Functional Analysis of Human Hub Proteins and Their Interactors Involved in the Intrinsic Disorder-Enriched Interactions





Abstract
:1. Introduction
2. Results and Discussion
2.1. Intrinsic Disorder in Hub Protein-Protein Interactions
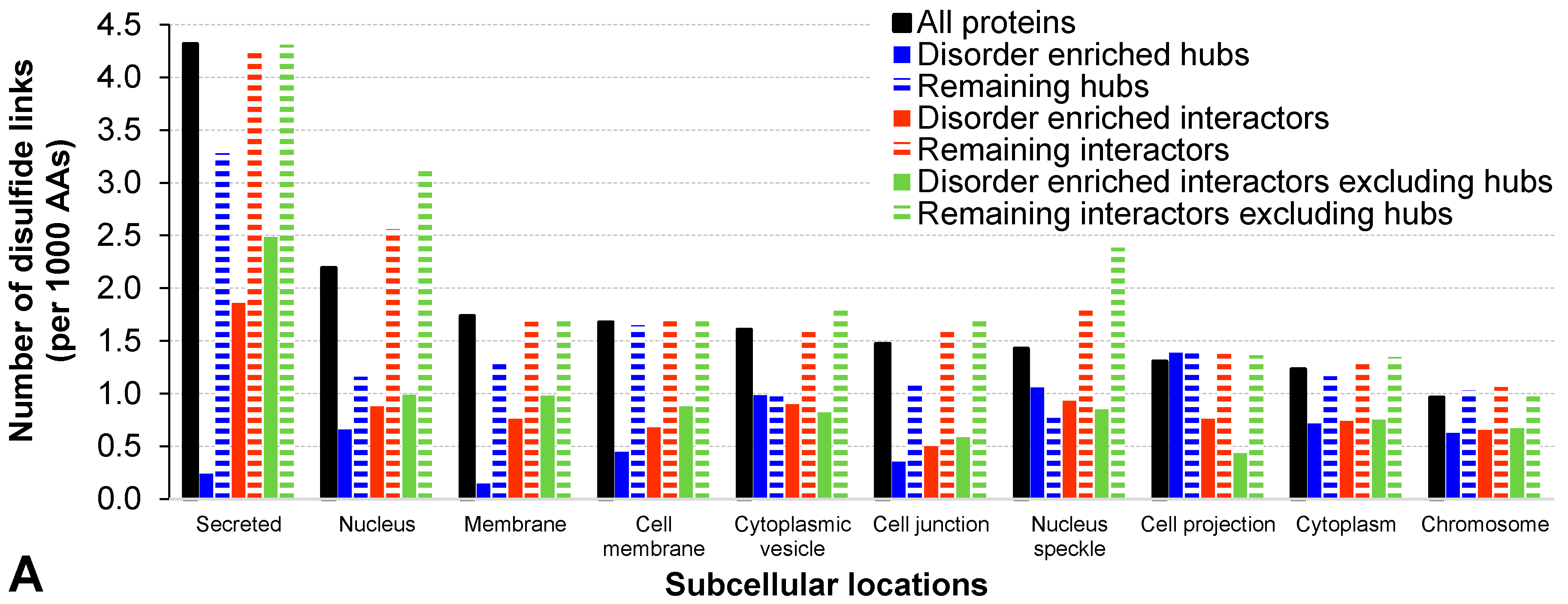
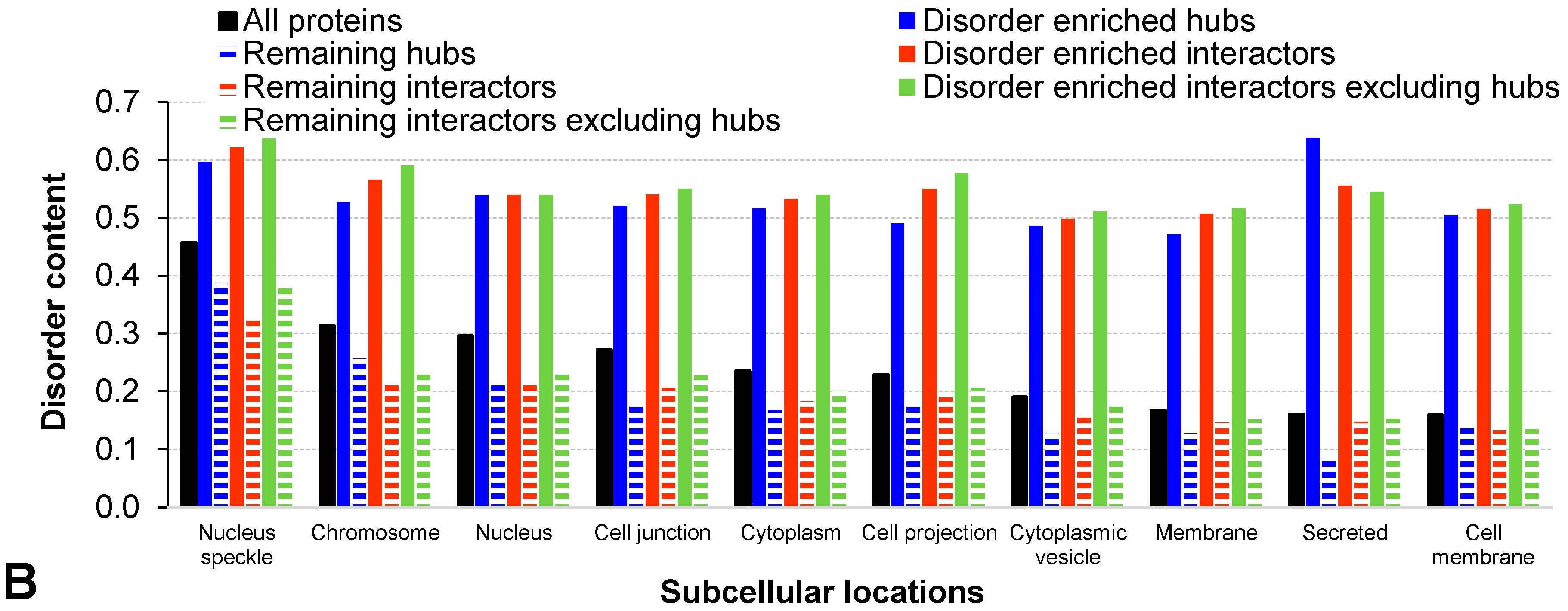
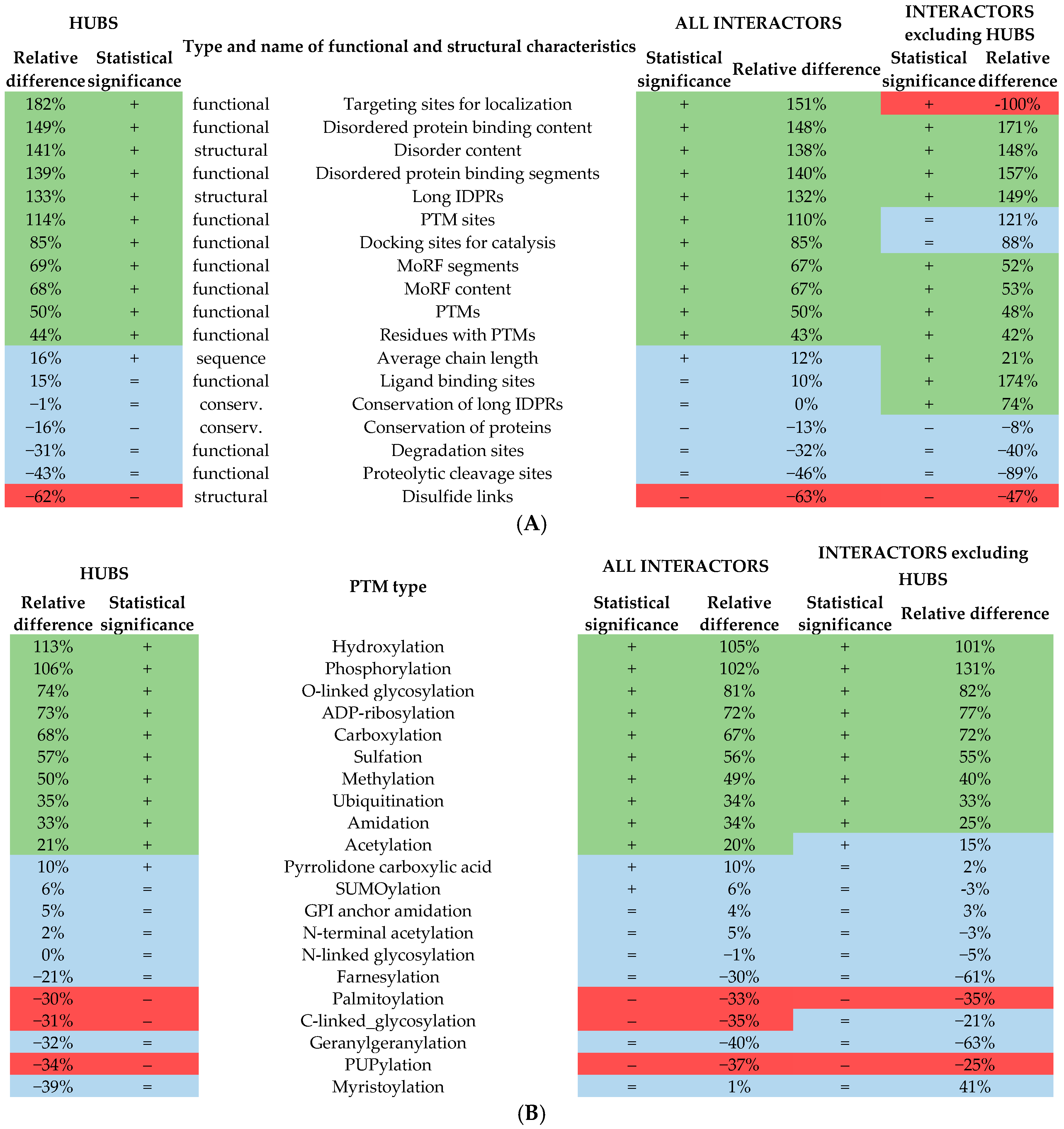
2.2. Functional and Structural Characteristics of Proteins Involved in the Disorder-Enriched Hub Protein-Protein Interactions
2.3. Proteins Involved in the Disorder-Enriched Hub Protein-Protein Interactions and Human Diseases
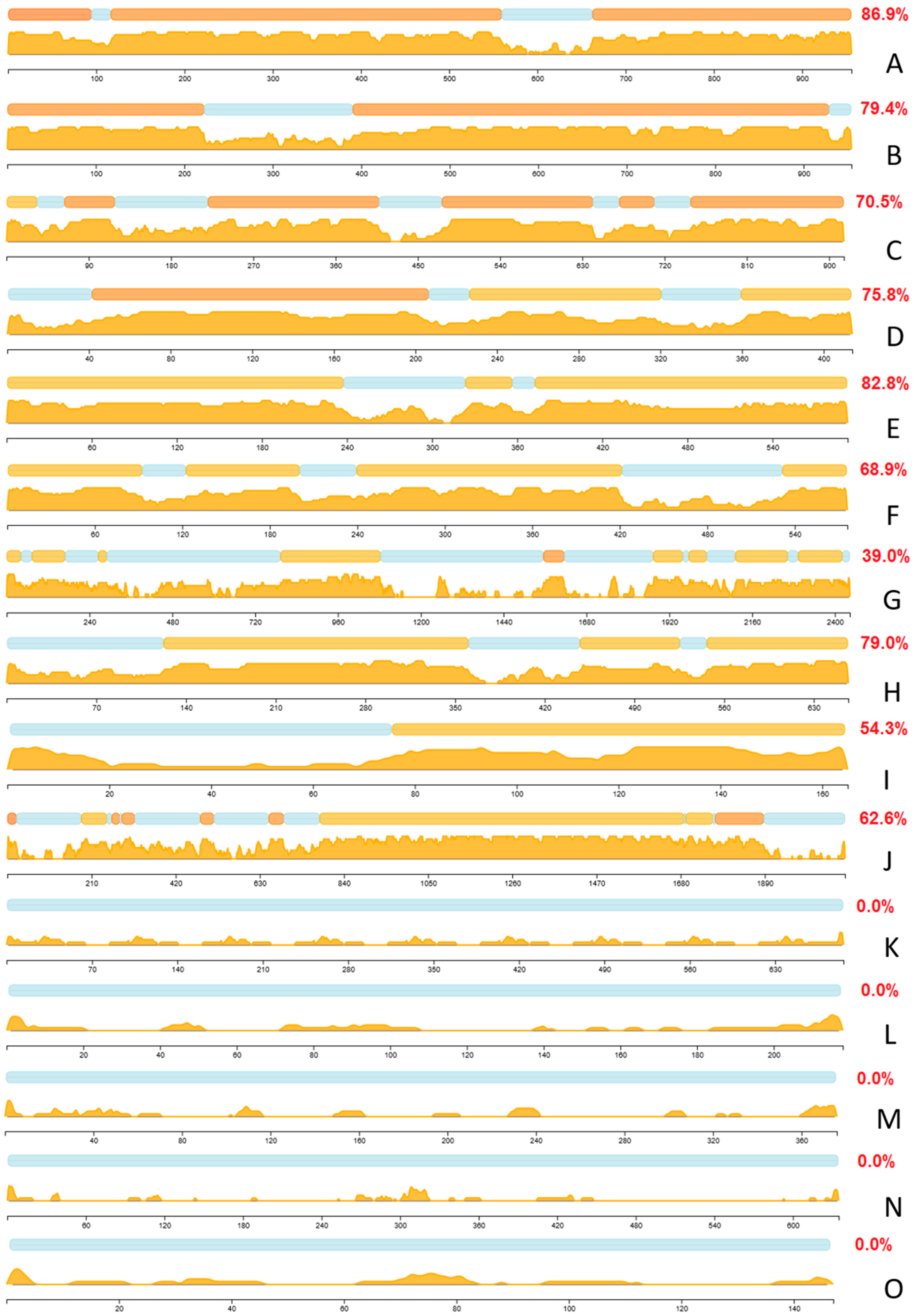
2.4. Several Illustrative Examples of Proteins Involved in the Disorder-Enriched Hub Protein-Protein Interactions
2.4.1. The Most Disordered Hubs
2.4.2. Highly Connected Hubs Enriched in Disorder
2.4.3. Highly Connected Hubs Depleted in Disorder
3. Materials and Methods
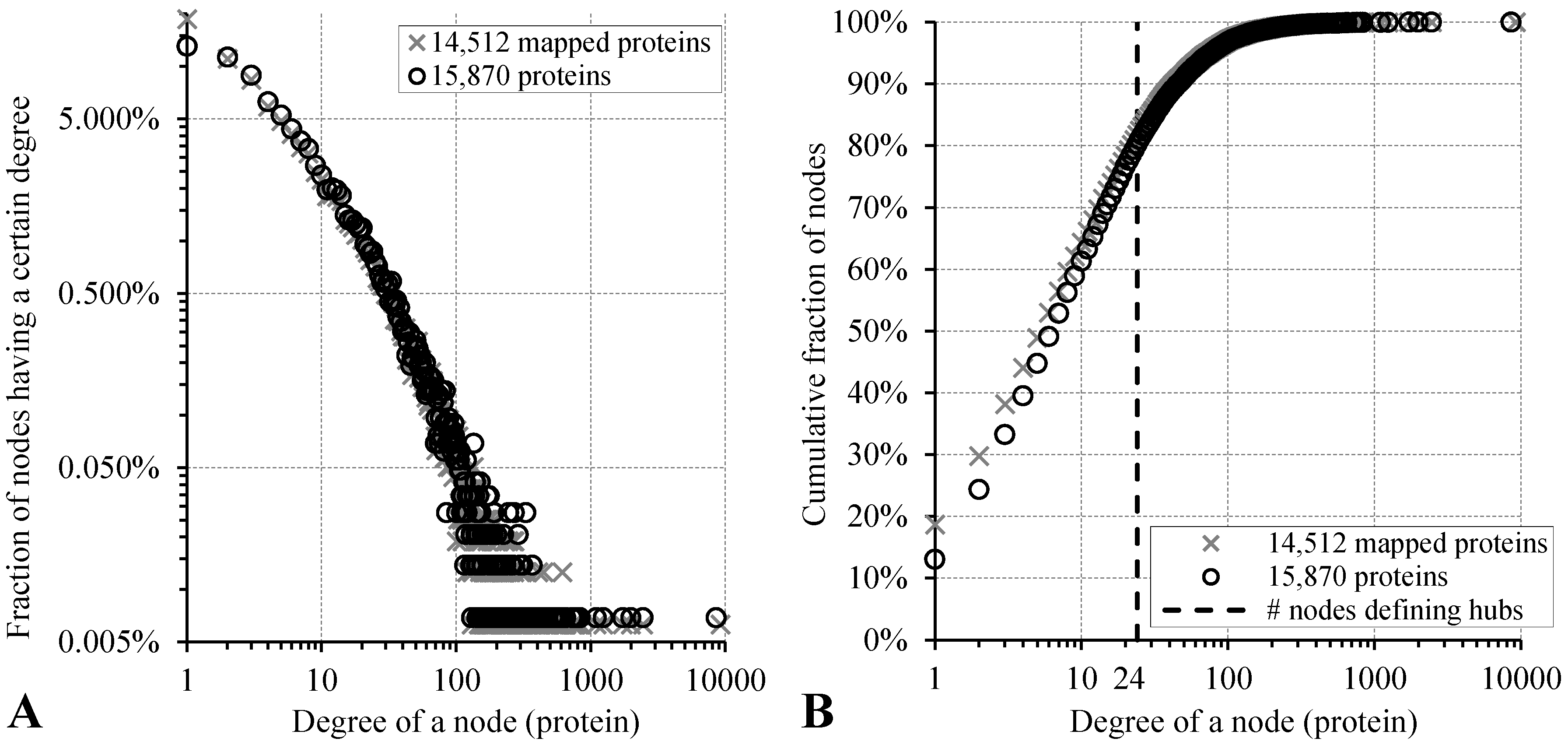
3.1. Dataset and Annotation of Hubs
3.2. Functional and Structural Characterization of Proteins
3.3. Annotation of Disease-Linked Proteins
3.4. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- De Las Rivas, J.; Fontanillo, C. Protein-protein interactions essentials: Key concepts to building and analyzing interactome networks. PLoS Comput. Biol. 2010, 6, e1000807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cusick, M.E.; Klitgord, N.; Vidal, M.; Hill, D.E. Interactome: Gateway into systems biology. Hum. Mol. Genet. 2005, 14, R171–R181. [Google Scholar] [CrossRef] [PubMed]
- Erdös, P.; Rényi, A. On the evolution of random graphs. Publ. Math. Inst. Hung. Acad. Sci. 1960, 5, 17–61. [Google Scholar]
- Barabasi, A.L.; Bonabeau, E. Scale-free networks. Sci. Am. 2003, 288, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Goh, K.I.; Oh, E.; Jeong, H.; Kahng, B.; Kim, D. Classification of scale-free networks. Proc. Natl. Acad. Sci. USA 2002, 99, 12583–12588. [Google Scholar] [CrossRef] [PubMed]
- Barabasi, A.L.; Albert, R. Emergence of scaling in random networks. Science 1999, 286, 509–512. [Google Scholar] [PubMed]
- Watts, D.J.; Strogatz, S.H. Collective dynamics of ′small-world’ networks. Nature 1998, 393, 440–442. [Google Scholar] [CrossRef] [PubMed]
- Wuchty, S. Scale-free behavior in protein domain networks. Mol. Biol. Evol. 2001, 18, 1694–1702. [Google Scholar] [CrossRef] [PubMed]
- Han, J.D.; Bertin, N.; Hao, T.; Goldberg, D.S.; Berriz, G.F.; Zhang, L.V.; Dupuy, D.; Walhout, A.J.; Cusick, M.E.; Roth, F.P.; et al. Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature 2004, 430, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.; Oldfield, C.J.; Ji, F.; Klitgord, N.; Cusick, M.E.; Radivojac, P.; Uversky, V.N.; Vidal, M.; Iakoucheva, L.M. Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput. Biol. 2006, 2, 890–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batada, N.N.; Reguly, T.; Breitkreutz, A.; Boucher, L.; Breitkreutz, B.J.; Hurst, L.D.; Tyers, M. Stratus not altocumulus: A new view of the yeast protein interaction network. PLoS Biol. 2006, 4, e317. [Google Scholar] [CrossRef] [PubMed]
- Aragues, R.; Sali, A.; Bonet, J.; Marti-Renom, M.A.; Oliva, B. Characterization of protein hubs by inferring interacting motifs from protein interactions. PLoS Comput. Biol. 2007, 3, 1761–1771. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Zhang, S.; Zhang, X.S.; Chen, L. Hubs with network motifs organize modularity dynamically in the protein-protein interaction network of yeast. PLoS ONE 2007, 2, e1207. [Google Scholar] [CrossRef] [PubMed]
- Hartwell, L.H.; Hopfield, J.J.; Leibler, S.; Murray, A.W. From molecular to modular cell biology. Nature 1999, 402, C47–C52. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Garner, E.; Guilliot, S.; Romero, P.; Albrecht, K.; Hart, J.; Obradovic, Z.; Kissinger, C.; Villafranca, J.E. Protein disorder and the evolution of molecular recognition: Theory, predictions and observations. Pac. Symp. Biocomput. 1998, 3, 473–484. [Google Scholar]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Daughdrill, G.W.; Pielak, G.J.; Uversky, V.N.; Cortese, M.S.; Dunker, A.K. Natively disordered proteins. In Handbook of Protein Folding; Buchner, J., Kiefhaber, T., Eds.; Wiley-VCH: Weinheim, Germany; Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005; pp. 271–353. [Google Scholar]
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim. Biophys. Acta 2010, 1804, 1231–1264. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Obradovic, Z.; Romero, P.; Garner, E.C.; Brown, C.J. Intrinsic protein disorder in complete genomes. Genome Inform. 2000, 11, 161–171. [Google Scholar]
- Uversky, V.N. The mysterious unfoldome: Structureless, underappreciated, yet vital part of any given proteome. J. Biomed. Biotechnol. 2010, 2010, 568068. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Yan, J.; Fan, X.; Mizianty, M.J.; Xue, B.; Wang, K.; Hu, G.; Uversky, V.N.; Kurgan, L. Exceptionally abundant exceptions: Comprehensive characterization of intrinsic disorder in all domains of life. Cell. Mol. Life Sci. 2015, 72, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Mizianty, M.J.; Kurgan, L. Genome-scale prediction of proteins with long intrinsically disordered regions. Proteins 2014, 82, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Radivojac, P.; Iakoucheva, L.M.; Oldfield, C.J.; Obradovic, Z.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder and functional proteomics. Biophys. J. 2007, 92, 1439–1456. [Google Scholar] [CrossRef] [PubMed]
