Major Challenges and Potential Microenvironment-Targeted Therapies in Glioblastoma


 ,
,
Abstract
:1. Glioblastoma Statistics
2. Overarching Challenges
2.1. Challenges Due to Hypoxia and Hyper-Vasculaturity in the Microenvironment
2.2. Challenges Due to Microenvironment-Driven Resistance to Antiangiogenic Therapy
2.3. Challenges Due to Microenvironment-Driven Alternative Vascularization
2.4. Challenges Due to an Immune Suppressive Microenvironment Following Standard Therapy
2.5. Challenges Due to Molecular and Genetic Heterogeneity in GBM Tumors
3. Potential Adjuvant Therapies against GBM
3.1. Anti-Myeloid Therapies
3.2. Immune Therapies
3.3. Anti-CYP4A Therapy
4. Conclusions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 20-HETE | 20-Hydroxyeicosatetraenoic acid |
| CD202b | Angiopoietin-1 receptor |
| AGPT2 | Angiopoietin-2 |
| AAT | Antiangiogenic therapy |
| BMDCs | Bone marrow-derived cells |
| CSC | Cancer stem cell |
| CBTRUS | Central Brain Tumor Registry of the United States |
| CNS | Central nervous system |
| CXCL7 | Chemokine (C-X-C motif) ligand 7 |
| CXCL8 | Chemokine (C-X-C motif) ligand 7 |
| CD | Clusters of differentiation |
| COL1A1 | Collagen, type 1, alpha 1 |
| CSF | Colony-stimulating factor |
| CSF1R | Colony-stimulating factor receptor 1 |
| CD45 | Common leukocyte antigen |
| CXCR4 | C-X-C Motif Chemokine Receptor 4 |
| CYP4A and CYP4F | Cytochromes P450 family enzymes |
| DLL | Delta-like ligand |
| ECs | Endothelial cells |
| EPCs | Endothelial progenitor cells |
| Eph | Ephrin |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| FGF | Fibroblast growth factor |
| GBM | Glioblastoma |
| GBM-O | Glioblastoma with oligodendroglioma component |
| GSCs | Glioma stem cell-like cells |
| G-CIMP | Glioma-CpG island methylator phenotype |
| G-CSF | Granulocyte-colony stimulating factor |
| gMDSC | Granulocytic myeloid-derived suppressor cells |
| GFP | Green fluorescent protein |
| CD34 | Hematopoietic progenitor cell antigen |
| HGF | Hepatocyte growth factor |
| hTERT | human telomerase reverse transcriptase |
| HIF1-α | Hypoxia-inducible factor- alpha |
| IGF | Insulin-like growth factor |
| IL8 | Interleukin 8 |
| IDH | Isocitrate dehydrogenase |
| MMPs | Matrix metalloproteinases |
| MAPK | Mitogen-activated protein kinase |
| MCP-1 | Monocyte chemoattractant protein 1 |
| MDM2 | Mouse double minute 2 homolog |
| MDSC | Myeloid-derived suppressor cells |
| NG2 | Neuron-glial antigen 2 |
| HET0016 | N-Hydroxy-N′-(4-butyl-2-methylphenyl)-formamidine |
| NO | Nitric oxide |
| NFκB | Nuclear factor-kappa beta |
| MGMT | O-6-Methylguanine-DNA Methyltransferase |
| PAS | Periodic Acid Schiff |
| PTEN | Phosphatase and tensin homolog |
| PI3K | Phosphatidylinositide 3-kinase |
| PLGF | Placental growth factor |
| CD31/PECAM-1 | Platelet endothelial cell adhesion molecule |
| PDGF | Platelet-derived growth factor |
| PDGFR | Platelet-derived growth factor receptor |
| RT | Radiotherapy |
| CD133 | Stem-cell marker (Prominin-1) |
| SDF-1α | Stromal-derived factor 1 alpha |
| TMZ | Temozolomide |
| TN-C | Tenascin C |
| TGFβ | Transforming growth factor-beta |
| TME | Tumor microenvironment |
| TP53 | Tumor p53 |
| TAM | Tumor-associated macrophage |
| TAMCs | Tumor-associated myeloid cells |
| TDEC | Tumor-derived endothelial cells |
| US FDA | United States Food and Drug Administration |
| VEGF | Vascular endothelial growth factor |
| VEGFR | Vascular endothelial growth factor receptor |
| VM | Vascular mimicry |
| WHO | World Health Organization |
References
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomarkers Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Liao, P.; Vecchione-Koval, T.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro Oncol. 2017, 19 (Suppl. 5), v1–v88. [Google Scholar] [CrossRef] [PubMed]
- Oliver, L.; Olivier, C.; Marhuenda, F.B.; Campone, M.; Vallette, F.M. Hypoxia and the malignant glioma microenvironment: Regulation and implications for therapy. Curr. Mol. Pharmacol. 2009, 2, 263–284. [Google Scholar] [CrossRef] [PubMed]
- Brem, S.; Cotran, R.; Folkman, J. Tumor angiogenesis: A quantitative method for histologic grading. J. Natl. Cancer Inst. 1972, 48, 347–356. [Google Scholar] [PubMed]
- Wang, N.; Jain, R.K.; Batchelor, T.T. New Directions in Anti-Angiogenic Therapy for Glioblastoma. Neurotherapeutics 2017, 14, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Hanash, S.; Schliekelman, M. Proteomic profiling of the tumor microenvironment: Recent insights and the search for biomarkers. Genome Med. 2014, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Charles, N.A.; Holland, E.C.; Gilbertson, R.; Glass, R.; Kettenmann, H. The brain tumor microenvironment. Glia 2011, 59, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Tugues, S.; Li, X.; Gualandi, L.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Biochem. J. 2011, 437, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegue, E.; Song, H.; Vandenberg, S.; Johnson, R.S.; Werb, Z.; et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 2008, 13, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Plate, K.H.; Breier, G.; Weich, H.A.; Risau, W. Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature 1992, 359, 845–848. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.K.; Itani, R.M.; Wang, H.; Tomikawa, M.; Sarfeh, I.J.; Szabo, S.; Tarnawski, A.S. Activation of VEGF and Ras genes in gastric mucosa during angiogenic response to ethanol injury. Am. J. Physiol. 1999, 276, G1345–G1355. [Google Scholar] [CrossRef]
