Modulation of Cell Death Pathways by Hepatitis C Virus Proteins in Huh7.5 Hepatoma Cells

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Results
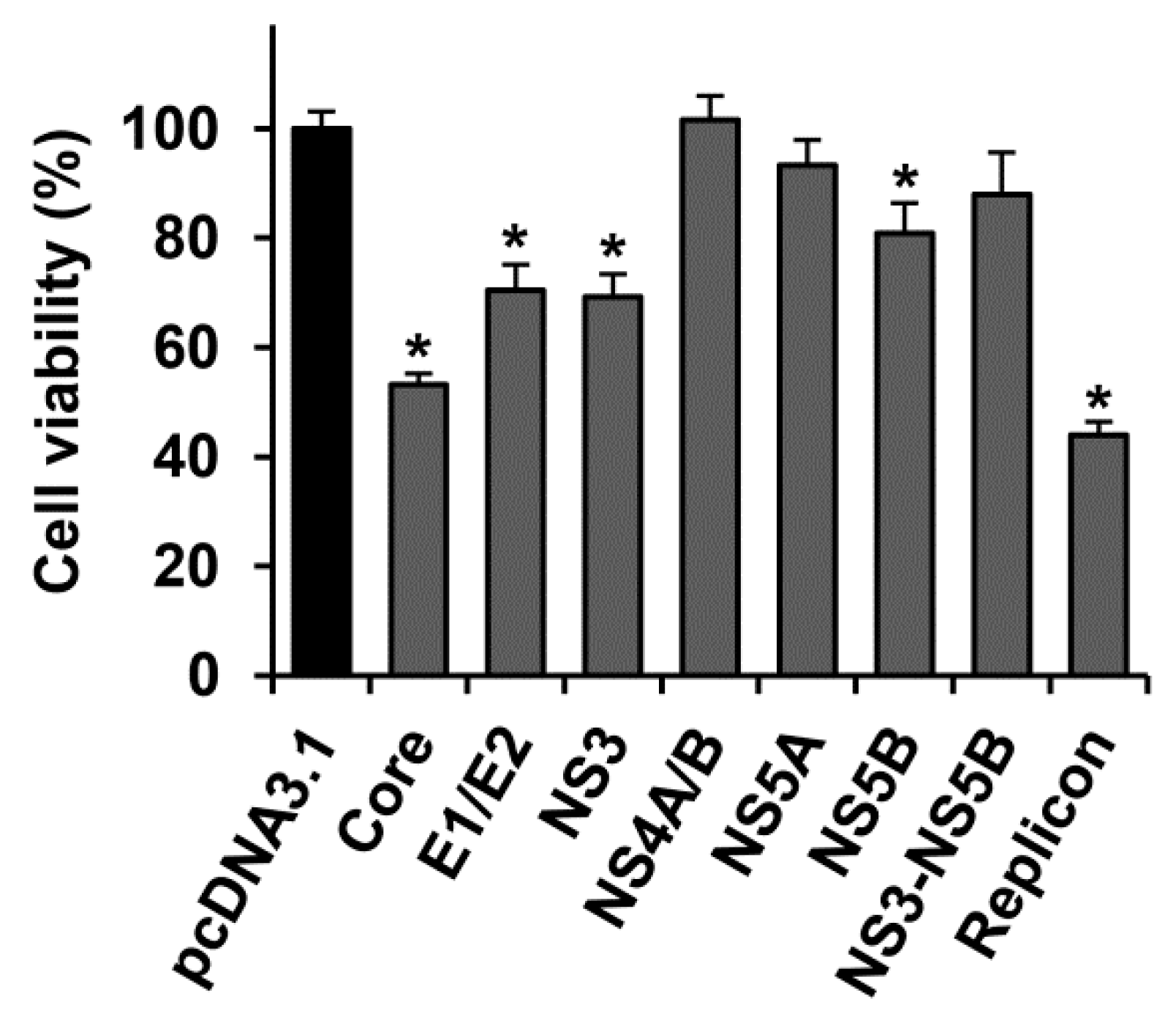
2.1. Several HCV Proteins Hamper Huh7.5 Cell Growth
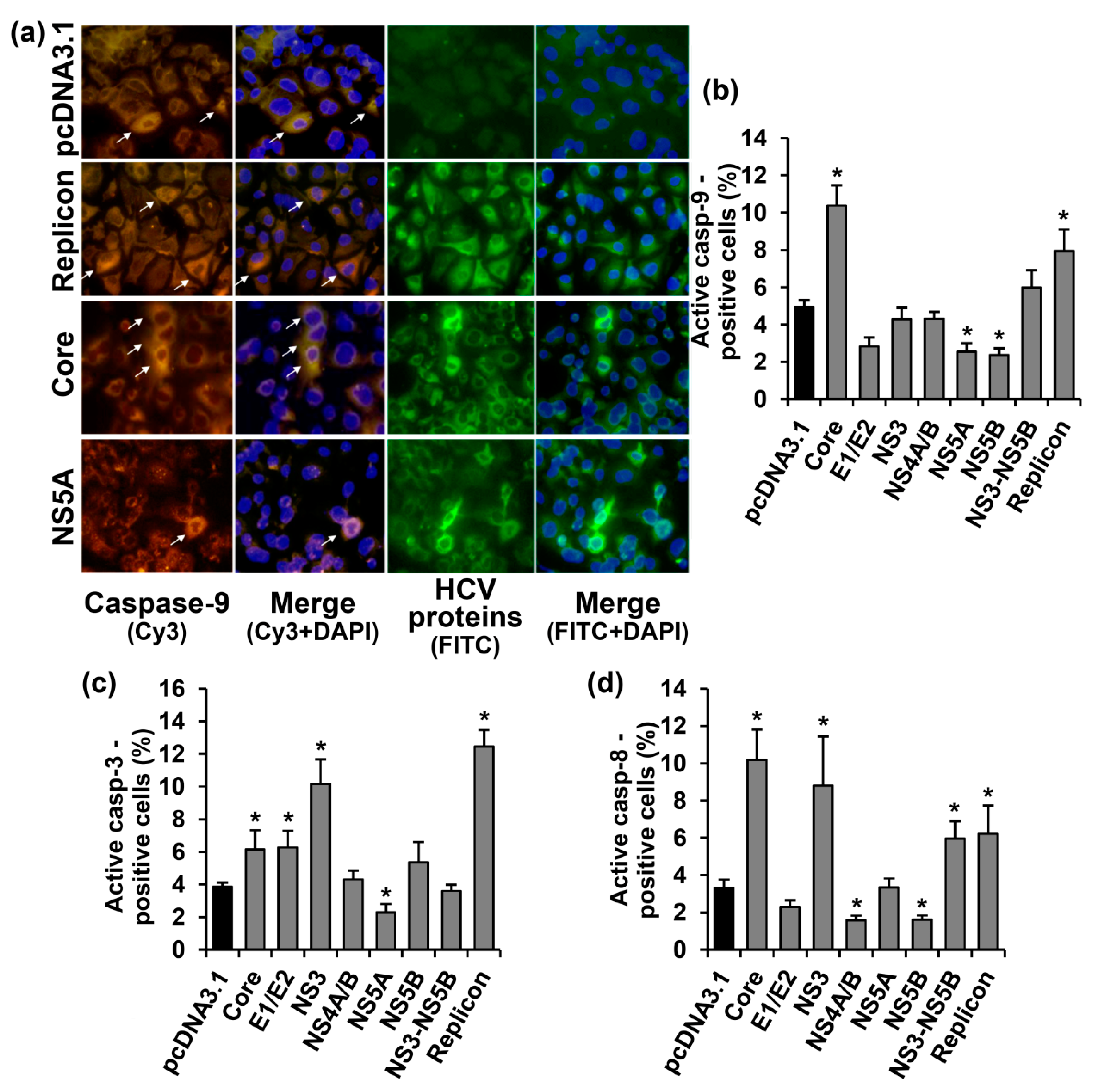
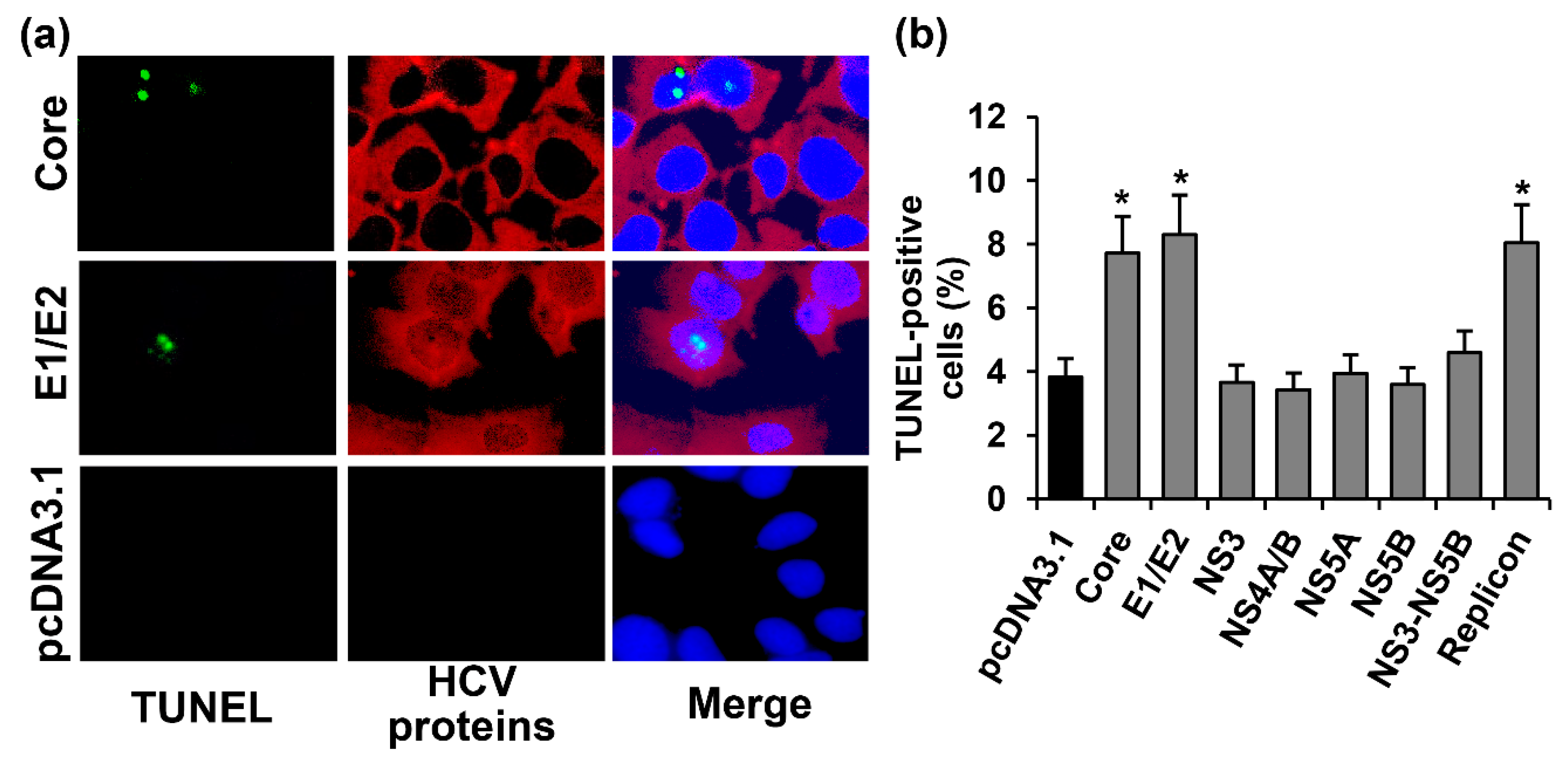
2.2. HCV Proteins Exhibit Different Regulatory Activity towards Apoptotic Pathways
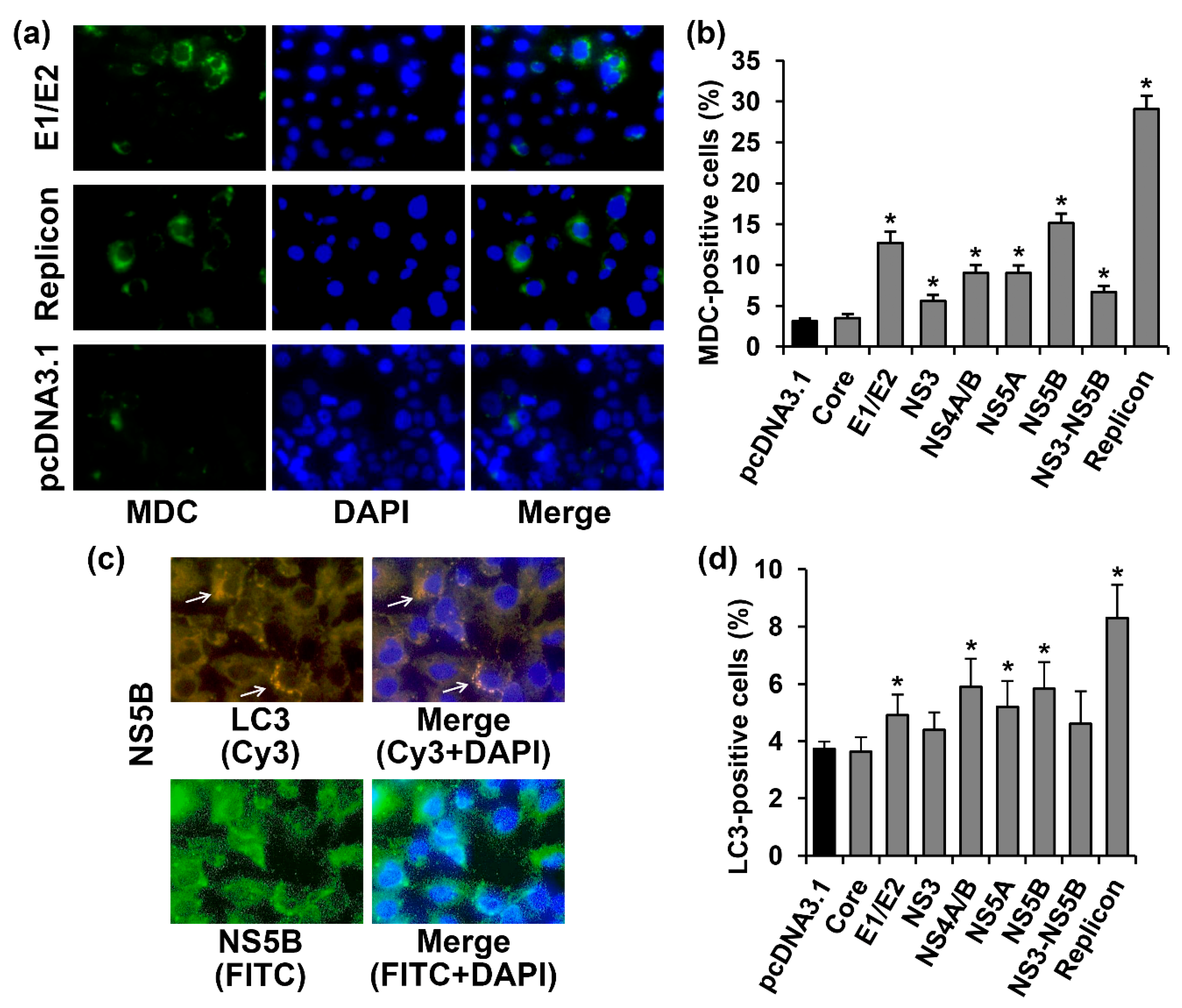
2.3. Several HCV Proteins Stimulate Autophagy in Huh7.5 Cells
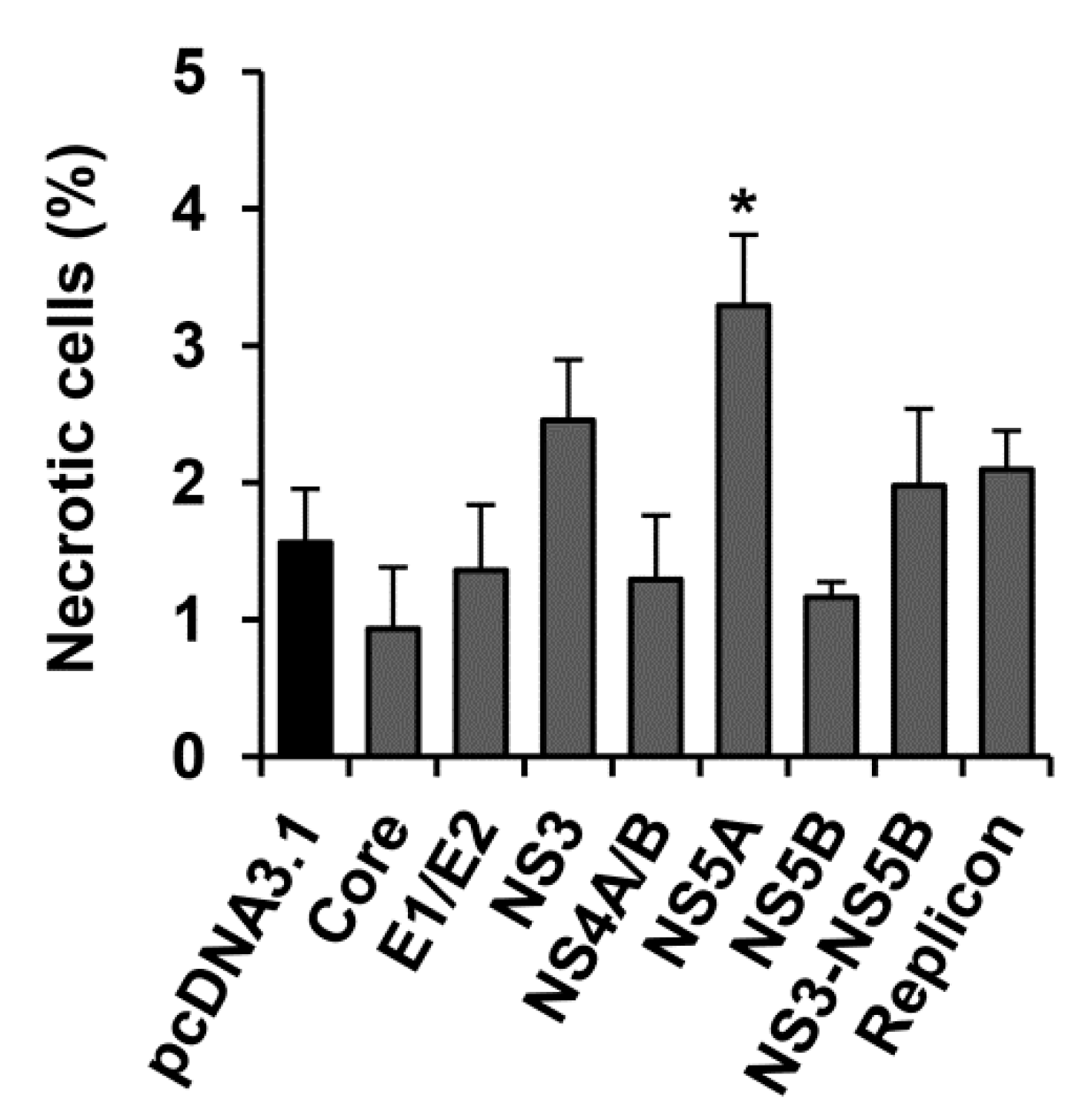
2.4. HCV NS5A Protein Moderately Increases the Incidence Rate of Necrosis
3. Discussion
4. Materials and Methods
4.1. Cells
4.2. Plasmids
4.3. Transfection
4.4. Immunocytochemical Analysis
4.5. Necrosis
4.6. Immunoblot Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dash, S.; Chava, S.; Aydin, Y.; Chandra, P.K.; Ferraris, P.; Chen, W.; Balart, L.A.; Wu, T.; Garry, R.F. Hepatitis c virus infection induces autophagy as a prosurvival mechanism to alleviate hepatic er-stress response. Viruses 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.Y.; Chen, S.S. Hepatitis c virus and cellular stress response: Implications to molecular pathogenesis of liver diseases. Viruses 2012, 4, 2251–2290. [Google Scholar] [CrossRef] [PubMed]
- Wang, K. Autophagy and apoptosis in liver injury. Cell. Cycle 2015, 14, 1631–1642. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Strasser, A.; McDunn, J.E.; Swanson, P.E. Cell death. N. Engl. J. Med. 2009, 361, 1570–1583. [Google Scholar] [CrossRef] [PubMed]
- Hajnoczky, G.; Davies, E.; Madesh, M. Calcium signaling and apoptosis. Biochem. Biophys. Res. Commun. 2003, 304, 445–454. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Tyurina, D.A.; Ivanova, O.N.; Kochetkov, S.N.; Bartosch, B.; Isaguliants, M.G. Oxidative stress, a trigger of hepatitis c and b virus-induced liver carcinogenesis. Oncotarget 2017, 8, 3895–3932. [Google Scholar] [CrossRef] [PubMed]
- Brenner, C.; Galluzzi, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in liver inflammation. J. Hepatol. 2013, 59, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Alibek, K.; Irving, S.; Sautbayeva, Z.; Kakpenova, A.; Bekmurzayeva, A.; Baiken, Y.; Imangali, N.; Shaimerdenova, M.; Mektepbayeva, D.; Balabiyev, A.; et al. Disruption of bcl-2 and bcl-xl by viral proteins as a possible cause of cancer. Infect. Agent Cancer 2014, 9, 44. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Kanda, T.; Wu, S.; Nakamoto, S.; Wakita, T.; Shirasawa, H.; Yokosuka, O. Hepatitis c virus nonstructural protein 5a inhibits thapsigargin-induced apoptosis. PLoS ONE 2014, 9, e113499. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Li, S.; Huang, M.; Xiong, Y.; Zhang, Q.; Ye, L.; Liu, J.; Zhu, X.; Sun, R.; Guo, Y. The roles of endoplasmic reticulum overload response induced by hcv and ns4b protein in human hepatocyte viability and virus replication. PLoS ONE 2015, 10, e0123190. [Google Scholar] [CrossRef] [PubMed]
- Aweya, J.J.; Sze, C.W.; Bayega, A.; Mohd-Ismail, N.K.; Deng, L.; Hotta, H.; Tan, Y.J. Ns5b induces up-regulation of the bh3-only protein, bik, essential for the hepatitis c virus rna replication and viral release. Virology 2015, 474, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Yeganeh, B.; Rezaei Moghadam, A.; Alizadeh, J.; Wiechec, E.; Alavian, S.M.; Hashemi, M.; Geramizadeh, B.; Samali, A.; Bagheri Lankarani, K.; Post, M.; et al. Hepatitis b and c virus-induced hepatitis: Apoptosis, autophagy, and unfolded protein response. World J. Gastroenterol. 2015, 21, 13225–13239. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Chandra, A. Autophagy modulation as a potential therapeutic target for liver diseases. J. Clin. Exp. Hepatol. 2014, 4, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Sir, D.; Kuo, C.F.; Tian, Y.; Liu, H.M.; Huang, E.J.; Jung, J.U.; Machida, K.; Ou, J.H. Replication of hepatitis c virus rna on autophagosomal membranes. J. Biol. Chem. 2012, 287, 18036–18043. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.Y.; Chen, S.S. Activation of the unfolded protein response and autophagy after hepatitis c virus infection suppresses innate antiviral immunity in vitro. J. Clin. Investig. 2011, 121, 37–56. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.; Raychoudhuri, A.; Steele, R.; Ray, R.; Ray, R.B. Knockdown of autophagy enhances the innate immune response in hepatitis c virus-infected hepatocytes. Hepatology 2011, 53, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Mohl, B.P.; Bartlett, C.; Mankouri, J.; Harris, M. Early events in the generation of autophagosomes are required for the formation of membrane structures involved in hepatitis c virus genome replication. J. Gen. Virol. 2016, 97, 680–693. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.; Bhanja Chowdhury, J.