- Galea, C.A.; Wang, Y.; Sivakolundu, S.G.; Kriwacki, R.W. Regulation of cell division by intrinsically unstructured proteins: Intrinsic flexibility, modularity, and signaling conduits. Biochemistry 2008, 47, 7598–7609. [Google Scholar] [CrossRef] [PubMed]
- Buljan, M.; Chalancon, G.; Dunker, A.K.; Bateman, A.; Balaji, S.; Fuxreiter, M.; Babu, M.M. Alternative splicing of intrinsically disordered regions and rewiring of protein interactions. Curr. Opin. Struct. Biol. 2013, 23, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsic disorder in proteins associated with neurodegenerative diseases. Front. Biosci. (Landmark Ed.) 2009, 14, 5188–5238. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Oldfield, C.J.; Midic, U.; Xie, H.; Xue, B.; Vucetic, S.; Iakoucheva, L.M.; Obradovic, Z.; Dunker, A.K. Unfoldomics of human diseases: Linking protein intrinsic disorder with diseases. BMC Genom. 2009, 10 (Suppl. 1), S7. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Targeting intrinsically disordered proteins in neurodegenerative and protein dysfunction diseases: Another illustration of the d(2) concept. Expert Rev. Proteom. 2010, 7, 543–564. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically disordered proteins and novel strategies for drug discovery. Expert Opin. Drug Discov. 2012, 7, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Wrecked regulation of intrinsically disordered proteins in diseases: Pathogenicity of deregulated regulators. Front. Mol. Biosci. 2014, 1, 6. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: Introducing the d2 concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Dave, V.; Iakoucheva, L.M.; Malaney, P.; Metallo, S.J.; Pathak, R.R.; Joerger, A.C. Pathological unfoldomics of uncontrolled chaos: Intrinsically disordered proteins and human diseases. Chem. Rev. 2014, 114, 6844–6879. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Showing your id: Intrinsic disorder as an id for recognition, regulation and cell signaling. J. Mol. Recognit. 2005, 18, 343–384. [Google Scholar] [CrossRef] [PubMed]
- Hasty, J.; Collins, J.J. Protein interactions. Unspinning the web. Nature 2001, 411, 30–31. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E. Einfluss der configuration auf die wirkung derenzyme. Ber. Dtsch. Chem. Ges. 1894, 27, 2985–2993. [Google Scholar] [CrossRef]
- Koshland, D.E., Jr.; Ray, W.J., Jr.; Erwin, M.J. Protein structure and enzyme action. Fed. Proc. 1958, 17, 1145–1150. [Google Scholar] [PubMed]
- Landsteiner, K. The Specificity of Serological Reactions; Courier Dover Publications: Mineola, NY, USA, 1936. [Google Scholar]
- Pauling, L. A theory of the structure and process of formation of antibodies. J. Am. Chem. Soc. 1940, 62, 2643–2657. [Google Scholar] [CrossRef]
- Karush, F. Heterogeneity of the binding sites of bovine serum albumin. J. Am. Chem. Soc. 1950, 72, 2705–2713. [Google Scholar] [CrossRef]
- Meador, W.E.; Means, A.R.; Quiocho, F.A. Modulation of calmodulin plasticity in molecular recognition on the basis of X-ray structures. Science 1993, 262, 1718–1721. [Google Scholar] [CrossRef] [PubMed]
- Kriwacki, R.W.; Hengst, L.; Tennant, L.; Reed, S.I.; Wright, P.E. Structural studies of p21Waf1/Cip1/Sdi1 in the free and cdk2-bound state: Conformational disorder mediates binding diversity. Proc. Natl. Acad. Sci. USA 1996, 93, 11504–11509. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. A protein-chameleon: Conformational plasticity of alpha-synuclein, a disordered protein involved in neurodegenerative disorders. J. Biomol. Struct. Dyn. 2003, 21, 211–234. [Google Scholar] [CrossRef] [PubMed]
- Patil, A.; Nakamura, H. Disordered domains and high surface charge confer hubs with the ability to interact with multiple proteins in interaction networks. FEBS Lett. 2006, 580, 2041–2045. [Google Scholar] [CrossRef] [PubMed]
- Dosztanyi, Z.; Chen, J.; Dunker, A.K.; Simon, I.; Tompa, P. Disorder and sequence repeats in hub proteins and their implications for network evolution. J. Proteome Res. 2006, 5, 2985–2995. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.P.; Ganapathi, M.; Sandhu, K.S.; Dash, D. Intrinsic unstructuredness and abundance of pest motifs in eukaryotic proteomes. Proteins 2006, 62, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Na, I.; Kurgan, L.; Uversky, V.N. Compartmentalization and functionality of nuclear disorder: Intrinsic disorder and protein-protein interactions in intra-nuclear compartments. Int. J. Mol. Sci. 2016, 17, 24. [Google Scholar] [CrossRef] [PubMed]
- Marinissen, M.J.; Gutkind, J.S. Scaffold proteins dictate rho gtpase-signaling specificity. Trends Biochem. Sci. 2005, 30, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, A.B.; Aspenstrom, P.; Hall, A. Human cnk1 acts as a scaffold protein, linking rho and ras signal transduction pathways. Mol. Cell. Biol. 2004, 24, 1736–1746. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, A.B.; Hall, A. Rho gtpases: Biochemistry and biology. Annu. Rev. Cell Dev. Biol. 2005, 21, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Hohenstein, P.; Giles, R.H. Brca1: A scaffold for p53 response? Trends Genet. 2003, 19, 489–494. [Google Scholar] [CrossRef]