- Gomez-Manzano, C.; Fueyo, J.; Jiang, H.; Glass, T.L.; Lee, H.Y.; Hu, M.; Liu, J.L.; Jasti, S.L.; Liu, T.J.; Conrad, C.A.; et al. Mechanisms underlying PTEN regulation of vascular endothelial growth factor and angiogenesis. Ann. Neurol. 2003, 53, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Shibuya, M. The 230 kDa mature form of KDR/Flk-1 (VEGF receptor-2) activates the PLC-gamma pathway and partially induces mitotic signals in NIH3T3 fibroblasts. Oncogene 1997, 14, 2079–2089. [Google Scholar] [CrossRef] [PubMed]
- Saino, M.; Maruyama, T.; Sekiya, T.; Kayama, T.; Murakami, Y. Inhibition of angiogenesis in human glioma cell lines by antisense RNA from the soluble guanylate cyclase genes, GUCY1A3 and GUCY1B3. Oncol. Rep. 2004, 12, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.C.; Goldman, C.K.; Gillespie, G.Y. Vascular endothelial growth factor in human glioma cell lines: Induced secretion by EGF, PDGF-BB, and bFGF. J. Neurosurg. 1995, 82, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Kerbel, R.S. Tumor angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Talasila, K.M.; Rosland, G.V.; Hagland, H.R.; Eskilsson, E.; Flones, I.H.; Fritah, S.; Azuaje, F.; Atai, N.; Harter, P.N.; Mittelbronn, M.; et al. The angiogenic switch leads to a metabolic shift in human glioblastoma. Neuro Oncol. 2017, 19, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [PubMed]
- Folkman, J.; Shing, Y. Angiogenesis. J. Biol. Chem. 1992, 267, 10931–10934. [Google Scholar] [PubMed]
- Ferrara, N. VEGF and Intraocular Neovascularization: From Discovery to Therapy. Transl. Vis. Sci. Technol. 2016, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J.; Long, D.M., Jr.; Becker, F.F. Growth and metastasis of tumor in organ culture. Cancer 1963, 16, 453–467. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Mittal, K.; Ebos, J.; Rini, B. Angiogenesis and the tumor microenvironment: Vascular endothelial growth factor and beyond. Semin. Oncol. 2014, 41, 235–251. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.; Pagani, A.; Magnani, E.; Rossi, T.; Noonan, D.M.; Cantelmo, A.R.; Albini, A. Inflammatory angiogenesis and the tumor microenvironment as targets for cancer therapy and prevention. Cancer Treat. Res. 2014, 159, 401–426. [Google Scholar] [PubMed]
- Samples, J.; Willis, M.; Klauber-Demore, N. Targeting angiogenesis and the tumor microenvironment. Surg. Oncol. Clin. N. Am. 2013, 22, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Saharinen, P.; Eklund, L.; Pulkki, K.; Bono, P.; Alitalo, K. VEGF and angiopoietin signaling in tumor angiogenesis and metastasis. Trends Mol. Med. 2011, 17, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Rahman, R.; Smith, S.; Rahman, C.; Grundy, R. Antiangiogenic therapy and mechanisms of tumor resistance in malignant glioma. J. Oncol. 2010, 2010, 251231. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, J.; Norden, A.D.; Wen, P.Y. Emerging antiangiogenic treatments for gliomas—Efficacy and safety issues. Curr. Opin. Neurol. 2008, 21, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Sweeney, C.J.; Sledge, G.W., Jr. Can tumor angiogenesis be inhibited without resistance? In Mechanisms of Angiogenesis; Matthias, C., Georg, B., Eds.; Birkhäuser Basel: Basel, Switzerland; pp. 95–112.
- Ali, M.M.; Kumar, S.; Shankar, A.; Varma, N.R.; Iskander, A.S.; Janic, B.; Chwang, W.B.; Jain, R.; Babajeni-Feremi, A.; Borin, T.F.; et al. Effects of tyrosine kinase inhibitors and CXCR4 antagonist on tumor growth and angiogenesis in rat glioma model: MRI and protein analysis study. Transl. Oncol. 2013, 6, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.M.; Janic, B.; Babajani-Feremi, A.; Varma, N.R.; Iskander, A.S.; Anagli, J.; Arbab, A.S. Changes in vascular permeability and expression of different angiogenic factors following anti-angiogenic treatment in rat glioma. PLoS ONE 2010, 5, e8727. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, T.T.; Duda, D.G.; di Tomaso, E.; Ancukiewicz, M.; Plotkin, S.R.; Gerstner, E.; Eichler, A.F.; Drappatz, J.; Hochberg, F.H.; Benner, T.; et al. Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. J. Clin. Oncol. 2010, 28, 2817–2823. [Google Scholar] [CrossRef] [PubMed]
- Kamoun, W.S.; Ley, C.D.; Farrar, C.T.; Duyverman, A.M.; Lahdenranta, J.; Lacorre, D.A.; Batchelor, T.T.; di Tomaso, E.; Duda, D.G.; Munn, L.L.; et al. Edema control by cediranib, a vascular endothelial growth factor receptor-targeted kinase inhibitor, prolongs survival despite persistent brain tumor growth in mice. J. Clin. Oncol. 2009, 27, 2542–2552. [Google Scholar] [CrossRef] [PubMed]