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis c virus upregulates beclin1 for induction of autophagy and activates mtor signaling. J. Virol. 2012, 86, 8705–8712. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, R.; Ploen, D.; Hildt, E. Hcv and oxidative stress: Implications for hcv life cycle and hcv-associated pathogenesis. Oxid. Med. Cell. Longev. 2016, 2016, 9012580. [Google Scholar] [CrossRef] [PubMed]
- Ploen, D.; Hildt, E. Hepatitis c virus comes for dinner: How the hepatitis c virus interferes with autophagy. World J. Gastroenterol. 2015, 21, 8492–8507. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.J.; El Khobar, K.; Chin, R.; Earnest-Silveira, L.; Angus, P.W.; Bock, C.T.; Nachbur, U.; Silke, J.; Torresi, J. Hepatitis c virus-induced hepatocyte cell death and protection by inhibition of apoptosis. J. Gen. Virol. 2014, 95, 2204–2215. [Google Scholar] [CrossRef] [PubMed]
- Ghosh Roy, S.; Sadigh, B.; Datan, E.; Lockshin, R.A.; Zakeri, Z. Regulation of cell survival and death during flavivirus infections. World J. Biol. Chem. 2014, 5, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Masalova, O.V.; Lesnova, E.I.; Permyakova, K.Y.; Samokhvalov, E.I.; Ivanov, A.V.; Kochetkov, S.N.; Kushch, A.A. Effect of hepatitis c virus proteins on the production of proinflammatory and profibrotic cytokines in huh7.5 human hepatoma cells. Mol. Biol. (Mosk) 2016, 50, 422–430. [Google Scholar] [CrossRef]
- Benali-Furet, N.L.; Chami, M.; Houel, L.; De Giorgi, F.; Vernejoul, F.; Lagorce, D.; Buscail, L.; Bartenschlager, R.; Ichas, F.; Rizzuto, R.; et al. Hepatitis c virus core triggers apoptosis in liver cells by inducing er stress and er calcium depletion. Oncogene 2005, 24, 4921–4933. [Google Scholar] [CrossRef] [PubMed]
- Rajalakshmy, A.R.; Malathi, J.; Madhavan, H.N. Hcv core and ns3 proteins mediate toll like receptor induced innate immune response in corneal epithelium. Exp. Eye Res. 2014, 128, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Soguero, C.; Joo, M.; Chianese-Bullock, K.A.; Nguyen, D.T.; Tung, K.; Hahn, Y.S. Hepatitis c virus core protein leads to immune suppression and liver damage in a transgenic murine model. J. Virol. 2002, 76, 9345–9354. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Lee, C.G.; Lee, C.W.; Choi, E.J.; Yoon, S.K.; Ahn, K.S.; Sung, Y.C. Hepatitis c virus core inhibits the fas-mediated p38 mitogen activated kinase signaling pathway in hepatocytes. Mol. Cells 2002, 13, 452–462. [Google Scholar] [PubMed]
