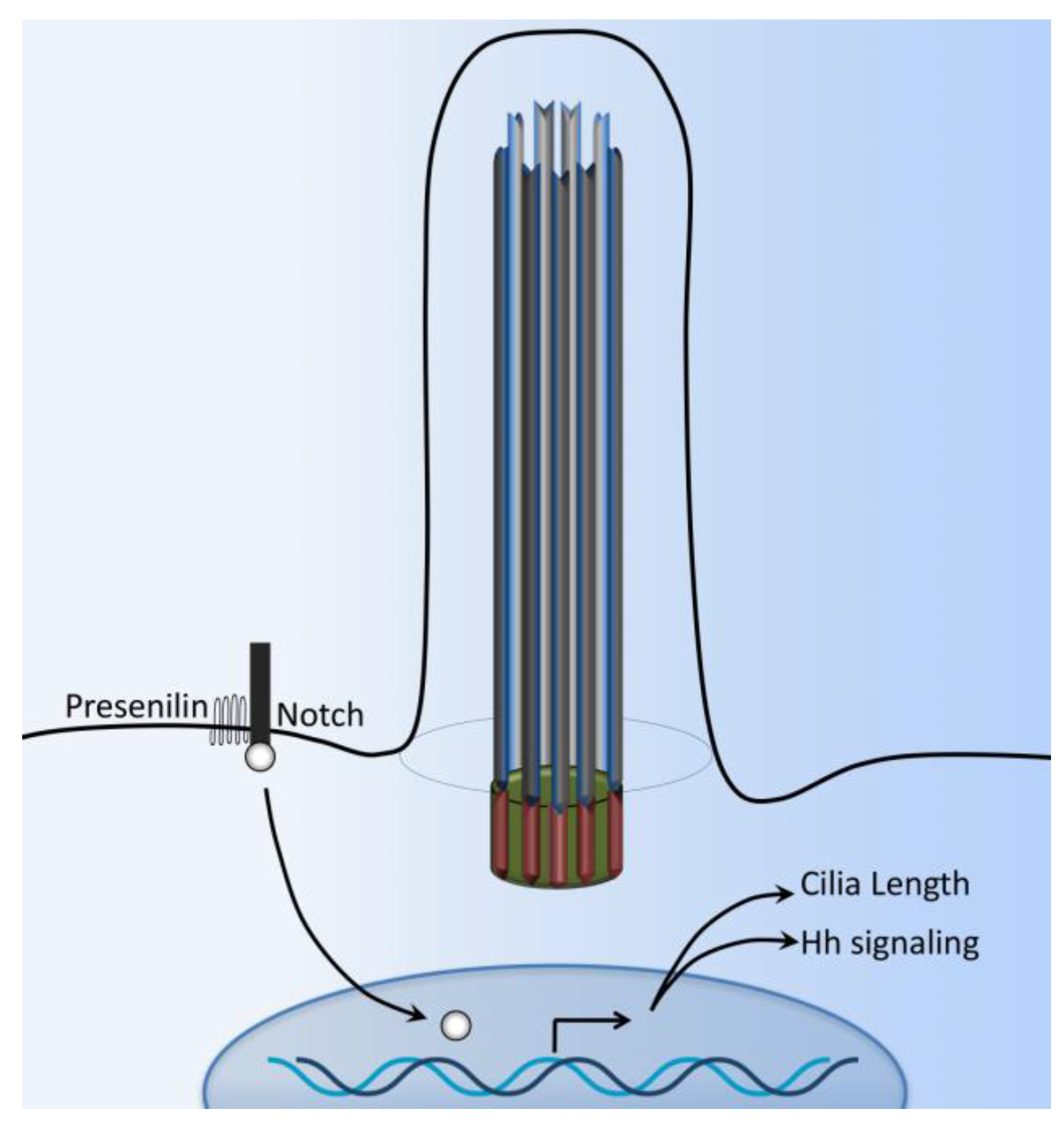
7. Notch Signaling
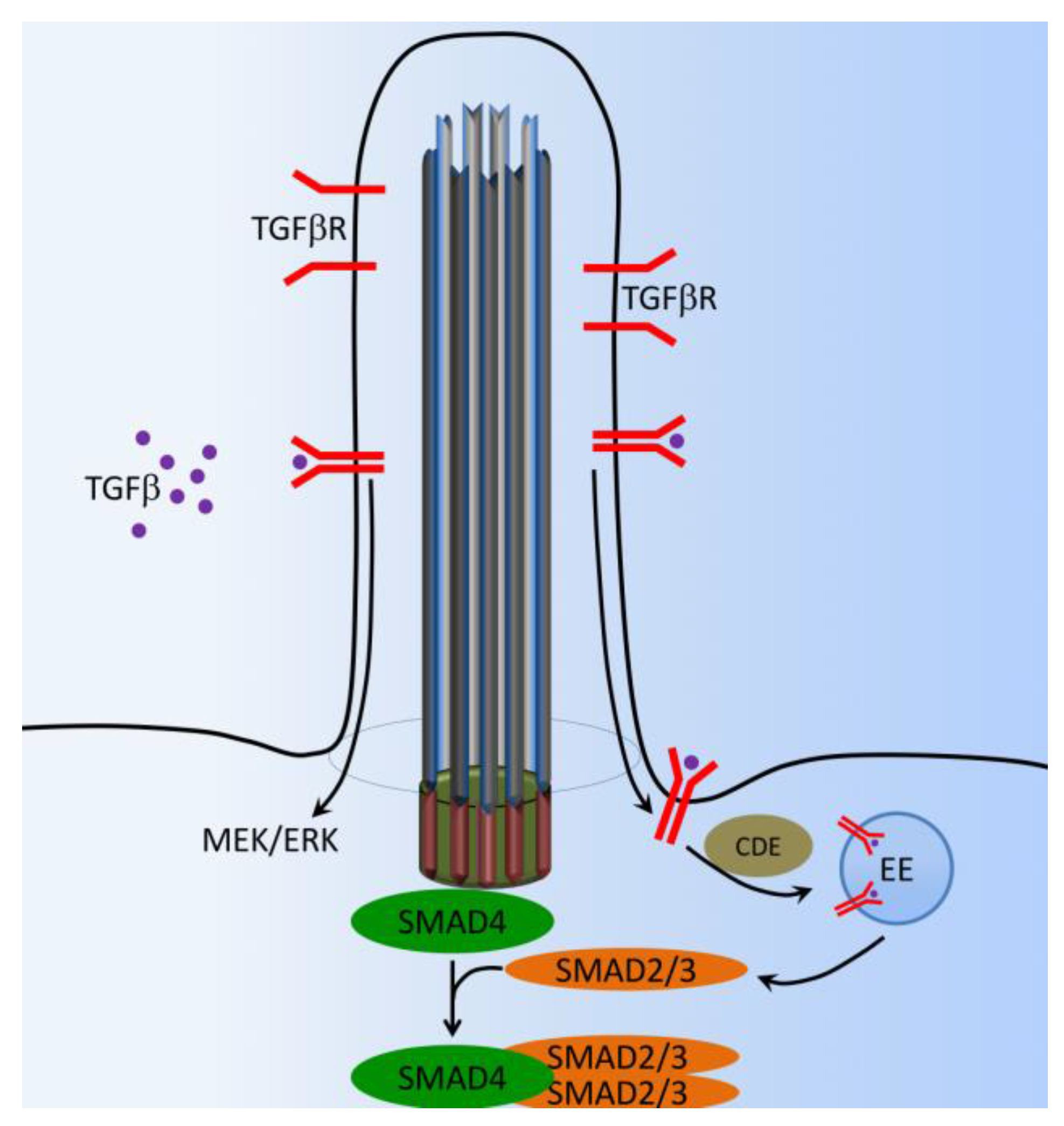
8. TGF-β Signaling
9. mTOR Signaling
10. OFD1 and Autophagy Signaling
11. Other Cilia-Dependent Signaling Pathways
12. Conclusions and Perspectives
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gherman, A.; Davis, E.E.; Katsanis, N. The ciliary proteome database: An integrated community resource for the genetic and functional dissection of cilia. Nat. Genet. 2006, 38, 961–962. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Thompson, J.; Yates, J.R., 3rd; Marshall, W.F. Proteomic analysis of mammalian primary cilia. Curr. Biol. 2012, 22, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Satir, P.; Christensen, S.T. Overview of structure and function of mammalian cilia. Annu. Rev. Physiol. 2007, 69, 377–400. [Google Scholar] [CrossRef] [PubMed]
- Lee, L. Mechanisms of mammalian ciliary motility: Insights from primary ciliary dyskinesia genetics. Gene 2011, 473, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, D. Primary cilia. J. Oral Maxillofac. Pathol. 2017, 21, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, K.; Chen, D.; Nishida, T.; Misaki, K.; Yonemura, S.; Hamada, H. Absence of radial spokes in mouse node cilia is required for rotational movement but confers ultrastructural instability as a trade-off. Dev. Cell 2015, 35, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Taschner, M.; Bhogaraju, S.; Lorentzen, E. Architecture and function of IFT complex proteins in ciliogenesis. Differentiation 2012, 83, S12–S22. [Google Scholar] [CrossRef] [PubMed]
- Silverman, M.A.; Leroux, M.R. Intraflagellar transport and the generation of dynamic, structurally and functionally diverse cilia. Trends Cell Biol. 2009, 19, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, J.L.; Witman, G.B. Intraflagellar transport. Nat. Rev. Mol. Cell Biol. 2002, 3, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Pazour, G.J.; Bloodgood, R.A. Targeting proteins to the ciliary membrane. Curr. Top. Dev. Biol. 2008, 85, 115–149. [Google Scholar] [PubMed]
- Marshall, W.F. Basal bodies platforms for building cilia. Curr. Top. Dev. Biol. 2008, 85, 1–22. [Google Scholar] [PubMed]
- Reiter, J.F.; Blacque, O.E.; Leroux, M.R. The base of the cilium: Roles for transition fibres and the transition zone in ciliary formation, maintenance and compartmentalization. EMBO Rep. 2012, 13, 608–618. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Nelson, W.J. Ciliary diffusion barrier: The gatekeeper for the primary cilium compartment. Cytoskeleton (Hoboken) 2011, 68, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Craige, B.; Tsao, C.C.; Diener, D.R.; Hou, Y.; Lechtreck, K.F.; Rosenbaum, J.L.; Witman, G.B. CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J. Cell Biol. 2010, 190, 927–940. [Google Scholar] [CrossRef] [PubMed]
- Schou, K.B.; Pedersen, L.B.; Christensen, S.T. Ins and outs of GPCR signaling in primary cilia. EMBO Rep. 2015, 16, 1099–1113. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.; Ni, T.T.; Sun, J.; Wan, H.; Amack, J.D.; Yu, G.; Fleming, J.; Chiang, C.; Li, W.; Papierniak, A.; et al. Prostaglandin signalling regulates ciliogenesis by modulating intraflagellar transport. Nat. Cell Biol. 2014, 16, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; White, S.R.; Shida, T.; Schulz, S.; Aguiar, M.; Gygi, S.P.; Bazan, J.F.; Nachury, M.V. The conserved bardet-biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell 2010, 141, 1208–1219. [Google Scholar] [CrossRef] [PubMed]
- Berbari, N.F.; Lewis, J.S.; Bishop, G.A.; Askwith, C.C.; Mykytyn, K. Bardet-biedl syndrome proteins are required for the localization of G protein-coupled receptors to primary cilia. Proc. Natl. Acad. Sci. USA 2008, 105, 4242–4246. [Google Scholar] [CrossRef] [PubMed]