- Rui, Y.; Xu, Z.; Lin, S.; Li, Q.; Rui, H.; Luo, W.; Zhou, H.M.; Cheung, P.Y.; Wu, Z.; Ye, Z.; et al. Axin stimulates p53 functions by activation of hipk2 kinase through multimeric complex formation. EMBO J. 2004, 23, 4583–4594. [Google Scholar] [CrossRef] [PubMed]
- Salahshor, S.; Woodgett, J.R. The links between axin and carcinogenesis. J. Clin. Pathol. 2005, 58, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.; Scott, J.D. Akap signalling complexes: Focal points in space and time. Nat. Rev. Mol. Cell Biol. 2004, 5, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Carpousis, A.J. The RNA degradosome of Escherichia coli: A multiprotein mRNA-degrading machine assembled on RNase E. Annu. Rev. Microbiol. 2007, 61, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Ekman, D.; Light, S.; Bjorklund, A.K.; Elofsson, A. What properties characterize the hub proteins of the protein-protein interaction network of saccharomyces cerevisiae? Genome Biol. 2006, 7, R45. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, C.J.; Meng, J.; Yang, J.Y.; Yang, M.Q.; Uversky, V.N.; Dunker, A.K. Flexible nets: Disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genom. 2008, 9 (Suppl. 1), S1. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. Flexible nets: The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005, 272, 5129–5148. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Lin, S.C. Axin: A master scaffold for multiple signaling pathways. Neurosignals 2004, 13, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Calderone, A.; Castagnoli, L.; Cesareni, G. Mentha: A resource for browsing integrated protein-interaction networks. Nat. Methods 2013, 10, 690–691. [Google Scholar] [CrossRef] [PubMed]
- Ota, M.; Gonja, H.; Koike, R.; Fukuchi, S. Multiple-localization and hub proteins. PLoS ONE 2016, 11, e0156455. [Google Scholar] [CrossRef] [PubMed]
- Bosnjak, I.; Bojovic, V.; Segvic-Bubic, T.; Bielen, A. Occurrence of protein disulfide bonds in different domains of life: A comparison of proteins from the protein data bank. Protein Eng. Des. Sel. 2014, 27, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Uversky, V.N.; Kurgan, L. Disordered nucleiome: Abundance of intrinsic disorder in the DNA- and RNA-binding proteins in 1121 species from Eukaryota, Bacteria and Archaea. Proteomics 2016, 16, 1486–1498. [Google Scholar] [CrossRef] [PubMed]
- Nguyen Ba, A.N.; Yeh, B.J.; van Dyk, D.; Davidson, A.R.; Andrews, B.J.; Weiss, E.L.; Moses, A.M. Proteome-wide discovery of evolutionary conserved sequences in disordered regions. Sci. Signal. 2012, 5, rs1. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T.; Garneau-Tsodikova, S.; Gatto, G.J. Protein posttranslational modifications: The chemistry of proteome diversifications. Angew. Chem. Int. Ed. Engl. 2005, 44, 7342–7372. [Google Scholar] [CrossRef] [PubMed]
- Deribe, Y.L.; Pawson, T.; Dikic, I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 2010, 17, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Jensen, O.N. Proteomic analysis of post-translational modifications. Nat. Biotechnol. 2003, 21, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Witze, E.S.; Old, W.M.; Resing, K.A.; Ahn, N.G. Mapping protein post-translational modifications with mass spectrometry. Nat. Methods 2007, 4, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Vucetic, S.; Iakoucheva, L.M.; Oldfield, C.J.; Dunker, A.K.; Obradovic, Z.; Uversky, V.N. Functional anthology of intrinsic disorder. 3. Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins. J. Proteome Res. 2007, 6, 1917–1932. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Brown, C.J.; Obradovic, Z. Identification and functions of usefully disordered proteins. Adv. Protein Chem. 2002, 62, 25–49. [Google Scholar] [PubMed]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradovic, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef] [PubMed]
- Iakoucheva, L.M.; Radivojac, P.; Brown, C.J.; O’Connor, T.R.; Sikes, J.G.; Obradovic, Z.; Dunker, A.K. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004, 32, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Radivojac, P.; Vacic, V.; Haynes, C.; Cocklin, R.R.; Mohan, A.; Heyen, J.W.; Goebl, M.G.; Iakoucheva, L.M. Identification, analysis, and prediction of protein ubiquitination sites. Proteins 2010, 78, 365–380. [Google Scholar] [CrossRef] [PubMed]
- Pejaver, V.; Hsu, W.L.; Xin, F.; Dunker, A.K.; Uversky, V.N.; Radivojac, P. The structural and functional signatures of proteins that undergo multiple events of post-translational modification. Protein Sci. 2014, 23, 1077–1093. [Google Scholar] [CrossRef] [PubMed]
- Midic, U.; Oldfield, C.J.; Dunker, A.K.; Obradovic, Z.; Uversky, V.N. Protein disorder in the human diseasome: Unfoldomics of human genetic diseases. BMC Genom. 2009, 10 (Suppl. 1), S12. [Google Scholar] [CrossRef] [PubMed]
- Midic, U.; Oldfield, C.J.; Dunker, A.K.; Obradovic, Z.; Uversky, V.N. Unfoldomics of human genetic diseases: Illustrative examples of ordered and intrinsically disordered members of the human diseasome. Protein Pept. Lett. 2009, 16, 1533–1547. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Vucetic, S.; Iakoucheva, L.M.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N.; Obradovic, Z. Functional anthology of intrinsic disorder. 1. Biological processes and functions of proteins with long disordered regions. J. Proteome Res. 2007, 6, 1882–1898. [Google Scholar] [CrossRef] [PubMed]