- Chang, M.L.; Chen, J.C.; Chang, M.Y.; Yeh, C.T.; Lin, W.P.; Liang, C.K.; Huang, S.F.; Dang, K.N.; Chiu, C.T.; Lin, D.Y. Acute expression of hepatitis c core protein in adult mouse liver: Mitochondrial stress and apoptosis. Scand. J. Gastroenterol. 2008, 43, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Smirnova, O.A.; Petrushanko, I.Y.; Ivanova, O.N.; Karpenko, I.L.; Alekseeva, E.; Sominskaya, I.; Makarov, A.A.; Bartosch, B.; Kochetkov, S.N.; et al. Hcv core protein uses multiple mechanisms to induce oxidative stress in human hepatoma huh7 cells. Viruses 2015, 7, 2745–2770. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Li, M.; Zhang, J.; Liu, S.; Wang, Q.; Quan, M.; Zhang, M.; Cheng, J. Regulation of hepg2 cell apoptosis by hepatitis c virus (hcv) core protein via the sirt1-p53-bax pathway. Virus Genes 2015, 51, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Qu, A.; Han, X.; Wang, Y. Hcv core protein represses the apoptosis and improves the autophagy of human hepatocytes. Int. J. Clin. Exp. Med. 2015, 8, 15787–15793. [Google Scholar] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Alnemri, E.S.; Altucci, L.; Andrews, D.; Annicchiarico-Petruzzelli, M.; et al. Essential versus accessory aspects of cell death: Recommendations of the nccd 2015. Cell. Death Differ. 2015, 22, 58–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbaczek, A.C.; Ribeiro, L.C.; Ximenes, V.F.; Afonso, A.; Nogueira, C.T.; Generoso, W.C.; Alberice, J.V.; Rudnicki, M.; Ferrer, R.; Fonseca, L.M.; et al. Inflammatory response of endothelial cells to hepatitis c virus recombinant envelope glycoprotein 2 protein exposure. Mem. Inst. Oswaldo Cruz 2014, 109, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Chiou, H.L.; Hsieh, Y.S.; Hsieh, M.R.; Chen, T.Y. Hcv e2 may induce apoptosis of huh-7 cells via a mitochondrial-related caspase pathway. Biochem. Biophys. Res. Commun. 2006, 345, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Zekri, A.N.; Sobhy, E.; Hussein, N.; Ahmed, O.S.; Hussein, A.; Shoman, S.; Soliman, A.H.; ElDin, H.M. E1/E2 of hepatitis c virus genotype4 and apoptosis. Asian Pac. J. Cancer Prev. 2016, 17, 3131–3138. [Google Scholar] [PubMed]