- Feistel, K.; Blum, M. Three types of cilia including a novel 9+4 axoneme on the notochordal plate of the rabbit embryo. Dev. Dyn. 2006, 235, 3348–3358. [Google Scholar] [CrossRef] [PubMed]
- Prensier, G.; Vivier, E.; Goldstein, S.; Schrevel, J. Motile flagellum with a “3+0” ultrastructure. Science 1980, 207, 1493–1494. [Google Scholar] [CrossRef] [PubMed]
- Adler, K.B.; Fand, I. Cilioinhibitory effect of phenothiazines in vitro and its antagonism by Ca++. Arch. Int. Pharmacodyn. Ther. 1977, 227, 309–323. [Google Scholar] [PubMed]
- Murakami, A.; Eckert, R. Cilia: Activation coupled to mechanical stimulation by calcium influx. Science 1972, 175, 1375–1377. [Google Scholar] [CrossRef] [PubMed]
- AbouAlaiwi, W.A.; Takahashi, M.; Mell, B.R.; Jones, T.J.; Ratnam, S.; Kolb, R.J.; Nauli, S.M. Ciliary polycystin-2 is a mechanosensitive calcium channel involved in nitric oxide signaling cascades. Circ. Res. 2009, 104, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Nauli, S.M.; Kawanabe, Y.; Kaminski, J.J.; Pearce, W.J.; Ingber, D.E.; Zhou, J. Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin-1. Circulation 2008, 117, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Masyuk, A.I.; Masyuk, T.V.; Splinter, P.L.; Huang, B.Q.; Stroope, A.J.; LaRusso, N.F. Cholangiocyte cilia detect changes in luminal fluid flow and transmit them into intracellular Ca2+ and camp signaling. Gastroenterology 2006, 131, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Nauli, S.M.; Alenghat, F.J.; Luo, Y.; Williams, E.; Vassilev, P.; Li, X.; Elia, A.E.; Lu, W.; Brown, E.M.; Quinn, S.J.; et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 2003, 33, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Rydholm, S.; Zwartz, G.; Kowalewski, J.M.; Kamali-Zare, P.; Frisk, T.; Brismar, H. Mechanical properties of primary cilia regulate the response to fluid flow. Am. J. Physiol. Renal. Physiol. 2010, 298, F1096–F1102. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Mohieldin, A.M.; Muntean, B.S.; Green, J.A.; Shah, J.V.; Mykytyn, K.; Nauli, S.M. Cilioplasm is a cellular compartment for calcium signaling in response to mechanical and chemical stimuli. Cell Mol. Life Sci. 2014, 71, 2165–2178. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.L.; Guevarra, M.D.; Nguyen, A.M.; Chua, M.C.; Wang, Y.; Jacobs, C.R. The primary cilium functions as a mechanical and calcium signaling nexus. Cilia 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Zhao, L.; Brueckner, M.; Sun, Z. Intraciliary calcium oscillations initiate vertebrate left-right asymmetry. Curr. Biol. 2015, 25, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Phua, S.C.; DeRose, R.; Chiba, S.; Narita, K.; Kalugin, P.N.; Katada, T.; Kontani, K.; Takeda, S.; Inoue, T. Genetically encoded calcium indicator illuminates calcium dynamics in primary cilia. Nat. Methods 2013, 10, 1105–1107. [Google Scholar] [CrossRef] [PubMed]
- Delling, M.; DeCaen, P.G.; Doerner, J.F.; Febvay, S.; Clapham, D.E. Primary cilia are specialized calcium signalling organelles. Nature 2013, 504, 311–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norris, D.P.; Jackson, P.K. Cell biology: Calcium contradictions in cilia. Nature 2016, 531, 582–583. [Google Scholar] [CrossRef] [PubMed]