- Vucetic, S.; Xie, H.; Iakoucheva, L.M.; Oldfield, C.J.; Dunker, A.K.; Obradovic, Z.; Uversky, V.N. Functional anthology of intrinsic disorder. 2. Cellular components, domains, technical terms, developmental processes, and coding sequence diversities correlated with long disordered regions. J. Proteome Res. 2007, 6, 1899–1916. [Google Scholar] [CrossRef] [PubMed]
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.D.; Obradovic, Z.; Dunker, A.K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002, 323, 573–584. [Google Scholar] [CrossRef]
- Baker, J.M.; Hudson, R.P.; Kanelis, V.; Choy, W.Y.; Thibodeau, P.H.; Thomas, P.J.; Forman-Kay, J.D. Cftr regulatory region interacts with nbd1 predominantly via multiple transient helices. Nat. Struct. Mol. Biol. 2007, 14, 738–745. [Google Scholar] [CrossRef] [PubMed]
- Wells, M.; Tidow, H.; Rutherford, T.J.; Markwick, P.; Jensen, M.R.; Mylonas, E.; Svergun, D.I.; Blackledge, M.; Fersht, A.R. Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proc. Natl. Acad. Sci. USA 2008, 105, 5762–5767. [Google Scholar] [CrossRef] [PubMed]
- Mark, W.Y.; Liao, J.C.; Lu, Y.; Ayed, A.; Laister, R.; Szymczyna, B.; Chakrabartty, A.; Arrowsmith, C.H. Characterization of segments from the central region of brca1: An intrinsically disordered scaffold for multiple protein-protein and protein-DNA interactions? J. Mol. Biol. 2005, 345, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.P.; Potikyan, G.; Savene, R.O.; Denny, C.T.; Uversky, V.N.; Lee, K.A. Multiple aromatic side chains within a disordered structure are critical for transcription and transforming activity of EWS family oncoproteins. Proc. Natl. Acad. Sci. USA 2007, 104, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Roman, A.; Oldfield, C.J.; Dunker, A.K. Protein intrinsic disorder and human papillomaviruses: Increased amount of disorder in E6 and E7 oncoproteins from high risk HPVs. J. Proteome Res. 2006, 5, 1829–1842. [Google Scholar] [CrossRef] [PubMed]
- Malaney, P.; Pathak, R.R.; Xue, B.; Uversky, V.N.; Dave, V. Intrinsic disorder in pten and its interactome confers structural plasticity and functional versatility. Sci. Rep. 2013, 3, 2035. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, K.; Mooney, S.M.; Parekh, N.; Getzenberg, R.H.; Kulkarni, P. A majority of the cancer/testis antigens are intrinsically disordered proteins. J. Cell. Biochem. 2011, 112, 3256–3267. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, N.; Alhothali, M.; Alfonso, M.H.; Breydo, L.; Uversky, V.N. Intrinsic disorder in proteins involved in amyotrophic lateral sclerosis. Cell. Mol. Life Sci. 2017, 74, 1297–1318. [Google Scholar] [CrossRef] [PubMed]
- Ferreon, A.C.; Ferreon, J.C.; Wright, P.E.; Deniz, A.A. Modulation of allostery by protein intrinsic disorder. Nature 2013, 498, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Cozzetto, D.; Jones, D.T. The contribution of intrinsic disorder prediction to the elucidation of protein function. Curr. Opin. Struct. Biol. 2013, 23, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.O.; Yu, L.; Campuzano, I.; Grant, S.G.; Choudhary, J.S. Phosphoproteomic analysis of the mouse brain cytosol reveals a predominance of protein phosphorylation in regions of intrinsic sequence disorder. Mol. Cell. Proteom. MCP 2008, 7, 1331–1348. [Google Scholar] [CrossRef] [PubMed]
- Kurotani, A.; Tokmakov, A.A.; Kuroda, Y.; Fukami, Y.; Shinozaki, K.; Sakurai, T. Correlations between predicted protein disorder and post-translational modifications in plants. Bioinformatics 2014, 30, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.R.; Zaidi, S.; Fang, Y.Y.; Uversky, V.N.; Radivojac, P.; Oldfield, C.J.; Cortese, M.S.; Sickmeier, M.; LeGall, T.; Obradovic, Z.; et al. Alternative splicing in concert with protein intrinsic disorder enables increased functional diversity in multicellular organisms. Proc. Natl. Acad. Sci. USA 2006, 103, 8390–8395. [Google Scholar] [CrossRef] [PubMed]
- Buljan, M.; Chalancon, G.; Eustermann, S.; Wagner, G.P.; Fuxreiter, M.; Bateman, A.; Babu, M.M. Tissue-specific splicing of disordered segments that embed binding motifs rewires protein interaction networks. Mol. Cell 2012, 46, 871–883. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsic disorder-based protein interactions and their modulators. Curr. Pharm. Des. 2013, 19, 4191–4213. [Google Scholar] [CrossRef] [PubMed]
- Colak, R.; Kim, T.; Michaut, M.; Sun, M.; Irimia, M.; Bellay, J.; Myers, C.L.; Blencowe, B.J.; Kim, P.M. Distinct types of disorder in the human proteome: Functional implications for alternative splicing. PLoS Comput. Biol. 2013, 9, e1003030. [Google Scholar] [CrossRef] [PubMed]
- Mitrea, D.M.; Kriwacki, R.W. Regulated unfolding of proteins in signaling. FEBS Lett. 2013, 587, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Follis, A.V.; Llambi, F.; Ou, L.; Baran, K.; Green, D.R.; Kriwacki, R.W. The DNA-binding domain mediates both nuclear and cytosolic functions of p53. Nat. Struct. Mol. Biol. 2014, 21, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fisher, J.C.; Mathew, R.; Ou, L.; Otieno, S.; Sublet, J.; Xiao, L.; Chen, J.; Roussel, M.F.; Kriwacki, R.W. Intrinsic disorder mediates the diverse regulatory functions of the cdk inhibitor p21. Nat. Chem. Biol. 2011, 7, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Ou, L.; Waddell, M.B.; Kriwacki, R.W. Mechanism of cell cycle entry mediated by the intrinsically disordered protein p27kip1. ACS Chem. Biol. 2012, 7, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Follis, A.V.; Galea, C.A.; Kriwacki, R.W. Intrinsic protein flexibility in regulation of cell proliferation: Advantages for signaling and opportunities for novel therapeutics. Adv. Exp. Med. Biol. 2012, 725, 27–49. [Google Scholar] [PubMed]