- Hitomi, J.; Katayama, T.; Eguchi, Y.; Kudo, T.; Taniguchi, M.; Koyama, Y.; Manabe, T.; Yamagishi, S.; Bando, Y.; Imaizumi, K.; et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and abeta-induced cell death. J. Cell. Biol. 2004, 165, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; Fitzgerald, U.; Samali, A. Caspase-12 and er-stress-mediated apoptosis: The story so far. Ann. N. Y. Acad. Sci. 2003, 1010, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.W.; Egan, P.A. Hepatitis c virus envelope proteins regulate chop via induction of the unfolded protein response. FASEB J. 2005, 19, 1510–1512. [Google Scholar] [CrossRef] [PubMed]
- Prikhod’ko, E.A.; Prikhod’ko, G.G.; Siegel, R.M.; Thompson, P.; Major, M.E.; Cohen, J.I. The ns3 protein of hepatitis c virus induces caspase-8-mediated apoptosis independent of its protease or helicase activities. Virology 2004, 329, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Nagano-Fujii, M.; Deng, L.; Ishido, S.; Sada, K.; Hotta, H. Single-point mutations of hepatitis c virus ns3 that impair p53 interaction and anti-apoptotic activity of ns3. Biochem. Biophys. Res. Commun. 2006, 340, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Siu, G.K.; Zhou, F.; Yu, M.K.; Zhang, L.; Wang, T.; Liang, Y.; Chen, Y.; Chan, H.C.; Yu, S. Hepatitis c virus ns5a protein cooperates with phosphatidylinositol 4-kinase iiialpha to induce mitochondrial fragmentation. Sci. Rep. 2016, 6, 23464. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; Waris, G.; Tanveer, R.; Siddiqui, A. Human hepatitis c virus ns5a protein alters intracellular calcium levels, induces oxidative stress, and activates stat-3 and nf-kappa b. Proc. Natl. Acad. Sci. USA 2001, 98, 9599–9604. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Smirnova, O.A.; Ivanova, O.N.; Masalova, O.V.; Kochetkov, S.N.; Isaguliants, M.G. Hepatitis c virus proteins activate nrf2/are pathway by distinct ros-dependent and independent mechanisms in huh7 cells. PLoS ONE 2011, 6, e24957. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, O.A.; Ivanova, O.N.; Bartosch, B.; Valuev-Elliston, V.T.; Mukhtarov, F.; Kochetkov, S.N.; Ivanov, A.V. Hepatitis c virus ns5a protein triggers oxidative stress by inducing nadph oxidases 1 and 4 and cytochrome p450 2e1. Oxid. Med. Cell. Longev. 2016, 2016, 8341937. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kang, W.; Ryu, S.W.; Kim, W.I.; Chang, D.Y.; Lee, D.H.; Park, D.Y.; Choi, Y.H.; Choi, K.; Shin, E.C.; et al. Hepatitis c virus infection enhances tnfalpha-induced cell death via suppression of nf-kappab. Hepatology 2012, 56, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Nomura-Takigawa, Y.; Nagano-Fujii, M.; Deng, L.; Kitazawa, S.; Ishido, S.; Sada, K.; Hotta, H. Non-structural protein 4a of hepatitis c virus accumulates on mitochondria and renders the cells prone to undergoing mitochondria-mediated apoptosis. J. Gen. Virol. 2006, 87, 1935–1945. [Google Scholar] [CrossRef] [PubMed]
- Targett-Adams, P.; Graham, E.J.; Middleton, J.; Palmer, A.; Shaw, S.M.; Lavender, H.; Brain, P.; Tran, T.D.; Jones, L.H.; Wakenhut, F.; et al. Small molecules targeting hepatitis c virus-encoded ns5a cause subcellular redistribution of their target: Insights into compound modes of action. J. Virol. 2011, 85, 6353–6368. [Google Scholar] [CrossRef] [PubMed]
- Silberstein, E.; Ulitzky, L.; Lima, L.A.; Cehan, N.; Teixeira-Carvalho, A.; Roingeard, P.; Taylor, D.R. Hcv-mediated apoptosis of hepatocytes in culture and viral pathogenesis. PLoS ONE 2016, 11, e0155708. [Google Scholar] [CrossRef] [PubMed]
- Biederbick, A.; Kern, H.F.; Elsasser, H.P. Monodansylcadaverine (mdc) is a specific in vivo marker for autophagic vacuoles. Eur. J. Cell. Biol. 1995, 66, 3–14. [Google Scholar] [PubMed]
- Klionsky, D.J.; Cuervo, A.M.; Seglen, P.O. Methods for monitoring autophagy from yeast to human. Autophagy 2007, 3, 181–206. [Google Scholar] [CrossRef] [PubMed]
- Chu, V.C.; Bhattacharya, S.; Nomoto, A.; Lin, J.; Zaidi, S.K.; Oberley, T.D.; Weinman, S.A.; Azhar, S.; Huang, T.T. Persistent Expression of Hepatitis C Virus Non-Structural Proteins Leads to Increased Autophagy and Mitochondrial Injury in Human Hepatoma Cells. PLoS ONE 2011, 6, e28551. [Google Scholar] [CrossRef] [PubMed]
- Quan, M.; Liu, S.; Li, G.; Wang, Q.; Zhang, J.; Zhang, M.; Li, M.; Gao, P.; Feng, S.; Cheng, J. A functional role for ns5atp9 in the induction of hcv ns5a-mediated autophagy. J. Viral Hepat. 2014, 21, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Su, W.C.; Chao, T.C.; Huang, Y.L.; Weng, S.C.; Jeng, K.S.; Lai, M.M. Rab5 and class iii phosphoinositide 3-kinase vps34 are involved in hepatitis c virus ns4b-induced autophagy. J. Virol. 2011, 85, 10561–10571. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kang, R.; Huang, H.; Xi, X.; Wang, B.; Zhao, Z. Hepatitis c virus core protein activates autophagy through eif2ak3 and atf6 upr pathway-mediated map1lc3b and atg12 expression. Autophagy 2014, 10, 766–784. [Google Scholar] [CrossRef] [PubMed]