- Delling, M.; Indzhykulian, A.A.; Liu, X.; Li, Y.; Xie, T.; Corey, D.P.; Clapham, D.E. Primary cilia are not calcium-responsive mechanosensors. Nature 2016, 531, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Nauli, S.M.; Pala, R.; Kleene, S.J. Calcium channels in primary cilia. Curr. Opin. Nephrol. Hypertens. 2016, 25, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Kottgen, M.; Buchholz, B.; Garcia-Gonzalez, M.A.; Kotsis, F.; Fu, X.; Doerken, M.; Boehlke, C.; Steffl, D.; Tauber, R.; Wegierski, T.; et al. TRPP2 and TRPV4 form a polymodal sensory channel complex. J. Cell. Biol. 2008, 182, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.X.; Giamarchi, A.; Rodat-Despoix, L.; Padilla, F.; Downs, T.; Tsiokas, L.; Delmas, P. Formation of a new receptor-operated channel by heteromeric assembly of TRPP2 and TRPC1 subunits. EMBO Rep. 2008, 9, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Pazour, G.J.; San Agustin, J.T.; Follit, J.A.; Rosenbaum, J.L.; Witman, G.B. Polycystin-2 localizes to kidney cilia and the ciliary level is elevated in orpk mice with polycystic kidney disease. Curr. Biol. 2002, 12, R378–R380. [Google Scholar] [CrossRef]
- DeCaen, P.G.; Liu, X.; Abiria, S.; Clapham, D.E. Atypical calcium regulation of the PKD2-L1 polycystin ion channel. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Xu, H.; Yao, Q.; Li, W.; Huang, Q.; Outeda, P.; Cebotaru, V.; Chiaravalli, M.; Boletta, A.; Piontek, K.; et al. Ciliary membrane proteins traffic through the golgi via a Rabep1/GGA1/Arl3-dependent mechanism. Nat. Commun. 2014, 5, 5482. [Google Scholar] [CrossRef] [PubMed]
- Hoffmeister, H.; Babinger, K.; Gurster, S.; Cedzich, A.; Meese, C.; Schadendorf, K.; Osten, L.; de Vries, U.; Rascle, A.; Witzgall, R. Polycystin-2 takes different routes to the somatic and ciliary plasma membrane. J. Cell Biol. 2011, 192, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, D.; Bulley, S.; Leo, M.D.; Burris, S.K.; Gabrick, K.S.; Boop, F.A.; Jaggar, J.H. Smooth muscle cell transient receptor potential polycystin-2 (TRPP2) channels contribute to the myogenic response in cerebral arteries. J. Physiol. 2013, 591, 5031–5046. [Google Scholar] [CrossRef] [PubMed]
- Yoshiba, S.; Shiratori, H.; Kuo, I.Y.; Kawasumi, A.; Shinohara, K.; Nonaka, S.; Asai, Y.; Sasaki, G.; Belo, J.A.; Sasaki, H.; et al. Cilia at the node of mouse embryos sense fluid flow for left-right determination via PKD2. Science 2012, 338, 226–231. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.; Somlo, S.; Makova, S.; Tian, X.; Brueckner, M. Two populations of node monocilia initiate left-right asymmetry in the mouse. Cell 2003, 114, 61–73. [Google Scholar] [CrossRef]
- Jin, X.; Muntean, B.S.; Aal-Aaboda, M.S.; Duan, Q.; Zhou, J.; Nauli, S.M. l-type calcium channel modulates cystic kidney phenotype. Biochim. Biophys. Acta 2014, 1842, 1518–1526. [Google Scholar] [CrossRef] [PubMed]
- Muntean, B.S.; Jin, X.; Williams, F.E.; Nauli, S.M. Primary cilium regulates CaV1.2 expression through Wnt signaling. J. Cell Physiol. 2014, 229, 1926–1934. [Google Scholar] [CrossRef] [PubMed]
- Huangfu, D.; Liu, A.; Rakeman, A.S.; Murcia, N.S.; Niswander, L.; Anderson, K.V. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 2003, 426, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; von Kessler, D.P.; Parks, S.; Beachy, P.A. Secretion and localized transcription suggest a role in positional signaling for products of the segmentation gene hedgehog. Cell 1992, 71, 33–50. [Google Scholar] [CrossRef]
- Goetz, S.C.; Ocbina, P.J.; Anderson, K.V. The primary cilium as a hedgehog signal transduction machine. Methods Cell Biol. 2009, 94, 199–222. [Google Scholar] [PubMed]
- Bai, C.B.; Stephen, D.; Joyner, A.L. All mouse ventral spinal cord patterning by hedgehog is Gli dependent and involves an activator function of Gli3. Dev. Cell 2004, 6, 103–115. [Google Scholar] [CrossRef]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Corbit, K.C.; Aanstad, P.; Singla, V.; Norman, A.R.; Stainier, D.Y.; Reiter, J.F. Vertebrate smoothened functions at the primary cilium. Nature 2005, 437, 1018–1021. [Google Scholar] [CrossRef] [PubMed]