- Neyns, B.; Sadones, J.; Chaskis, C.; Dujardin, M.; Everaert, H.; Lv, S.; Duerinck, J.; Tynninen, O.; Nupponen, N.; Michotte, A.; et al. Phase II study of sunitinib malate in patients with recurrent high-grade glioma. J. Neurooncol. 2011, 103, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Vredenburgh, J.J.; Coan, A.; Desjardins, A.; Peters, K.B.; Gururangan, S.; Sathornsumetee, S.; Rich, J.N.; Herndon, J.E.; Friedman, H.S. Phase I study of sunitinib and irinotecan for patients with recurrent malignant glioma. J. Neurooncol. 2011, 105, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Egorin, M.J.; Desjardins, A.; Vredenburgh, J.J.; Beumer, J.H.; Lagattuta, T.F.; Gururangan, S.; Herndon, J.E., 2nd; Salvado, A.J.; Friedman, H.S. Phase I pharmacokinetic study of the vascular endothelial growth factor receptor tyrosine kinase inhibitor vatalanib (PTK787) plus imatinib and hydroxyurea for malignant glioma. Cancer 2009, 115, 2188–2198. [Google Scholar] [CrossRef] [PubMed]
- Achyut, B.R.; Shankar, A.; Iskander, A.S.; Ara, R.; Knight, R.A.; Scicli, A.G.; Arbab, A.S. Chimeric Mouse model to track the migration of bone marrow derived cells in glioblastoma following anti-angiogenic treatments. Cancer Biol. Ther. 2016, 17, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Shaaban, S.; Alsulami, M.; Arbab, S.A.; Ara, R.; Shankar, A.; Iskander, A.; Angara, K.; Jain, M.; Bagher-Ebadian, H.; Achyut, B.R.; et al. Targeting Bone Marrow to Potentiate the Anti-Tumor Effect of Tyrosine Kinase Inhibitor in Preclinical Rat Model of Human Glioblastoma. Int. J. Cancer Res. 2016, 12, 69–81. [Google Scholar] [PubMed]
- Achyut, B.R.; Shankar, A.; Iskander, A.S.; Ara, R.; Angara, K.; Zeng, P.; Knight, R.A.; Scicli, A.G.; Arbab, A.S. Bone marrow derived myeloid cells orchestrate antiangiogenic resistance in glioblastoma through coordinated molecular networks. Cancer Lett. 2015, 369, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Achyut, B.R.; Angara, K.; Jain, M.; Borin, T.F.; Rashid, M.H.; Iskander, A.S.M.; Ara, R.; Kolhe, R.; Howard, S.; Venugopal, N.; et al. Canonical NFkappaB signaling in myeloid cells is required for the glioblastoma growth. Sci. Rep. 2017, 7, 13754. [Google Scholar] [CrossRef] [PubMed]
- Rosen, L.S. Clinical experience with angiogenesis signaling inhibitors: Focus on vascular endothelial growth factor (VEGF) blockers. Cancer Control 2002, 9 (Suppl. 2), 36–44. [Google Scholar] [CrossRef] [PubMed]
- Norden, A.D.; Young, G.S.; Setayesh, K.; Muzikansky, A.; Klufas, R.; Ross, G.L.; Ciampa, A.S.; Ebbeling, L.G.; Levy, B.; Drappatz, J.; et al. Bevacizumab for recurrent malignant gliomas: Efficacy, toxicity, and patterns of recurrence. Neurology 2008, 70, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Kreisl, T.N.; Zhang, W.; Odia, Y.; Shih, J.H.; Butman, J.A.; Hammoud, D.; Iwamoto, F.M.; Sul, J.; Fine, H.A. A phase II trial of single-agent bevacizumab in patients with recurrent anaplastic glioma. Neuro Oncol. 2011, 13, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Desjardins, A.; Peters, K.B.; Gururangan, S.; Sampson, J.H.; McLendon, R.E.; Herndon, J.E., 2nd; Bulusu, A.; Threatt, S.; Friedman, A.H.; et al. Phase II study of carboplatin, irinotecan, and bevacizumab for bevacizumab naive, recurrent glioblastoma. J. Neurooncol. 2012, 107, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Arbab, A.S. Neovascularization in Glioblastoma: Current Pitfall in Anti-angiogenic therapy. Zhong Liu Za Zhi 2013, 1, 16–19. [Google Scholar] [PubMed]
- Hardee, M.E.; Zagzag, D. Mechanisms of glioma-associated neovascularization. Am. J. Pathol. 2012, 181, 1126–1141. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Angara, K.; Rashid, M.H.; Shankar, A.; Ara, R.; Iskander, A.; Borin, T.F.; Jain, M.; Achyut, B.R.; Arbab, A.S. Vascular mimicry in glioblastoma following anti-angiogenic and anti-20-HETE therapies. Histol. Histopathol. 2017, 32, 917–928. [Google Scholar] [PubMed]
- Angara, K.; Borin, T.F.; Arbab, A.S. Vascular Mimicry: A Novel Neovascularization Mechanism Driving Anti-Angiogenic Therapy (AAT) Resistance in Glioblastoma. Transl. Oncol. 2017, 10, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Arbab, A.S.; Jain, M.; Achyut, B.R. Vascular Mimicry: The Next Big Glioblastoma Target. Biochem. Physiol. 2015, 4, e410. [Google Scholar] [PubMed]
- Folberg, R.; Hendrix, M.J.; Maniotis, A.J. Vasculogenic mimicry and tumor angiogenesis. Am. J. Pathol. 2000, 156, 361–381. [Google Scholar] [CrossRef]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef]
- Sullivan, J.P.; Nahed, B.V.; Madden, M.W.; Oliveira, S.M.; Springer, S.; Bhere, D.; Chi, A.S.; Wakimoto, H.; Rothenberg, S.M.; Sequist, L.V.; et al. Brain tumor cells in circulation are enriched for mesenchymal gene expression. Cancer Discov. 2014, 4, 1299–1309. [Google Scholar] [CrossRef] [PubMed]
- Ortensi, B.; Setti, M.; Osti, D.; Pelicci, G. Cancer stem cell contribution to glioblastoma invasiveness. Stem Cell Res. Ther. 2013, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Chen, C.; Shi, Y.; Wu, Q.; Gimple, R.C.; Fang, X.; Huang, Z.; Zhai, K.; Ke, S.Q.; Ping, Y.F.; et al. Targeting Glioma Stem Cell-Derived Pericytes Disrupts the Blood-Tumor Barrier and Improves Chemotherapeutic Efficacy. Cell Stem Cell 2017, 21, 591–603. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Hsieh, I.Y.; Huang, X.; Li, J.; Zhao, W. Glioblastoma Stem-Like Cells: Characteristics, Microenvironment, and Therapy. Front. Pharmacol. 2016, 7, 477. [Google Scholar] [CrossRef] [PubMed]
- Liebelt, B.D.; Shingu, T.; Zhou, X.; Ren, J.; Shin, S.A.; Hu, J. Glioma Stem Cells: Signaling, Microenvironment, and Therapy. Stem Cells Int. 2016, 2016, 7849890. [Google Scholar] [CrossRef] [PubMed]