- Yoon, M.K.; Mitrea, D.M.; Ou, L.; Kriwacki, R.W. Cell cycle regulation by the intrinsically disordered proteins p21 and p27. Biochem. Soc. Trans. 2012, 40, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Mitrea, D.M.; Yoon, M.K.; Ou, L.; Kriwacki, R.W. Disorder-function relationships for the cell cycle regulatory proteins p21 and p27. Biol. Chem. 2012, 393, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Moldoveanu, T.; Grace, C.R.; Llambi, F.; Nourse, A.; Fitzgerald, P.; Gehring, K.; Kriwacki, R.W.; Green, D.R. Bid-induced structural changes in bak promote apoptosis. Nat. Struct. Mol. Biol. 2013, 20, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Frye, J.J.; Brown, N.G.; Petzold, G.; Watson, E.R.; Grace, C.R.; Nourse, A.; Jarvis, M.A.; Kriwacki, R.W.; Peters, J.M.; Stark, H.; et al. Electron microscopy structure of human APC/C(CDH1)-EMI1 reveals multimodal mechanism of E3 ligase shutdown. Nat. Struct. Mol. Biol. 2013, 20, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Su, M.; Soni, G.; Salem, S.; Colbert, C.L.; Sinha, S.C. Intrinsically disordered regions in autophagy proteins. Proteins 2014, 82, 565–578. [Google Scholar] [CrossRef] [PubMed]
- Chakrabortee, S.; Tripathi, R.; Watson, M.; Schierle, G.S.; Kurniawan, D.P.; Kaminski, C.F.; Wise, M.J.; Tunnacliffe, A. Intrinsically disordered proteins as molecular shields. Mol. Biosyst. 2012, 8, 210–219. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, N.; Garcia-Pino, A.; Buts, L.; Haesaerts, S.; Charlier, D.; Zangger, K.; Wyns, L.; De Greve, H.; Loris, R. Rejuvenation of ccdb-poisoned gyrase by an intrinsically disordered protein domain. Mol. Cell 2009, 35, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Norholm, A.B.; Hendus-Altenburger, R.; Bjerre, G.; Kjaergaard, M.; Pedersen, S.F.; Kragelund, B.B. The intracellular distal tail of the Na+/H+ exchanger NHE1 is intrinsically disordered: Implications for NHE1 trafficking. Biochemistry 2011, 50, 3469–3480. [Google Scholar] [CrossRef] [PubMed]
- Follis, A.V.; Chipuk, J.E.; Fisher, J.C.; Yun, M.K.; Grace, C.R.; Nourse, A.; Baran, K.; Ou, L.; Min, L.; White, S.W.; et al. Puma binding induces partial unfolding within bcl-xl to disrupt p53 binding and promote apoptosis. Nat. Chem. Biol. 2013, 9, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Rudiger, S.; Freund, S.M.; Veprintsev, D.B.; Fersht, A.R. Crinept-trosy nmr reveals p53 core domain bound in an unfolded form to the chaperone hsp90. Proc. Natl. Acad. Sci. USA 2002, 99, 11085–11090. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Yamout, M.A.; Venkitakrishnan, R.P.; Preece, N.E.; Kroon, G.; Wright, P.E.; Dyson, H.J. Localization of sites of interaction between p23 and hsp90 in solution. J. Biol. Chem. 2006, 281, 14457–14464. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, F.; Arsene-Ploetze, F.; Rist, W.; Rudiger, S.; Schneider-Mergener, J.; Mayer, M.P.; Bukau, B. Molecular basis for regulation of the heat shock transcription factor sigma32 by the dnak and dnaj chaperones. Mol. Cell 2008, 32, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Didenko, T.; Duarte, A.M.; Karagoz, G.E.; Rudiger, S.G. Hsp90 structure and function studied by NMR spectroscopy. Biochim. Biophys. Acta 2012, 1823, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Karagoz, G.E.; Duarte, A.M.; Akoury, E.; Ippel, H.; Biernat, J.; Moran Luengo, T.; Radli, M.; Didenko, T.; Nordhues, B.A.; Veprintsev, D.B.; et al. Hsp90-Tau complex reveals molecular basis for specificity in chaperone action. Cell 2014, 156, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Reuven, N.; Shaul, Y. The nanny model for IDPs. Nat. Chem. Biol. 2009, 5, 778–781. [Google Scholar] [CrossRef] [PubMed]
- Demarest, S.J.; Martinez-Yamout, M.; Chung, J.; Chen, H.; Xu, W.; Dyson, H.J.; Evans, R.M.; Wright, P.E. Mutual synergistic folding in recruitment of CBP/p300 by p160 nuclear receptor coactivators. Nature 2002, 415, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.O.; Bae, S.H.; Dyson, H.J.; Wright, P.E. NMR relaxation study of the complex formed between CBP and the activation domain of the nuclear hormone receptor coactivator ACTR. Biochemistry 2008, 47, 1299–1308. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Fisher, J.C.; Dillon, C.P.; Kriwacki, R.W.; Kuwana, T.; Green, D.R. Mechanism of apoptosis induction by inhibition of the anti-apoptotic BCL-2 proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 20327–20332. [Google Scholar] [CrossRef] [PubMed]
- Ferreon, J.C.; Lee, C.W.; Arai, M.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Cooperative regulation of p53 by modulation of ternary complex formation with CBP/p300 and hdm2. Proc. Natl. Acad. Sci. USA 2009, 106, 6591–6596. [Google Scholar] [CrossRef] [PubMed]