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the caspase-8-flip(l) complex inhibits ripk3-dependent necrosis. Nature 2011, 471, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Pietschmann, T.; Lohmann, V.; Rutter, G.; Kurpanek, K.; Bartenschlager, R. Characterization of Cell Lines Carrying Self-Replicating Hepatitis C Virus RNAs. J. Virol. 2001, 75, 1252–1264. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.J.; Vanags, D.M.; Clouston, A.D.; Richardson, M.M.; Purdie, D.M.; Jonsson, J.R.; Powell, E.E. Steatosis and Liver Cell Apoptosis in Chronic Hepatitis C: A Mechanism for Increased Liver Injury. Hepatology 2004, 39, 1230–1238. [Google Scholar] [CrossRef] [PubMed]
- Blight, K.J.; McKeating, J.A.; Rice, C.M. Highly permissive cell lines for subgenomic and genomic hepatitis c virus rna replication. J. Virol. 2002, 76, 13001–13014. [Google Scholar] [CrossRef] [PubMed]
- Frese, M.; Barth, K.; Kaul, A.; Lohmann, V.; Schwarzle, V.; Bartenschlager, R. Hepatitis c virus rna replication is resistant to tumour necrosis factor-alpha. J. Gen. Virol. 2003, 84, 1253–1259. [Google Scholar] [CrossRef] [PubMed]
- Masalova, O.V.; Lesnova, E.I.; Grabovetskii, V.V.; Smirnova, O.A.; Ulanova, T.I.; Burkov, A.N.; Ivanov, A.V.; Zaberezhnyi, A.D.; Ataullakhanov, R.I.; Kushch, A.A. DNA immunization with a plasmid carrying the gene of hepatitis c virus protein 5a (ns5a) induces an effective cellular immune response. Mol. Biol. Mosk 2010, 44, 245–253. [Google Scholar] [CrossRef]
- Masalova, O.V.; Lesnova, E.I.; Ivanov, A.V.; Pichugin, A.V.; Permiakova, K.; Smirnova, O.A.; Tynitskaia, V.L.; Ulanova, T.I.; Burkov, A.N.; Kochetkov, S.N.; et al. Comparative analysis of the immune response to DNA constructions encoding hepatitis c virus nonstructural proteins. Vopr. Virusol. 2013, 58, 21–28. [Google Scholar] [PubMed]
- Permyakova, K.Y.; Lesnova, E.I.; Masalova, O.V.; Ivanov, A.V.; Ataullakhanov, R.I.; Kushch, A.A. Immunogenic properties of DNA constructions containing structural and nonstructural regions of hepatitis c virus. Immunologiya 2015, 36, 162–167. [Google Scholar]
- Rechkina, E.A.; Denisova, G.F.; Masalova, O.V.; Lideman, L.F.; Denisov, D.A.; Lesnova, E.I.; Ataullakhanov, R.I.; Gur’ianova, S.V.; Kushch, A.A. Mapping of antigenic determinants of hepatitis c virus proteins using phage display. Mol. Biol. (Mosk) 2006, 40, 312–323. [Google Scholar] [CrossRef]