- Audia, A.; Conroy, S.; Glass, R.; Bhat, K.P.L. The Impact of the Tumor Microenvironment on the Properties of Glioma Stem-Like Cells. Front. Oncol. 2017, 7, 143. [Google Scholar] [CrossRef] [PubMed]
- Soda, Y.; Marumoto, T.; Friedmann-Morvinski, D.; Soda, M.; Liu, F.; Michiue, H.; Pastorino, S.; Yang, M.; Hoffman, R.M.; Kesari, S.; et al. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4274–4280. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Y.; Ke, Y.Q.; Lu, G.H.; Song, Z.H.; Yu, L.; Xiao, S.; Sun, X.L.; Jiang, X.D.; Yang, Z.L.; Hu, C.C. Vasculogenic mimicry is a prognostic factor for postoperative survival in patients with glioblastoma. J. Neurooncol. 2013, 112, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.M.; Zhang, Q.P.; Mu, Y.G.; Zhang, X.H.; Sai, K.; Pang, J.C.; Ng, H.K.; Chen, Z.P. Clinical significance of vasculogenic mimicry in human gliomas. J. Neurooncol. 2011, 105, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Malmstrom, A.; Gronberg, B.H.; Marosi, C.; Stupp, R.; Frappaz, D.; Schultz, H.; Abacioglu, U.; Tavelin, B.; Lhermitte, B.; Hegi, M.E.; et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: The Nordic randomised, phase 3 trial. Lancet Oncol. 2012, 13, 916–926. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Ellor, S.V.; Pagano-Young, T.A.; Avgeropoulos, N.G. Glioblastoma: Background, standard treatment paradigms, and supportive care considerations. J. Law Med. Ethics 2014, 42, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Walid, M.S. Prognostic factors for long-term survival after glioblastoma. Perm. J. 2008, 12, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Van Meir, E.G.; Hadjipanayis, C.G.; Norden, A.D.; Shu, H.K.; Wen, P.Y.; Olson, J.J. Exciting new advances in neuro-oncology: The avenue to a cure for malignant glioma. CA Cancer J. Clin. 2010, 60, 166–193. [Google Scholar] [CrossRef] [PubMed]
- Woehrer, A.; Bauchet, L.; Barnholtz-Sloan, J.S. Glioblastoma survival: Has it improved? Evidence from population-based studies. Curr. Opin. Neurol. 2014, 27, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Swartz, A.M.; Li, Q.J.; Sampson, J.H. Rindopepimut: A promising immunotherapeutic for the treatment of glioblastoma multiforme. Immunotherapy 2014, 6, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Swartz, A.M.; Batich, K.A.; Fecci, P.E.; Sampson, J.H. Peptide vaccines for the treatment of glioblastoma. J. Neurooncol. 2014, 123, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Venur, V.A.; Peereboom, D.M.; Ahluwalia, M.S. Current medical treatment of glioblastoma. Cancer Treat. Res. 2015, 163, 103–115. [Google Scholar] [PubMed]
- Hoffermann, M.; Bruckmann, L.; Kariem Mahdy, A.; Asslaber, M.; Payer, F.; von Campe, G. Treatment results and outcome in elderly patients with glioblastoma multiforme—A retrospective single institution analysis. Clin. Neurol. Neurosurg. 2015, 128, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Tsang, D.S.; Khan, L.; Perry, J.R.; Soliman, H.; Sahgal, A.; Keith, J.L.; Mainprize, T.G.; Das, S.; Zhang, L.; Tsao, M.N. Survival Outcomes in Elderly Patients with Glioblastoma. Clin. Oncol. 2015, 27, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Lau, D.; Magill, S.T.; Aghi, M.K. Molecularly targeted therapies for recurrent glioblastoma: Current and future targets. Neurosurg. Focus 2014, 37, E15. [Google Scholar] [CrossRef] [PubMed]
- Multhoff, G.; Radons, J. Radiation, Inflammation, and Immune Responses in Cancer. Front. Oncol. 2012, 2, 58. [Google Scholar] [CrossRef] [PubMed]
- Yeung, Y.T.; McDonald, K.L.; Grewal, T.; Munoz, L. Interleukins in glioblastoma pathophysiology: Implications for therapy. Br. J. Pharmacol. 2013, 168, 591–606. [Google Scholar] [CrossRef] [PubMed]
- Hardee, M.E.; Marciscano, A.E.; Medina-Ramirez, C.M.; Zagzag, D.; Narayana, A.; Lonning, S.M.; Barcellos-Hoff, M.H. Resistance of Glioblastoma-Initiating Cells to Radiation Mediated by the Tumor Microenvironment Can Be Abolished by Inhibiting Transforming Growth Factor-β. Cancer Res. 2012, 72, 4119–4129. [Google Scholar] [CrossRef] [PubMed]
- Lu-Emerson, C.; Snuderl, M.; Kirkpatrick, N.D.; Goveia, J.; Davidson, C.; Huang, Y.; Riedemann, L.; Taylor, J.; Ivy, P.; Duda, D.G.; et al. Increase in tumor-associated macrophages after antiangiogenic therapy is associated with poor survival among patients with recurrent glioblastoma. Neuro Oncol. 2013, 15, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Allavena, P.; Garlanda, C.; Borrello, M.G.; Sica, A.; Mantovani, A. Pathways connecting inflammation and cancer. Curr. Opin. Genet. Dev. 2008, 18, 3–10. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Fadul, C.E.; Fisher, J.L.; Gui, J.; Hampton, T.H.; Cote, A.L.; Ernstoff, M.S. Immune modulation effects of concomitant temozolomide and radiation therapy on peripheral blood mononuclear cells in patients with glioblastoma multiforme. Neuro Oncol. 2011, 13, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Marrinan, J.; Frishman, C.; Sampath, P. Impact of temozolomide on immune response during malignant glioma chemotherapy. Clin. Dev. Immunol. 2012, 2012, 831090. [Google Scholar] [CrossRef] [PubMed]
- Achyut, B.R.; Arbab, A.S. Myeloid cell signatures in tumor microenvironment predicts therapeutic response in cancer. OncoTargets Ther. 2016, 9, 1047–1055. [Google Scholar]