- Robbins, D.J.; Fei, D.L.; Riobo, N.A. The Hedgehog signal transduction network. Sci. Signal. 2012, 5, re6. [Google Scholar] [CrossRef] [PubMed]
- Tukachinsky, H.; Lopez, L.V.; Salic, A. A mechanism for vertebrate hedgehog signaling: Recruitment to cilia and dissociation of SuFu-Gli protein complexes. J. Cell Biol. 2010, 191, 415–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fliegauf, M.; Benzing, T.; Omran, H. When cilia go bad: Cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 2007, 8, 880–893. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, N.; Thiyagarajan, S.; Elcheva, I.; Saleem, M.; Dlugosz, A.; Mukhtar, H.; Spiegelman, V.S. Gli2 is targeted for ubiquitination and degradation by β-TrCP ubiquitin ligase. J. Biol. Chem. 2006, 281, 19320–19326. [Google Scholar] [CrossRef] [PubMed]
- Smelkinson, M.G.; Zhou, Q.; Kalderon, D. Regulation of Ci-SCFSlimb binding, Ci proteolysis, and Hedgehog pathway activity by Ci phosphorylation. Dev. Cell 2007, 13, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, L.; Wang, B.; Ou, C.Y.; Chien, C.T.; Jiang, J. A hedgehog-induced BTB protein modulates hedgehog signaling by degrading Ci/Gli transcription factor. Dev. Cell 2006, 10, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Tempe, D.; Casas, M.; Karaz, S.; Blanchet-Tournier, M.F.; Concordet, J.P. Multisite protein kinase a and glycogen synthase kinase 3β phosphorylation leads to Gli3 ubiquitination by SCFβTrCP. Mol. Cell Biol. 2006, 26, 4316–4326. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, Y. Evidence for the direct involvement of βTrCP in Gli3 protein processing. Proc. Natl. Acad. Sci. USA 2006, 103, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Wang, C.; Wang, B. Phosphorylation of Gli2 by protein kinase A is required for Gli2 processing and degradation and the Sonic Hedgehog-regulated mouse development. Dev. Biol. 2009, 326, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Haycraft, C.J.; Banizs, B.; Aydin-Son, Y.; Zhang, Q.; Michaud, E.J.; Yoder, B.K. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005, 1, e53. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, J.M.; Katsanis, N. Ciliary function and Wnt signal modulation. Curr. Top. Dev. Biol. 2008, 85, 175–195. [Google Scholar] [PubMed]
- Bergmann, C.; Fliegauf, M.; Bruchle, N.O.; Frank, V.; Olbrich, H.; Kirschner, J.; Schermer, B.; Schmedding, I.; Kispert, A.; Kranzlin, B.; et al. Loss of nephrocystin-3 function can cause embryonic lethality, Meckel-Gruber-like syndrome, situs inversus, and renal-hepatic-pancreatic dysplasia. Am. J. Hum. Genet. 2008, 82, 959–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbit, K.C.; Shyer, A.E.; Dowdle, W.E.; Gaulden, J.; Singla, V.; Chen, M.H.; Chuang, P.T.; Reiter, J.F. Kif3a constrains β-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat. Cell Biol. 2008, 10, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.J.; May-Simera, H.; Eichers, E.R.; Kai, M.; Hill, J.; Jagger, D.J.; Leitch, C.C.; Chapple, J.P.; Munro, P.M.; Fisher, S.; et al. Disruption of bardet-biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat. Genet. 2005, 37, 1135–1140. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Gloy, J.; Ganner, A.; Bullerkotte, A.; Bashkurov, M.; Kronig, C.; Schermer, B.; Benzing, T.; Cabello, O.A.; Jenny, A.; et al. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat. Genet. 2005, 37, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Bisgrove, B.W.; Yost, H.J. The roles of cilia in developmental disorders and disease. Development 2006, 133, 4131–4143. [Google Scholar] [CrossRef] [PubMed]