- Perry, S.W.; Epstein, L.G.; Gelbard, H.A. Simultaneous in situ detection of apoptosis and necrosis in monolayer cultures by tunel and trypan blue staining. Biotechniques 1997, 22, 1102–1106. [Google Scholar] [PubMed]
- Schlueter, N.; Lussi, A.; Ganss, C.; Gruber, R. L929 fibroblast bioassay on the in vitro toxicity of sncl2, h3po4, clearfil se primer and combinations thereof. Swiss Dent. J. 2016, 126, 566–572. [Google Scholar] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Markers of Cell Death | HCV Proteins | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Core | E1/E2 | NS3 | NS4A/B | NS5A | NS5B | NS3-NS5B | HCV Replicon | ||
| Apoptosis | Caspase-8 | 3.1 1 | ~ 2 | 2.7 | 0.5 | ~ | 0.5 | 1.8 | 1.9 |
| Caspase-9 | 2.1 | ~ | ~ | ~ | 0.5 | 0.5 | ~ | 1.6 | |
| Caspase-3 | 1.6 | 1.6 | 2.6 | ~ | 0.6 | ~ | ~ | 3.2 | |
| TUNEL | 2.0 | 2.2 | ~ | ~ | ~ | ~ | ~ | 2.1 | |
| Auto-phagy | MDC | ~ | 4.1 | 1.8 | 2.9 | 2.9 | 4.8 | 2.1 | 9.3 |
| LC3-II | ~ | 1.3 | ~ | 1.6 | 1.4 | 1.6 | ~ | 2.2 | |
| Necrosis | ~ | ~ | ~ | ~ | 2.1 | ~ | ~ | ~ | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masalova, O.V.; Lesnova, E.I.; Solyev, P.N.; Zakirova, N.F.; Prassolov, V.S.; Kochetkov, S.N.; Ivanov, A.V.; Kushch, A.A. Modulation of Cell Death Pathways by Hepatitis C Virus Proteins in Huh7.5 Hepatoma Cells. Int. J. Mol. Sci. 2017, 18, 2346. https://doi.org/10.3390/ijms18112346
Masalova OV, Lesnova EI, Solyev PN, Zakirova NF, Prassolov VS, Kochetkov SN, Ivanov AV, Kushch AA. Modulation of Cell Death Pathways by Hepatitis C Virus Proteins in Huh7.5 Hepatoma Cells. International Journal of Molecular Sciences. 2017; 18(11):2346. https://doi.org/10.3390/ijms18112346
Chicago/Turabian StyleMasalova, Olga V., Ekaterina I. Lesnova, Pavel N. Solyev, Natalia F. Zakirova, Vladimir S. Prassolov, Sergey N. Kochetkov, Alexander V. Ivanov, and Alla A. Kushch. 2017. "Modulation of Cell Death Pathways by Hepatitis C Virus Proteins in Huh7.5 Hepatoma Cells" International Journal of Molecular Sciences 18, no. 11: 2346. https://doi.org/10.3390/ijms18112346
APA StyleMasalova, O. V., Lesnova, E. I., Solyev, P. N., Zakirova, N. F., Prassolov, V. S., Kochetkov, S. N., Ivanov, A. V., & Kushch, A. A. (2017). Modulation of Cell Death Pathways by Hepatitis C Virus Proteins in Huh7.5 Hepatoma Cells. International Journal of Molecular Sciences, 18(11), 2346. https://doi.org/10.3390/ijms18112346