- Bruchard, M.; Mignot, G.; Derangere, V.; Chalmin, F.; Chevriaux, A.; Vegran, F.; Boireau, W.; Simon, B.; Ryffel, B.; Connat, J.L.; et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat. Med. 2013, 19, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, J.X.; Liu, J.P.; You, C.; Liu, Y.H.; Mao, Q. Gain of function of mutant TP53 in glioblastoma: Prognosis and response to temozolomide. Ann. Surg. Oncol. 2014, 21, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Brazdova, M.; Quante, T.; Togel, L.; Walter, K.; Loscher, C.; Tichy, V.; Cincarova, L.; Deppert, W.; Tolstonog, G.V. Modulation of gene expression in U251 glioblastoma cells by binding of mutant p53 R273H to intronic and intergenic sequences. Nucleic Acids Res. 2009, 37, 1486–1500. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef] [PubMed]
- Olar, A.; Aldape, K.D. Using the molecular classification of glioblastoma to inform personalized treatment. J. Pathol. 2014, 232, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Hoshide, R.; Jandial, R. 2016 World Health Organization Classification of Central Nervous System Tumors: An Era of Molecular Biology. World Neurosurg. 2016, 94, 561–562. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Broniscer, A.; Tatevossian, R.G.; Sabin, N.D.; Klimo, P., Jr.; Dalton, J.; Lee, R.; Gajjar, A.; Ellison, D.W. Clinical, radiological, histological and molecular characteristics of paediatric epithelioid glioblastoma. Neuropathol. Appl. Neurobiol. 2014, 40, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Kleinschmidt-DeMasters, B.K.; Aisner, D.L.; Foreman, N.K. BRAF VE1 immunoreactivity patterns in epithelioid glioblastomas positive for BRAF V600E mutation. Am. J. Surg. Pathol. 2015, 39, 528–540. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Appin, C.L.; Gao, J.; Chisolm, C.; Torian, M.; Alexis, D.; Vincentelli, C.; Schniederjan, M.J.; Hadjipanayis, C.; Olson, J.J.; Hunter, S.; et al. Glioblastoma with oligodendroglioma component (GBM-O): Molecular genetic and clinical characteristics. Brain Pathol. 2013, 23, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Hinrichs, B.H.; Newman, S.; Appin, C.L.; Dunn, W.; Cooper, L.; Pauly, R.; Kowalski, J.; Rossi, M.R.; Brat, D.J. Farewell to GBM-O: Genomic and transcriptomic profiling of glioblastoma with oligodendroglioma component reveals distinct molecular subgroups. Acta Neuropathol. Commun. 2016, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Labussiere, M.; Idbaih, A.; Wang, X.W.; Marie, Y.; Boisselier, B.; Falet, C.; Paris, S.; Laffaire, J.; Carpentier, C.; Criniere, E.; et al. All the 1p19q codeleted gliomas are mutated on IDH1 or IDH2. Neurology 2010, 74, 1886–1890. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Liu, Y.; Wang, Y.; Ke, R.H.; Mao, Y.; Ye, Z.R. Chromosome 1p/19q status combined with expression of p53 protein improves the diagnostic and prognostic evaluation of oligodendrogliomas. Chin. Med. J. (Engl.) 2010, 123, 3566–3573. [Google Scholar] [PubMed]
- Eoli, M.; Menghi, F.; Bruzzone, M.G.; De Simone, T.; Valletta, L.; Pollo, B.; Bissola, L.; Silvani, A.; Bianchessi, D.; D’Incerti, L.; et al. Methylation of O6-methylguanine DNA methyltransferase and loss of heterozygosity on 19q and/or 17p are overlapping features of secondary glioblastomas with prolonged survival. Clin. Cancer Res. 2007, 13, 2606–2613. [Google Scholar] [CrossRef] [PubMed]
- Achyut, B.R. Impact of Microenvironment in Therapy-Induced Neovascularization of Glioblastoma. Biochem. Physiol. 2013, 2, e121. [Google Scholar] [CrossRef]
- Achyut, B.R.; Yang, L. Transforming growth factor-beta in the gastrointestinal and hepatic tumor microenvironment. Gastroenterology 2011, 141, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Lahmar, Q.; Keirsse, J.; Laoui, D.; Movahedi, K.; Van Overmeire, E.; Van Ginderachter, J.A. Tissue-resident versus monocyte-derived macrophages in the tumor microenvironment. Biochim. Biophys. Acta 2016, 1865, 23–34. [Google Scholar] [CrossRef] [PubMed]
- El Gazzar, M. microRNAs as potential regulators of myeloid-derived suppressor cell expansion. Innate Immun. 2014, 20, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Nakasone, E.S.; Askautrud, H.A.; Kees, T.; Park, J.H.; Plaks, V.; Ewald, A.J.; Fein, M.; Rasch, M.G.; Tan, Y.X.; Qiu, J.; et al. Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell 2012, 21, 488–503. [Google Scholar] [CrossRef] [PubMed]
- De Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.; Tonlaar, N.; Garwood, E.; Huo, D.; Moore, D.; Khramtsov, A.; Au, A.; Baehner, F.; Chen, Y.; Malaka, D.; et al. Proliferating macrophages associated with high grade, hormone receptor negative breast cancer and poor clinical outcome. Breast Cancer Res. Treat. 2011, 128, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Jinushi, M.; Baghdadi, M.; Chiba, S.; Yoshiyama, H. Regulation of cancer stem cell activities by tumor-associated macrophages. Am. J. Cancer Res. 2012, 2, 529–539. [Google Scholar] [PubMed]
- Shostak, K.; Chariot, A. NF-κB, stem cells and breast cancer: The links get stronger. Breast Cancer Res. BCR 2011, 13, 214. [Google Scholar] [CrossRef] [PubMed]
- Andrade, J.; Ge, S.; Symbatyan, G.; Rosol, M.S.; Olch, A.J.; Crooks, G.M. Effects of sublethal irradiation on patterns of engraftment after murine bone marrow transplantation. Biol. Blood Marrow Transplant. 2011, 17, 608–619. [Google Scholar] [CrossRef] [PubMed]