- Davenport, J.R.; Yoder, B.K. An incredible decade for the primary cilium: A look at a once-forgotten organelle. Am. J. Physiol. Renal Physiol. 2005, 289, F1159–F1169. [Google Scholar] [CrossRef] [PubMed]
- Wagner, U.; Brownlees, J.; Irving, N.G.; Lucas, F.R.; Salinas, P.C.; Miller, C.C. Overexpression of the mouse dishevelled-1 protein inhibits GSK-3β-mediated phosphorylation of tau in transfected mammalian cells. FEBS Lett. 1997, 411, 369–372. [Google Scholar] [CrossRef]
- Jenny, A.; Mlodzik, M. Planar cell polarity signaling: A common mechanism for cellular polarization. Mt. Sinai J. Med. 2006, 73, 738–750. [Google Scholar] [PubMed]
- May-Simera, H.L.; Kelley, M.W. Cilia, Wnt signaling, and the cytoskeleton. Cilia 2012, 1, 7. [Google Scholar] [CrossRef] [PubMed]
- Park, T.J.; Mitchell, B.J.; Abitua, P.B.; Kintner, C.; Wallingford, J.B. Dishevelled controls apical docking and planar polarization of basal bodies in ciliated epithelial cells. Nat. Genet. 2008, 40, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Otto, E.A.; Schermer, B.; Obara, T.; O’Toole, J.F.; Hiller, K.S.; Mueller, A.M.; Ruf, R.G.; Hoefele, J.; Beekmann, F.; Landau, D.; et al. Mutations in invs encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat. Genet. 2003, 34, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, J.M.; Davis, E.E.; Katsanis, N. The vertebrate primary cilium in development, homeostasis, and disease. Cell 2009, 137, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Nie, H.; Nesin, V.; Tran, U.; Outeda, P.; Bai, C.X.; Keeling, J.; Maskey, D.; Watnick, T.; Wessely, O.; et al. The polycystin complex mediates Wnt/Ca2+ signalling. Nat. Cell Biol. 2016, 18, 752–764. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.T.; Clement, C.A.; Satir, P.; Pedersen, L.B. Primary cilia and coordination of receptor tyrosine kinase (RTK) signalling. J. Pathol. 2012, 226, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008, 22, 1276–1312. [Google Scholar] [CrossRef] [PubMed]
- Schneider, L.; Cammer, M.; Lehman, J.; Nielsen, S.K.; Guerra, C.F.; Veland, I.R.; Stock, C.; Hoffmann, E.K.; Yoder, B.K.; Schwab, A.; et al. Directional cell migration and chemotaxis in wound healing response to PDGF-AA are coordinated by the primary cilium in fibroblasts. Cell Physiol. Biochem. 2010, 25, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Schneider, L.; Clement, C.A.; Teilmann, S.C.; Pazour, G.J.; Hoffmann, E.K.; Satir, P.; Christensen, S.T. PDGFRαα signaling is regulated through the primary cilium in fibroblasts. Curr. Biol. 2005, 15, 1861–1866. [Google Scholar] [CrossRef] [PubMed]
- Mans, D.A.; Voest, E.E.; Giles, R.H. All along the watchtower: Is the cilium a tumor suppressor organelle? Biochim. Biophys. Acta 2008, 1786, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Theisen, C.S.; Wahl, J.K., 3rd; Johnson, K.R.; Wheelock, M.J. NHERF links the N-cadherin/catenin complex to the platelet-derived growth factor receptor to modulate the actin cytoskeleton and regulate cell motility. Mol. Biol. Cell 2007, 18, 1220–1232. [Google Scholar] [CrossRef] [PubMed]
- Veland, I.R.; Awan, A.; Pedersen, L.B.; Yoder, B.K.; Christensen, S.T. Primary cilia and signaling pathways in mammalian development, health and disease. Nephron Physiol. 2009, 111, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Hori, K.; Sen, A.; Artavanis-Tsakonas, S. Notch signaling at a glance. J. Cell Sci. 2013, 126, 2135–2140. [Google Scholar] [CrossRef] [PubMed]
- Guruharsha, K.G.; Kankel, M.W.; Artavanis-Tsakonas, S. The notch signalling system: Recent insights into the complexity of a conserved pathway. Nat. Rev. Genet. 2012, 13, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Pierfelice, T.; Alberi, L.; Gaiano, N. Notch in the vertebrate nervous system: An old dog with new tricks. Neuron 2011, 69, 840–855. [Google Scholar] [CrossRef] [PubMed]