- Wojciak, J.M.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Structural basis for recruitment of CBP/p300 coactivators by stat1 and stat2 transactivation domains. EMBO J. 2009, 28, 948–958. [Google Scholar] [CrossRef] [PubMed]
- Ferreon, J.C.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Structural basis for subversion of cellular control mechanisms by the adenoviral e1a oncoprotein. Proc. Natl. Acad. Sci. USA 2009, 106, 13260–13265. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Structure of the p53 transactivation domain in complex with the nuclear receptor coactivator binding domain of creb binding protein. Biochemistry 2010, 49, 9964–9971. [Google Scholar] [CrossRef] [PubMed]
- Kostic, M.; Matt, T.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Solution structure of the HDM2 C2H2C4 RING, a domain critical for ubiquitination of p53. J. Mol. Biol. 2006, 363, 433–450. [Google Scholar] [CrossRef] [PubMed]
- Grimmler, M.; Wang, Y.; Mund, T.; Cilensek, Z.; Keidel, E.M.; Waddell, M.B.; Jakel, H.; Kullmann, M.; Kriwacki, R.W.; Hengst, L. Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell 2007, 128, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.; Easton, J.B.; Huang, S.; Mishra, A.; Xiao, L.; Lacy, E.R.; Kriwacki, R.W.; Houghton, P.J. Negative regulation of ASK1 by p21Cip1 involves a small domain that includes serine 98 that is phosphorylated by ASK1 in vivo. Mol. Cell. Biol. 2007, 27, 3530–3541. [Google Scholar] [CrossRef] [PubMed]
- Galea, C.A.; Nourse, A.; Wang, Y.; Sivakolundu, S.G.; Heller, W.T.; Kriwacki, R.W. Role of intrinsic flexibility in signal transduction mediated by the cell cycle regulator, p27kip1. J. Mol. Biol. 2008, 376, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Mitrea, D.M.; Grace, C.R.; Buljan, M.; Yun, M.K.; Pytel, N.J.; Satumba, J.; Nourse, A.; Park, C.G.; Madan Babu, M.; White, S.W.; et al. Structural polymorphism in the N-terminal oligomerization domain of NPM1. Proc. Natl. Acad. Sci. USA 2014, 111, 4466–4471. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Tsvetkov, P.; Kahana, C.; Shaul, Y. A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes Dev. 2005, 19, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Asher, G.; Paz, A.; Reuven, N.; Sussman, J.L.; Silman, I.; Shaul, Y. Operational definition of intrinsically unstructured protein sequences based on susceptibility to the 20s proteasome. Proteins 2008, 70, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Reuven, N.; Prives, C.; Shaul, Y. Susceptibility of p53 unstructured n terminus to 20 s proteasomal degradation programs the stress response. J. Biol. Chem. 2009, 284, 26234–26242. [Google Scholar] [CrossRef] [PubMed]
- Wiggins, C.M.; Tsvetkov, P.; Johnson, M.; Joyce, C.L.; Lamb, C.A.; Bryant, N.J.; Komander, D.; Shaul, Y.; Cook, S.J. BIM(El), an intrinsically disordered protein, is degraded by 20S proteasomes in the absence of poly-ubiquitylation. J. Cell Sci. 2011, 124, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Suskiewicz, M.J.; Sussman, J.L.; Silman, I.; Shaul, Y. Context-dependent resistance to proteolysis of intrinsically disordered proteins. Protein Sci. 2011, 20, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Myers, N.; Moscovitz, O.; Sharon, M.; Prilusky, J.; Shaul, Y. Thermo-resistant intrinsically disordered proteins are efficient 20s proteasome substrates. Mol. Biosyst. 2012, 8, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Kurgan, L. High-throughput prediction of RNA, DNA and protein binding regions mediated by intrinsic disorder. Nucleic Acids Res. 2015, 43, e121. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Wang, C.; Uversky, V.N.; Kurgan, L. Prediction of disordered RNA, DNA, and protein binding regions using disordpbind. Methods Mol. Biol. 2017, 1484, 187–203. [Google Scholar] [PubMed]
- Dosztanyi, Z.; Meszaros, B.; Simon, I. Anchor: Web server for predicting protein binding regions in disordered proteins. Bioinformatics 2009, 25, 2745–2746. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, T.; Walsh, I.; Martin, A.J.M.; Tosatto, S.C.E. Mobidb: A comprehensive database of intrinsic protein disorder annotations. Bioinformatics 2012, 28, 2080–2081. [Google Scholar] [CrossRef] [PubMed]
- Potenza, E.; Di Domenico, T.; Walsh, I.; Tosatto, S.C. Mobidb 2.0: An improved database of intrinsically disordered and mobile proteins. Nucleic Acids Res. 2015, 43, D315–D320. [Google Scholar] [CrossRef] [PubMed]
- Necci, M.; Piovesan, D.; Dosztanyi, Z.; Tosatto, S.C.E. Mobidb-lite: Fast and highly specific consensus prediction of intrinsic disorder in proteins. Bioinformatics 2017, 33, 1402–1404. [Google Scholar] [CrossRef] [PubMed]
- Dosztanyi, Z.; Csizmok, V.; Tompa, P.; Simon, I. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J. Mol. Biol. 2005, 347, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Walsh, I.; Martin, A.J.; Di Domenico, T.; Tosatto, S.C. Espritz: Accurate and fast prediction of protein disorder. Bioinformatics 2012, 28, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Hsu Ia, W.; Tarn, W.Y. Trap150 activates pre-mRNA splicing and promotes nuclear mRNA degradation. Nucleic Acids Res. 2010, 38, 3340–3350. [Google Scholar] [CrossRef] [PubMed]