- Achyut, B.R.; Arbab, A.S. Myeloid Derived Suppressor Cells: Fuel the Fire. Biochem. Physiol. 2014, 3, e123. [Google Scholar] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, F.; Wu, X.; Qu, X.; Kowanetz, M.; Yu, L.; Tan, M.; Meng, Y.G.; Ferrara, N. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc. Natl. Acad. Sci. USA 2009, 106, 6742–6747. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, F.; Wu, X.; Malik, A.K.; Zhong, C.; Baldwin, M.E.; Schanz, S.; Fuh, G.; Gerber, H.P.; Ferrara, N. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat. Biotechnol. 2007, 25, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Piao, Y.; Liang, J.; Holmes, L.; Zurita, A.J.; Henry, V.; Heymach, J.V.; de Groot, J.F. Glioblastoma resistance to anti-VEGF therapy is associated with myeloid cell infiltration, stem cell accumulation, and a mesenchymal phenotype. Neuro Oncol. 2012, 14, 1379–1392. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.S.; Wu, X.; Zhuang, G.; Ngu, H.; Kasman, I.; Zhang, J.; Vernes, J.M.; Jiang, Z.; Meng, Y.G.; Peale, F.V.; et al. An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nat. Med. 2013, 19, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Sawanobori, Y.; Ueha, S.; Kurachi, M.; Shimaoka, T.; Talmadge, J.E.; Abe, J.; Shono, Y.; Kitabatake, M.; Kakimi, K.; Mukaida, N.; et al. Chemokine-mediated rapid turnover of myeloid-derived suppressor cells in tumor-bearing mice. Blood 2008, 111, 5457–5466. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.Y.; Nguyen, A.V.; Russell, R.G.; Pollard, J.W. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J. Exp. Med. 2001, 193, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Movahedi, K.; Guilliams, M.; Van den Bossche, J.; Van den Bergh, R.; Gysemans, C.; Beschin, A.; De Baetselier, P.; Van Ginderachter, J.A. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 2008, 111, 4233–4244. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Escamilla, J.; Mok, S.; David, J.; Priceman, S.; West, B.; Bollag, G.; McBride, W.; Wu, L. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013, 73, 2782–2794. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, K.P.; Rowe, V.; Bofinger, H.M.; Thomas, R.; Sasmono, T.; Hume, D.A.; Hill, G.R. The colony-stimulating factor 1 receptor is expressed on dendritic cells during differentiation and regulates their expansion. J. Immunol. 2005, 175, 1399–1405. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.A. Colony-stimulating factors in inflammation and autoimmunity. Nat. Rev. Immunol. 2008, 8, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Hume, D.A.; MacDonald, K.P. Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood 2012, 119, 1810–1820. [Google Scholar] [CrossRef] [PubMed]
- Priceman, S.J.; Sung, J.L.; Shaposhnik, Z.; Burton, J.B.; Torres-Collado, A.X.; Moughon, D.L.; Johnson, M.; Lusis, A.J.; Cohen, D.A.; Iruela-Arispe, M.L.; et al. Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: Combating tumor evasion of antiangiogenic therapy. Blood 2010, 115, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef] [PubMed]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Bowman, R.L.; Akkari, L.; Quick, M.L.; Schuhmacher, A.J.; Huse, J.T.; Holland, E.C.; Sutton, J.C.; Joyce, J.A. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 2016, 352, aad3018. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Molecular Pathways: Deciphering Mechanisms of Resistance to Macrophage-Targeted Therapies. Clin. Cancer Res. 2017, 23, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Couzin-Frankel, J. Breakthrough of the year 2013. Cancer immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef] [PubMed]
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer immunotherapy: The beginning of the end of cancer? BMC Med. 2016, 14, 73. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.G.; Gupta, S.; Goel, S. Immunotherapy: Incorporation in the evolving paradigm of renal cancer management and future prospects. Oncotarget 2017, 8, 17313–17327. [Google Scholar] [CrossRef] [PubMed]
- Thallinger, C.; Fureder, T.; Preusser, M.; Heller, G.; Mullauer, L.; Holler, C.; Prosch, H.; Frank, N.; Swierzewski, R.; Berger, W.; et al. Review of cancer treatment with immune checkpoint inhibitors: Current concepts, expectations, limitations and pitfalls. Wien. Klin. Wochenschr. 2017, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Restifo, N.P.; Smyth, M.J.; Snyder, A. Acquired resistance to immunotherapy and future challenges. Nat. Rev. Cancer 2016, 16, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Zhang, H.; Gu, L.; Ye, B.; Jian, Z.; Stary, C.; Xiong, X. Advances in Immunotherapy for Glioblastoma Multiforme. J. Immunol. Res. 2017, 2017, 3597613. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.M.; Sheng, J.; Scicli, G.M.; Arbab, A.S.; Lehman, N.L.; Edwards, P.A.; Falck, J.R.; Roman, R.J.; Scicli, A.G. Expression of CYP4A1 in U251 human glioma cell induces hyperproliferative phenotype in vitro and rapidly growing tumors in vivo. J. Pharmacol. Exp. Ther. 2008, 327, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Roman, R.J.; Fenstermacher, J.D.; Brown, S.L.; Falck, J.R.; Arbab, A.S.; Edwards, P.A.; Scicli, A.G. 9L gliosarcoma cell proliferation and tumor growth in rats are suppressed by N-hydroxy-N’-(4-butyl-2-methylphenol) formamidine (HET0016), a selective inhibitor of CYP4A. J. Pharmacol. Exp. Ther. 2006, 317, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Shankar, A.; Borin, T.F.; Iskander, A.; Varma, N.R.; Achyut, B.R.; Jain, M.; Mikkelsen, T.; Guo, A.M.; Chwang, W.B.; Ewing, J.R.; et al. Combination of vatalanib and a 20-HETE synthesis inhibitor results in decreased tumor growth in an animal model of human glioma. OncoTargets Ther. 2016, 9, 1205–1219. [Google Scholar]