- Louvi, A.; Artavanis-Tsakonas, S. Notch signalling in vertebrate neural development. Nat. Rev. Neurosci. 2006, 7, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Henrique, D.; Adam, J.; Myat, A.; Chitnis, A.; Lewis, J.; Ish-Horowicz, D. Expression of a Delta homologue in prospective neurons in the chick. Nature 1995, 375, 787–790. [Google Scholar] [CrossRef] [PubMed]
- Okigawa, S.; Mizoguchi, T.; Okano, M.; Tanaka, H.; Isoda, M.; Jiang, Y.J.; Suster, M.; Higashijima, S.; Kawakami, K.; Itoh, M. Different combinations of notch ligands and receptors regulate V2 interneuron progenitor proliferation and V2a/V2b cell fate determination. Dev. Biol. 2014, 391, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Leitch, C.C.; Lodh, S.; Prieto-Echague, V.; Badano, J.L.; Zaghloul, N.A. Basal body proteins regulate notch signaling through endosomal trafficking. J. Cell Sci. 2014, 127, 2407–2419. [Google Scholar] [CrossRef] [PubMed]
- Ezratty, E.J.; Stokes, N.; Chai, S.; Shah, A.S.; Williams, S.E.; Fuchs, E. A role for the primary cilium in notch signaling and epidermal differentiation during skin development. Cell 2011, 145, 1129–1141. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.F.; Schock, E.N.; Attia, A.C.; Stottmann, R.W.; Brugmann, S.A. The ciliary baton: Orchestrating neural crest cell development. Curr. Top. Dev. Biol. 2015, 111, 97–134. [Google Scholar] [PubMed]
- Lopes, S.S.; Lourenco, R.; Pacheco, L.; Moreno, N.; Kreiling, J.; Saude, L. Notch signalling regulates left-right asymmetry through ciliary length control. Development 2010, 137, 3625–3632. [Google Scholar] [CrossRef] [PubMed]
- Krebs, L.T.; Iwai, N.; Nonaka, S.; Welsh, I.C.; Lan, Y.; Jiang, R.; Saijoh, Y.; O’Brien, T.P.; Hamada, H.; Gridley, T. Notch signaling regulates left-right asymmetry determination by inducing nodal expression. Genes Dev. 2003, 17, 1207–1212. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.H.; Yang, L.; Dessaud, E.; Chuang, K.; Moore, D.M.; Rohatgi, R.; Briscoe, J.; Novitch, B.G. Notch activity modulates the responsiveness of neural progenitors to sonic hedgehog signaling. Dev. Cell 2015, 33, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Wang, X.F. Signaling cross-talk between TGF-β/BMP and other pathways. Cell Res. 2009, 19, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Janssens, K.; ten Dijke, P.; Janssens, S.; Van Hul, W. Transforming growth factor-β1 to the bone. Endocr. Rev. 2005, 26, 743–774. [Google Scholar] [CrossRef] [PubMed]
- Sarahrudi, K.; Thomas, A.; Mousavi, M.; Kaiser, G.; Kottstorfer, J.; Kecht, M.; Hajdu, S.; Aharinejad, S. Elevated transforming growth factor-beta 1 (TGF-β1) levels in human fracture healing. Injury 2011, 42, 833–837. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wu, X.; Lei, W.; Pang, L.; Wan, C.; Shi, Z.; Zhao, L.; Nagy, T.R.; Peng, X.; Hu, J.; et al. TGF-β1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 2009, 15, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Cho, T.J.; Gerstenfeld, L.C.; Einhorn, T.A. Differential temporal expression of members of the transforming growth factor β superfamily during murine fracture healing. J. Bone Miner. Res. 2002, 17, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Mohtai, M.; Iwamoto, Y. Fluid shear stress increases transforming growth factor β 1 expression in human osteoblast-like cells: Modulation by cation channel blockades. Calcif. Tissue Int. 1998, 63, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Rys, J.P.; DuFort, C.C.; Monteiro, D.A.; Baird, M.A.; Oses-Prieto, J.A.; Chand, S.; Burlingame, A.L.; Davidson, M.W.; Alliston, T.N. Discrete spatial organization of TGF β receptors couples receptor multimerization and signaling to cellular tension. Elife 2015, 4, e09300. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, M.L.; Awan, A.; Warzecha, C.B.; Christensen, S.T.; Andersen, C.Y. Immunofluorescence microscopy and mRNA analysis of human embryonic stem cells (hESCs) including primary cilia associated signaling pathways. Methods Mol. Biol. 2016, 1307, 123–140. [Google Scholar] [PubMed]