- Heyd, F.; Lynch, K.W. Phosphorylation-dependent regulation of PSF by GSK3 controls CD45 alternative splicing. Mol. Cell 2010, 40, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Beli, P.; Lukashchuk, N.; Wagner, S.A.; Weinert, B.T.; Olsen, J.V.; Baskcomb, L.; Mann, M.; Jackson, S.P.; Choudhary, C. Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol. Cell 2012, 46, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Okano, H.J.; Darnell, R.B.; Roeder, R.G. The TRAP100 component of the TRAP/Mediator complex is essential in broad transcriptional events and development. EMBO J. 2002, 21, 3464–3475. [Google Scholar] [CrossRef] [PubMed]
- Katano-Toki, A.; Satoh, T.; Tomaru, T.; Yoshino, S.; Ishizuka, T.; Ishii, S.; Ozawa, A.; Shibusawa, N.; Tsuchiya, T.; Saito, T.; et al. THRAP3 interacts with HELZ2 and plays a novel role in adipocyte differentiation. Mol. Endocrinol. 2013, 27, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Lande-Diner, L.; Boyault, C.; Kim, J.Y.; Weitz, C.J. A positive feedback loop links circadian clock factor clock-bmal1 to the basic transcriptional machinery. Proc. Natl. Acad. Sci. USA 2013, 110, 16021–16026. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, K.; Kawamura, T.; Iwanari, H.; Ohashi, R.; Naito, M.; Kodama, T.; Hamakubo, T. Identification of wilms’ tumor 1-associating protein complex and its role in alternative splicing and the cell cycle. J. Biol. Chem. 2013, 288, 33292–33302. [Google Scholar] [CrossRef] [PubMed]
- Ino, Y.; Arakawa, N.; Ishiguro, H.; Uemura, H.; Kubota, Y.; Hirano, H.; Toda, T. Phosphoproteome analysis demonstrates the potential role of THRAP3 phosphorylation in androgen-independent prostate cancer cell growth. Proteomics 2016, 16, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Strasser, K.; Masuda, S.; Mason, P.; Pfannstiel, J.; Oppizzi, M.; Rodriguez-Navarro, S.; Rondon, A.G.; Aguilera, A.; Struhl, K.; Reed, R.; et al. Trex is a conserved complex coupling transcription with messenger RNA export. Nature 2002, 417, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Chi, B.; Wang, K.; Du, Y.; Gui, B.; Chang, X.; Wang, L.; Fan, J.; Chen, S.; Wu, X.; Li, G.; et al. A Sub-Element in PRE enhances nuclear export of intronless mRNAs by recruiting the TREX complex via ZC3H18. Nucleic Acids Res. 2014, 42, 7305–7318. [Google Scholar] [CrossRef] [PubMed]
- Garee, J.P.; Oesterreich, S. Safb1’s multiple functions in biological control-lots still to be done! J. Cell. Biochem. 2010, 109, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Nayler, O.; Stratling, W.; Bourquin, J.P.; Stagljar, I.; Lindemann, L.; Jasper, H.; Hartmann, A.M.; Fackelmayer, F.O.; Ullrich, A.; Stamm, S. SAF-B protein couples transcription and pre-mRNA splicing to SAR/MAR elements. Nucleic Acids Res. 1998, 26, 3542–3549. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L.; Koonin, E.V. Sap—A putative DNA-binding motif involved in chromosomal organization. Trends Biochem. Sci. 2000, 25, 112–114. [Google Scholar] [CrossRef]
- Nery, F.C.; Rui, E.; Kuniyoshi, T.M.; Kobarg, J. Evidence for the interaction of the regulatory protein ki-1/57 with p53 and its interacting proteins. Biochem. Biophys. Res. Commun. 2006, 341, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Nery, F.C.; Passos, D.O.; Garcia, V.S.; Kobarg, J. Ki-1/57 interacts with RACK1 and is a substrate for the phosphorylation by phorbol 12-myristate 13-acetate-activated protein kinase C. J. Biol. Chem. 2004, 279, 11444–11455. [Google Scholar] [CrossRef] [PubMed]
- Jobert, L.; Argentini, M.; Tora, L. PRMT1 mediated methylation of TAF15 is required for its positive gene regulatory function. Exp. Cell Res. 2009, 315, 1273–1286. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A.; Lutz, Y.; Heard, D.J.; Chambon, P.; Tora, L. Htaf(II)68, a novel RNA/ssDNA-binding protein with homology to the pro-oncoproteins TLS/FUS and EWS is associated with both TFIID and RNA polymerase II. EMBO J. 1996, 15, 5022–5031. [Google Scholar] [PubMed]
- Law, W.J.; Cann, K.L.; Hicks, G.G. TLS, EWS and TAF15: A model for transcriptional integration of gene expression. Brief. Funct. Genom. Proteom. 2006, 5, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Arvand, A.; Denny, C.T. Biology of EWS/ETS fusions in Ewing’s family tumors. Oncogene 2001, 20, 5747–5754. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A.; Bell, B.; Tora, L. The N-terminal domain of human TAFII68 displays transactivation and oncogenic properties. Oncogene 1999, 18, 8000–8010. [Google Scholar] [CrossRef] [PubMed]
- Takayama, S.; Xie, Z.; Reed, J.C. An evolutionarily conserved family of HSP70/HSC70 molecular chaperone regulators. J. Biol. Chem. 1999, 274, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Rauch, J.N.; Zuiderweg, E.R.; Gestwicki, J.E. Non-canonical interactions between heat shock cognate protein 70 (HSC70) and BCL2-associated anthanogene (BAG) co-chaperones are important for client release. J. Biol. Chem. 2016, 291, 19848–19857. [Google Scholar] [CrossRef] [PubMed]
- Ingham, R.J.; Colwill, K.; Howard, C.; Dettwiler, S.; Lim, C.S.; Yu, J.; Hersi, K.; Raaijmakers, J.; Gish, G.; Mbamalu, G.; et al. Ww domains provide a platform for the assembly of multiprotein networks. Mol. Cell. Biol. 2005, 25, 7092–7106. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.M.; La Thangue, N.B. P300/CBP proteins: Hats for transcriptional bridges and scaffolds. J. Cell Sci. 2001, 114, 2363–2373. [Google Scholar] [PubMed]