- Borin, T.F.; Shankar, A.; Angara, K.; Rashid, M.H.; Jain, M.; Iskander, A.; Ara, R.; Lebedyeva, I.; Korkaya, H.; Achyut, B.R.; et al. HET0016 decreases lung metastasis from breast cancer in immune-competent mouse model. PLoS ONE 2017, 12, e0178830. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.M.; Janic, B.; Sheng, J.; Falck, J.R.; Roman, R.J.; Edwards, P.A.; Arbab, A.S.; Scicli, A.G. The cytochrome P450 4A/F-20-hydroxyeicosatetraenoic acid system: A regulator of endothelial precursor cells derived from human umbilical cord blood. J. Pharmacol. Exp. Ther. 2011, 338, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Gamage, N.H.; Alsulami, M.; Shankar, A.; Achyut, B.R.; Angara, K.; Rashid, M.H.; Iskander, A.; Borin, T.F.; Wenbo, Z.; et al. Intravenous Formulation of HET0016 Decreased Human Glioblastoma Growth and Implicated Survival Benefit in Rat Xenograft Models. Sci. Rep. 2017, 7, 41809. [Google Scholar] [CrossRef] [PubMed]
- Achyut, B.R.; Bader, D.A.; Robles, A.I.; Wangsa, D.; Harris, C.C.; Ried, T.; Yang, L. Inflammation-mediated genetic and epigenetic alterations drive cancer development in the neighboring epithelium upon stromal abrogation of TGF-beta signaling. PLoS Genet. 2013, 9, e1003251. [Google Scholar] [CrossRef] [PubMed]
- Dondossola, E.; Rangel, R.; Guzman-Rojas, L.; Barbu, E.M.; Hosoya, H.; St John, L.S.; Molldrem, J.J.; Corti, A.; Sidman, R.L.; Arap, W.; et al. CD13-positive bone marrow-derived myeloid cells promote angiogenesis, tumor growth, and metastasis. Proc. Natl. Acad. Sci. USA 2013, 110, 20717–20722. [Google Scholar] [CrossRef] [PubMed]
- Seton-Rogers, S. Tumour microenvironment: Means of resistance. Nat. Rev. Cancer 2013, 13, 607. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, Y.; Chen, H.; Zhang, J.; Zhang, J.; Qin, T.; Duan, C.; Chen, X.; Liu, Y.; Zhou, X.; et al. Inhibition of CYP4A by a novel flavonoid FLA-16 prolongs survival and normalizes tumor vasculature in glioma. Cancer Lett. 2017, 402, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.W.; Yu, T.J.; Zhang, J.; Li, Y.; Chen, H.L.; Yang, G.F.; Yu, W.; Liu, Y.Z.; Liu, X.X.; Duan, C.F.; et al. CYP4A in tumor-associated macrophages promotes pre-metastatic niche formation and metastasis. Oncogene 2017, 36, 5045–5057. [Google Scholar] [CrossRef] [PubMed]
- Borin, T.F.A.K.; Rashid, M.H.; Achyut, B.R.; Arbab, A.S. Arachidonic Acid Metabolite as a Novel Therapeutic Target in Breast Cancer Metastasis. Int. J. Mol. Sci. 2017, 18, 2661. [Google Scholar] [CrossRef]
- Acharyya, S.; Oskarsson, T.; Vanharanta, S.; Malladi, S.; Kim, J.; Morris, P.G.; Manova-Todorova, K.; Leversha, M.; Hogg, N.; Seshan, V.E.; et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 2012, 150, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Fakhrejahani, E.; Toi, M. Antiangiogenesis therapy for breast cancer: An update and perspectives from clinical trials. Jpn. J. Clin. Oncol. 2014, 44, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Arbab, A.S. Activation of alternative pathways of angiogenesis and involvement of stem cells following anti-angiogenesis treatment in glioma. Histol. Histopathol. 2012, 27, 549–557. [Google Scholar] [PubMed]
- Polivka, J., Jr.; Polivka, J.; Holubec, L.; Kubikova, T.; Priban, V.; Hes, O.; Pivovarcikova, K.; Treskova, I. Advances in Experimental Targeted Therapy and Immunotherapy for Patients with Glioblastoma Multiforme. Anticancer Res. 2017, 37, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Lenting, K.; Verhaak, R.; Ter Laan, M.; Wesseling, P.; Leenders, W. Glioma: Experimental models and reality. Acta Neuropathol. 2017, 133, 263–282. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, Y.; Yang, J.; Hagan, J.P.; Li, M. Vertebrate animal models of glioma: Understanding the mechanisms and developing new therapies. Biochim. Biophys. Acta 2013, 1836, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Ha, G.; Tseng, Y.Y.; Greenwald, N.F.; Oh, C.; Shih, J.; McFarland, J.M.; Wong, B.; Boehm, J.S.; Beroukhim, R.; et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat. Genet. 2017, 49, 1567–1575. [Google Scholar] [CrossRef] [PubMed]


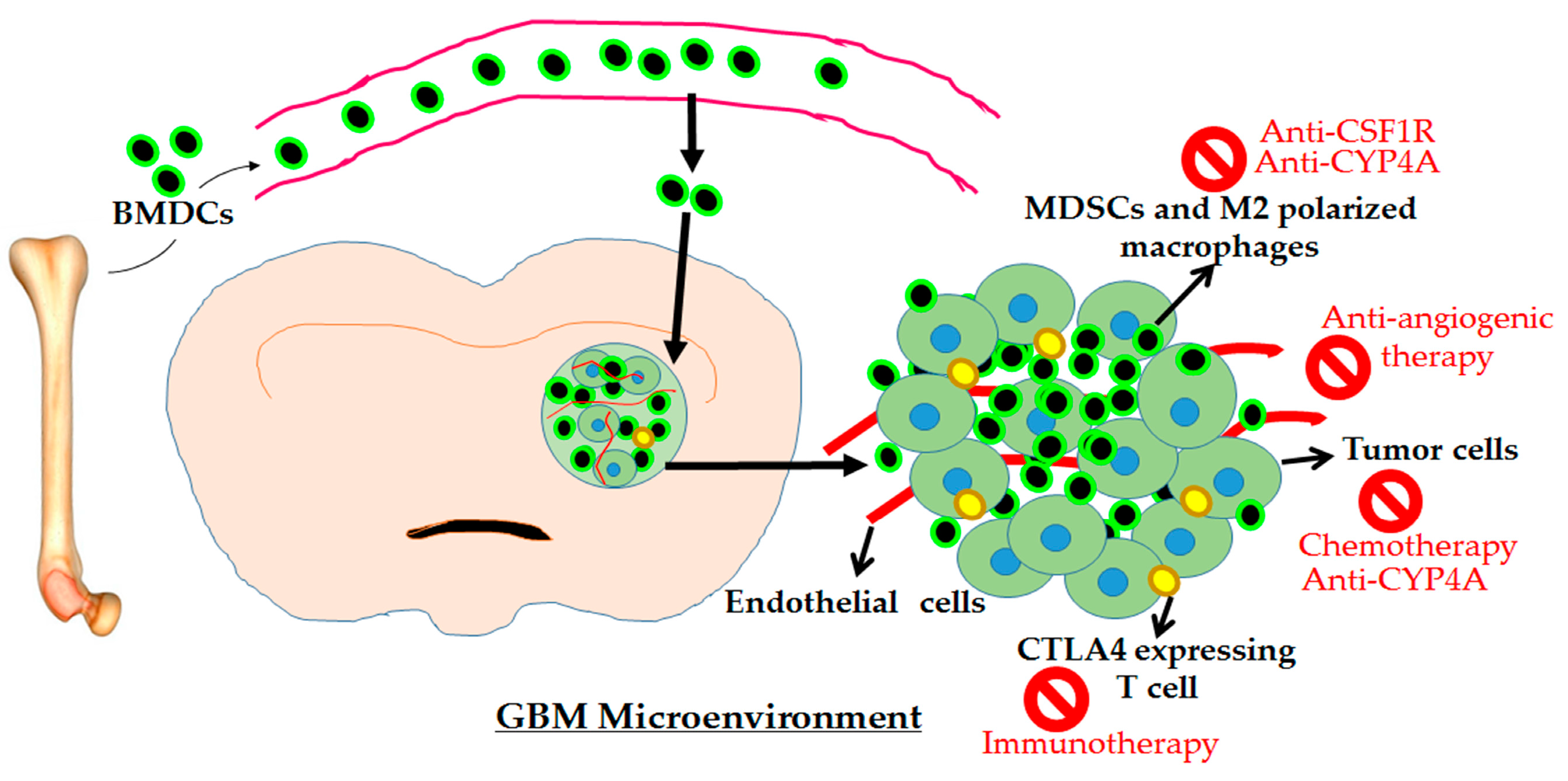{kind=link}
| Key Protein | Full Name | Category | Key Function(s) |
|---|---|---|---|
| ANGPT2 | Angiopoietin-2 | Growth factor | Tumor neovascularization, metastasis, and inflammation |
| COL1A1 | Collagen, type 1, alpha (α) 1 | Structural protein, part of connective tissue | Tumor neovascularization |
| CD31/PECAM-1 | Platelet endothelial cell adhesion molecule | Endothelial cell marker | Leukocyte transmigration, neovascularization, and integrin activation |
| CD34 | Hematopoietic progenitor cell antigen | Hematopoietic stem cell marker | Attachment of stem cells to bone marrow ECM, stromal cells, facilitates cell migration |