- Clement, C.A.; Ajbro, K.D.; Koefoed, K.; Vestergaard, M.L.; Veland, I.R.; Henriques de Jesus, M.P.; Pedersen, L.B.; Benmerah, A.; Andersen, C.Y.; Larsen, L.A.; et al. TGF-β signaling is associated with endocytosis at the pocket region of the primary cilium. Cell Rep. 2013, 3, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Labour, M.N.; Riffault, M.; Christensen, S.T.; Hoey, D.A. TGFβ1—Induced recruitment of human bone mesenchymal stem cells is mediated by the primary cilium in a SMAD3-dependent manner. Sci. Rep. 2016, 6, 35542. [Google Scholar] [CrossRef] [PubMed]
- Ibraghimov-Beskrovnaya, O.; Natoli, T.A. mTOR signaling in polycystic kidney disease. Trends Mol. Med. 2011, 17, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Weimbs, T. Polycystic kidney disease and renal injury repair: Common pathways, fluid flow, and the function of polycystin-1. Am. J. Physiol. Renal. Physiol. 2007, 293, F1423–F1432. [Google Scholar] [CrossRef] [PubMed]
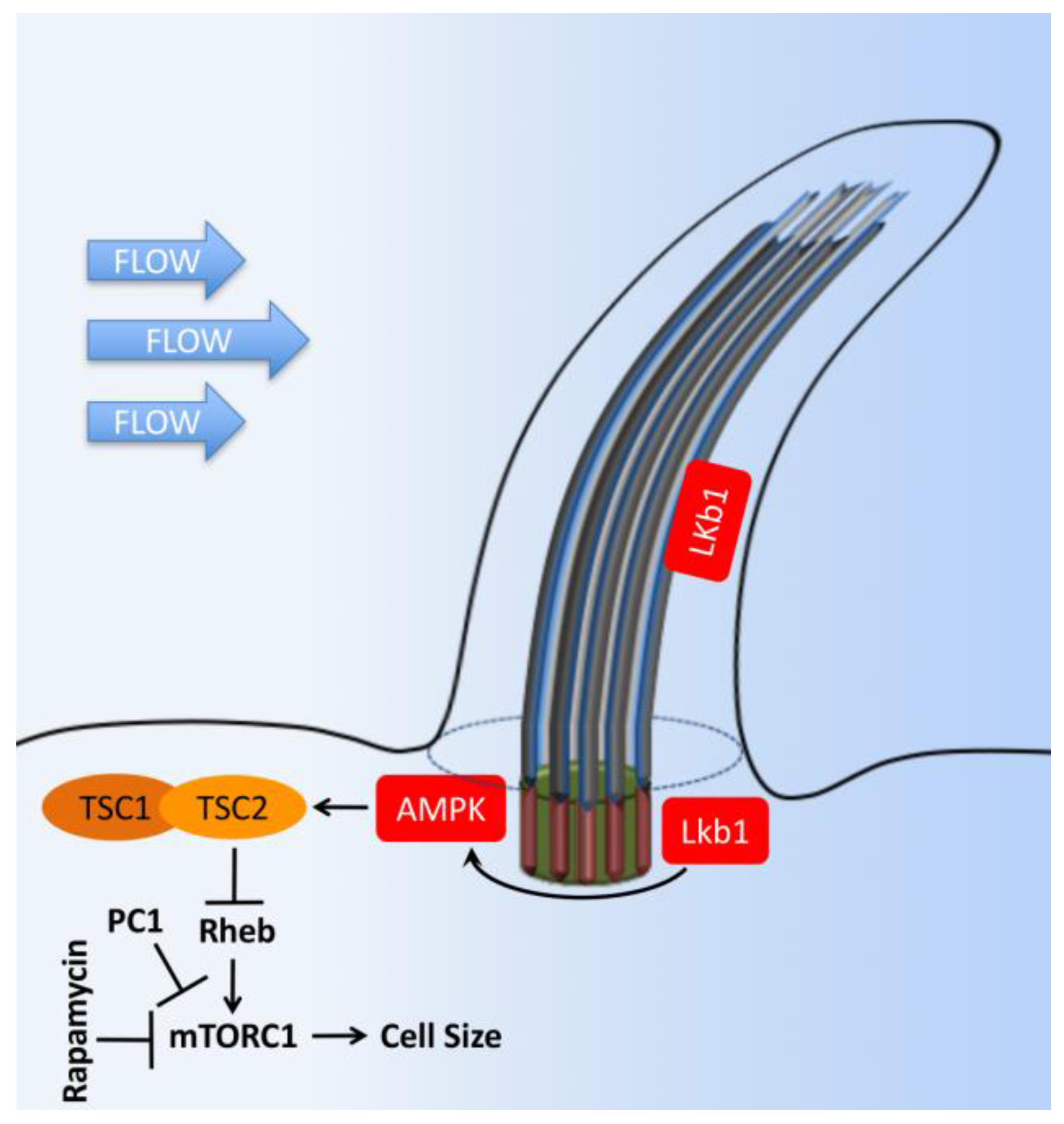
- Hartman, T.R.; Liu, D.; Zilfou, J.T.; Robb, V.; Morrison, T.; Watnick, T.; Henske, E.P. The tuberous sclerosis proteins regulate formation of the primary cilium via a rapamycin-insensitive and polycystin 1-independent pathway. Hum. Mol. Genet. 2009, 18, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Boehlke, C.; Kotsis, F.; Patel, V.; Braeg, S.; Voelker, H.; Bredt, S.; Beyer, T.; Janusch, H.; Hamann, C.; Godel, M.; et al. Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nat. Cell Biol. 2010, 12, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Bell, P.D.; Fitzgibbon, W.; Sas, K.; Stenbit, A.E.; Amria, M.; Houston, A.; Reichert, R.; Gilley, S.; Siegal, G.P.; Bissler, J.; et al. Loss of primary cilia upregulates renal hypertrophic signaling and promotes cystogenesis. J. Am. Soc. Nephrol. 2011, 22, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Dere, R.; Wilson, P.D.; Sandford, R.N.; Walker, C.L. Carboxy terminal tail of polycystin-1 regulates localization of TSC2 to repress mTOR. PLoS ONE 2010, 5, e9239. [Google Scholar] [CrossRef] [PubMed]
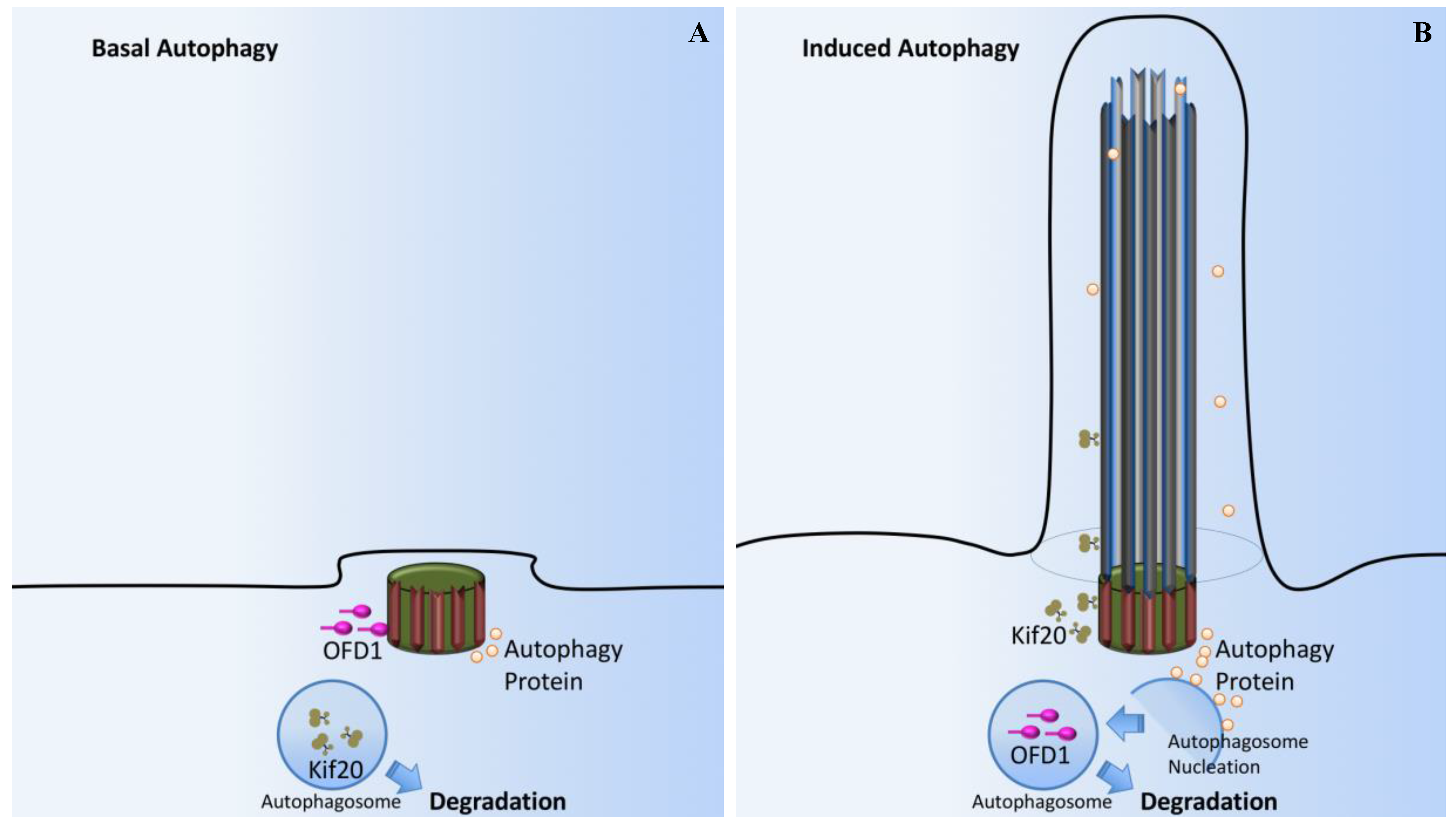
- Tang, Z.; Lin, M.G.; Stowe, T.R.; Chen, S.; Zhu, M.; Stearns, T.; Franco, B.; Zhong, Q. Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature 2013, 502, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Pampliega, O.; Orhon, I.; Patel, B.; Sridhar, S.; Diaz-Carretero, A.; Beau, I.; Codogno, P.; Satir, B.H.; Satir, P.; Cuervo, A.M. Functional interaction between autophagy and ciliogenesis. Nature 2013, 502, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Brailov, I.; Bancila, M.; Brisorgueil, M.J.; Miquel, M.C.; Hamon, M.; Verge, D. Localization of 5-HT6 receptors at the plasma membrane of neuronal cilia in the rat brain. Brain Res. 2000, 872, 271–275. [Google Scholar] [CrossRef]
- Handel, M.; Schulz, S.; Stanarius, A.; Schreff, M.; Erdtmann-Vourliotis, M.; Schmidt, H.; Wolf, G.; Hollt, V. Selective targeting of somatostatin receptor 3 to neuronal cilia. Neuroscience 1999, 89, 909–926. [Google Scholar] [CrossRef]
- Koemeter-Cox, A.I.; Sherwood, T.W.; Green, J.A.; Steiner, R.A.; Berbari, N.F.; Yoder, B.K.; Kauffman, A.S.; Monsma, P.C.; Brown, A.; Askwith, C.C.; et al. Primary cilia enhance kisspeptin receptor signaling on gonadotropin-releasing hormone neurons. Proc. Natl. Acad. Sci. USA 2014, 111, 10335–10340. [Google Scholar] [CrossRef] [PubMed]