| CD45 | Protein tyrosine phosphatase, receptor type, C (also known as Common leukocyte antigen) | Pan-leukocyte marker | Signal transduction in hematopoiesis |
| CD133 | Prominin-1 | Stem-cell marker | Cancer stem cells with CD133 undergo self-renewal and differentiation |
| CD202b | Angiopoietin-1 receptor | Endothelial-cell marker | Promotes neovascularization |
| CSF | Colony-stimulating factor 1/Macrophage colony-stimulating factor | Cytokine | proliferation, differentiation, and survival of monocytes, macrophages, and bone marrow progenitor cells |
| CSF-1R | Colony-stimulating factor receptor-1 | Cytokine receptor | Cytokine receptor that facilitates the actions of CSF-1 |
| CYP4A and CYP4F | Cytochromes P450 family of enzymes | Enzymes involved in arachidonic acid metabolism | Production of 20-HETE, an eicosanoid metabolite that promotes neovascularization, migration, inflammation, and metastasis |
| CXCL7 | Chemokine (C-X-C motif) ligand 7 | Cytokine | mitogenesis, synthesis of extracellular matrix, glucose metabolism and synthesis of plasminogen activator, recruitment of CXCR2+ myeloid cells |
| CXCL8 (IL-8) | Chemokine (C-X-C motif) ligand 8 (Interleukin-8) | Chemokine | Neutrophil chemotactic factor, chemotaxis of other granulocytic cells and CXCR2+ myeloid cells, potent pro-neovasculogenic chemokine |
| EGF | Epithelial Growth Fator | Growth factor | Cellular proliferation, differentiation, and survival |
| Eph A1 and A2 | Ephrin A1 and A2 | Receptor tyrosine kinase | Embryonic development, post-natal angiogenesis, stem cell differentiation and migration |
| FGF | Fibroblast growth factor | Growth factor | Angiogenesis, wound healing, embryonic development, and various endocrine signaling pathways, proliferation, and differentiation of various cell types |
| G-CSF | Granulocyte-colony stimulating factor | Cytokine | Survival, proliferation, differentiation, and function of neutrophil precursors and mature neutrophils |
| HGF | Hepatocyte growth factor | Growth, motility and morphogenic factor | Embryonic organ development, specifically in myogenesis, in adult organ regeneration, and in wound healing, mediates pro-tumorigenic roles in growing tumors |
| HIF-1α | Hypoxia-inducible factor 1 α | Transcription factor | Released in response to hypoxia, neovascularization, energy metabolism, cell survival, and tumor invasion |
| IGF | Insulin-like growth factor | Growth factor | Promotes growth and survival of tumor cells |
| MCP-1/CCL2 | Monocyte chemoattractant protein 1 | Chemokine | Recruitment of several inflammatory monocytes, memory T cells, and dendritic cells to the tumor |
| MGMT | O-6-methylguanine-DNA methyltransferase | Enzyme/protein | DNA Repair promotes resistance of tumor cells to chemotherapy (esp. Temozolomide (TMZ)) |
| MMP-2 and 9 | Matrix Metalloproteinases-2 and 9 | Proteinase enzymes | Degradation of extra-cellular matrix (ECM) proteins, promotes angiogenesis by ECM remodeling |
| NG2 | Neuron-glial antigen 2/Chondroitin sulfate proteoglycan 4 | Chondroitin sulfate proteoglycan | Tumor cell metastasis and invasion |
| PDGF | Platelet-derived growth factor | Growth factor | Pro-angiogenic molecule |
| PLGF | Placental growth factor | Growth factor | Pro-angiogenic molecule |
| SDF-1α | Stromal-derived factor 1 α | Chemokine | Chemotactic protein to facilitate recruitment of bone marrow-derived cells and endothelial progenitor cells |
| VEGF | Vascular endothelial growth factor | Growth factor | Promotes neovascularisation by facilitating survival and development of endothelial cells and proliferation of endothelial progenitor cells |
| VEGFR | Vascular endothelial growth factor receptor | Receptor tyrosine kinase | Receptor for VEGF to promote neovascularization |
| TN-C | Tenascin C | Glycoprotein | Tumor cell proliferation and migration |
| 20-HETE | 20-Hydroxyeicosatetraenoic acid | Eicosanoid metabolite of Arachidonic acid | Neovascularization, tumor cell growth, proliferation, migration, and recruitment of angiogenic myeloid cells to tumor microenvironment |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arbab, A.S.; Rashid, M.H.; Angara, K.; Borin, T.F.; Lin, P.-C.; Jain, M.; Achyut, B.R. Major Challenges and Potential Microenvironment-Targeted Therapies in Glioblastoma. Int. J. Mol. Sci. 2017, 18, 2732. https://doi.org/10.3390/ijms18122732
Arbab AS, Rashid MH, Angara K, Borin TF, Lin P-C, Jain M, Achyut BR. Major Challenges and Potential Microenvironment-Targeted Therapies in Glioblastoma. International Journal of Molecular Sciences. 2017; 18(12):2732. https://doi.org/10.3390/ijms18122732
Chicago/Turabian StyleArbab, Ali S., Mohammad H. Rashid, Kartik Angara, Thaiz F. Borin, Ping-Chang Lin, Meenu Jain, and Bhagelu R. Achyut. 2017. "Major Challenges and Potential Microenvironment-Targeted Therapies in Glioblastoma" International Journal of Molecular Sciences 18, no. 12: 2732. https://doi.org/10.3390/ijms18122732
APA StyleArbab, A. S., Rashid, M. H., Angara, K., Borin, T. F., Lin, P. -C., Jain, M., & Achyut, B. R. (2017). Major Challenges and Potential Microenvironment-Targeted Therapies in Glioblastoma. International Journal of Molecular Sciences, 18(12), 2732. https://doi.org/10.3390/ijms18122732