- Guadiana, S.M.; Semple-Rowland, S.; Daroszewski, D.; Madorsky, I.; Breunig, J.J.; Mykytyn, K.; Sarkisian, M.R. Arborization of dendrites by developing neocortical neurons is dependent on primary cilia and type 3 adenylyl cyclase. J. Neurosci. 2013, 33, 2626–2638. [Google Scholar] [CrossRef] [PubMed]
- Besschetnova, T.Y.; Kolpakova-Hart, E.; Guan, Y.; Zhou, J.; Olsen, B.R.; Shah, J.V. Identification of signaling pathways regulating primary cilium length and flow-mediated adaptation. Curr. Biol. 2010, 20, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Avasthi, P.; Marley, A.; Lin, H.; Gregori-Puigjane, E.; Shoichet, B.K.; von Zastrow, M.; Marshall, W.F. A chemical screen identifies class a G-protein coupled receptors as regulators of cilia. ACS Chem. Biol. 2012, 7, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Verleyen, D.; Luyten, F.P.; Tylzanowski, P. Orphan G-protein coupled receptor 22 (Gpr22) regulates cilia length and structure in the zebrafish Kupffer’s vesicle. PLoS ONE 2014, 9, e110484. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, V.S.; Muntean, B.S.; Kathem, S.H.; Hwang, J.J.; Aboualaiwi, W.A.; Nauli, S.M. Roles of dopamine receptor on chemosensory and mechanosensory primary cilia in renal epithelial cells. Front. Physiol. 2014, 5, 72. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Majeed, S.; Nauli, S.M. Dopamine receptor type 5 in the primary cilia has dual chemo- and mechano-sensory roles. Hypertension 2011, 58, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Rbaibi, Y.; Cui, S.; Mo, D.; Carattino, M.; Rohatgi, R.; Satlin, L.M.; Szalinski, C.M.; Swanhart, L.M.; Folsch, H.; Hukriede, N.A.; et al. OCRL1 modulates cilia length in renal epithelial cells. Traffic 2012, 13, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Suh, Y.A.; Oh, J.H.; Lee, B.R.; Kim, J.; Jang, S.J. KIF3A binds to β-arrestin for suppressing Wnt/β -catenin signalling independently of primary cilia in lung cancer. Sci. Rep. 2016, 6, 32770. [Google Scholar] [CrossRef] [PubMed]
- Balmer, S.; Dussert, A.; Collu, G.M.; Benitez, E.; Iomini, C.; Mlodzik, M. Components of intraflagellar transport complex a function independently of the cilium to regulate canonical Wnt signaling in drosophila. Dev. Cell 2015, 34, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, C. The complex world of wnt receptor signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.C.; Katsanis, N. Context-dependent regulation of wnt signaling through the primary cilium. J. Am. Soc. Nephrol. 2013, 24, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.H.; Wilson, C.W.; Li, Y.J.; Law, K.K.; Lu, C.S.; Gacayan, R.; Zhang, X.; Hui, C.C.; Chuang, P.T. Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev. 2009, 23, 1910–1928. [Google Scholar] [CrossRef] [PubMed]









© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pala, R.; Alomari, N.; Nauli, S.M. Primary Cilium-Dependent Signaling Mechanisms. Int. J. Mol. Sci. 2017, 18, 2272. https://doi.org/10.3390/ijms18112272
Pala R, Alomari N, Nauli SM. Primary Cilium-Dependent Signaling Mechanisms. International Journal of Molecular Sciences. 2017; 18(11):2272. https://doi.org/10.3390/ijms18112272
Chicago/Turabian StylePala, Rajasekharreddy, Nedaa Alomari, and Surya M. Nauli. 2017. "Primary Cilium-Dependent Signaling Mechanisms" International Journal of Molecular Sciences 18, no. 11: 2272. https://doi.org/10.3390/ijms18112272
APA StylePala, R., Alomari, N., & Nauli, S. M. (2017). Primary Cilium-Dependent Signaling Mechanisms. International Journal of Molecular Sciences, 18(11), 2272. https://doi.org/10.3390/ijms18112272