- Cazzalini, O.; Sommatis, S.; Tillhon, M.; Dutto, I.; Bachi, A.; Rapp, A.; Nardo, T.; Scovassi, A.I.; Necchi, D.; Cardoso, M.C.; et al. CBP and p300 acetylate pcna to link its degradation with nucleotide excision repair synthesis. Nucleic Acids Res. 2014, 42, 8433–8448. [Google Scholar] [CrossRef] [PubMed]
- Imhof, A.; Yang, X.J.; Ogryzko, V.V.; Nakatani, Y.; Wolffe, A.P.; Ge, H. Acetylation of general transcription factors by histone acetyltransferases. Curr. Biol. 1997, 7, 689–692. [Google Scholar] [CrossRef]
- Zhang, W.; Bieker, J.J. Acetylation and modulation of erythroid kruppel-like factor (EKLF) activity by interaction with histone acetyltransferases. Proc. Natl. Acad. Sci. USA 1998, 95, 9855–9860. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, H.; Daitoku, H.; Hatta, M.; Aoyama, H.; Yoshimochi, K.; Fukamizu, A. Acetylation of FOXO1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc. Natl. Acad. Sci. USA 2005, 102, 11278–11283. [Google Scholar] [CrossRef] [PubMed]
- Snowden, A.W.; Perkins, N.D. Cell cycle regulation of the transcriptional coactivators p300 and CREB binding protein. Biochem. Pharmacol. 1998, 55, 1947–1954. [Google Scholar] [CrossRef]
- Andrisani, O.M. CREB-mediated transcriptional control. Crit. Rev. Eukaryot. Gene Expr. 1999, 9, 19–32. [Google Scholar] [PubMed]
- Dyson, H.J.; Wright, P.E. Role of intrinsic protein disorder in the function and interactions of the transcriptional coactivators CREB-binding protein (CBP) and p300. J. Biol. Chem. 2016, 291, 6714–6722. [Google Scholar] [CrossRef] [PubMed]
- Bedford, D.C.; Kasper, L.H.; Fukuyama, T.; Brindle, P.K. Target gene context influences the transcriptional requirement for the KAT3 family of CBP and p300 histone acetyltransferases. Epigenetics 2010, 5, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Zakaryan, R.P.; Gehring, H. Identification and characterization of the nuclear localization/retention signal in the EWS proto-oncoprotein. J. Mol. Biol. 2006, 363, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, L.E.; Fredericks, W.J.; Barr, F.G.; Rauscher, F.J., 3rd. Fusion of the EWS1 and WT1 genes as a result of the t(11;22)(p13;q12) translocation in desmoplastic small round cell tumors. Med. Pediatr. Oncol. 1996, 27, 434–439. [Google Scholar] [CrossRef]
- May, W.A.; Denny, C.T. Biology of EWS/FLI and related fusion genes in Ewing’s sarcoma and primitive neuroectodermal tumor. Curr. Top. Microbiol. Immunol. 1997, 220, 143–150. [Google Scholar] [PubMed]
- Erkizan, H.V.; Uversky, V.N.; Toretsky, J.A. Oncogenic partnerships: EWS-FLI1 protein interactions initiate key pathways of Ewing’s sarcoma. Clin. Cancer Res. 2010, 16, 4077–4083. [Google Scholar] [CrossRef] [PubMed]
- Hegyi, H.; Buday, L.; Tompa, P. Intrinsic structural disorder confers cellular viability on oncogenic fusion proteins. PLoS Comput. Biol. 2009, 5, e1000552. [Google Scholar] [CrossRef] [PubMed]
- Kriwacki, R.W.; Wu, J.; Tennant, L.; Wright, P.E.; Siuzdak, G. Probing protein structure using biochemical and biophysical methods. Proteolysis, matrix-assisted laser desorption/ionization mass spectrometry, high-performance liquid chromatography and size-exclusion chromatography of p21Waf1/Cip1/Sdi1. J. Chromatogr. A 1997, 777, 23–30. [Google Scholar] [CrossRef]
- LaBaer, J.; Garrett, M.D.; Stevenson, L.F.; Slingerland, J.M.; Sandhu, C.; Chou, H.S.; Fattaey, A.; Harlow, E. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997, 11, 847–862. [Google Scholar] [CrossRef] [PubMed]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair 2016, 42, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Kroker, A.J.; Bruning, J.B. P21 exploits residue Tyr151 as a tether for high-affinity PCNA binding. Biochemistry 2015, 54, 3483–3493. [Google Scholar] [CrossRef] [PubMed]
- Stucki, M.; Jackson, S.P. MDC1/NFBD1: A key regulator of the DNA damage response in higher eukaryotes. DNA Repair 2004, 3, 953–957. [Google Scholar] [CrossRef] [PubMed]
- Lou, Z.; Chini, C.C.; Minter-Dykhouse, K.; Chen, J. Mediator of DNA damage checkpoint protein 1 regulates brca1 localization and phosphorylation in DNA damage checkpoint control. J. Biol. Chem. 2003, 278, 13599–13602. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.S.; Wang, B.; Bignell, C.R.; Taylor, A.M.; Elledge, S.J. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 2003, 421, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Lou, Z.; Minter-Dykhouse, K.; Wu, X.; Chen, J. MDC1 is coupled to activated CHK2 in mammalian DNA damage response pathways. Nature 2003, 421, 957–961. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.; Stucki, M.; Falck, J.; D’Amours, D.; Rahman, D.; Pappin, D.; Bartek, J.; Jackson, S.P. MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature 2003, 421, 952–956. [Google Scholar] [CrossRef] [PubMed]
- Peng, A.; Chen, P.L. NFBD1, like 53BP1, is an early and redundant transducer mediating CHK2 phosphorylation in response to DNA damage. J. Biol. Chem. 2003, 278, 8873–8876. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Stern, D.F. NFBD1/KIAA0170 is a chromatin-associated protein involved in DNA damage signaling pathways. J. Biol. Chem. 2003, 278, 8795–8803. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.L.; Bodero, A.J.; Chen, P.L. NFBD1, a novel nuclear protein with signature motifs of FHA and BRCT, and an internal 41-amino acid repeat sequence, is an early participant in DNA damage response. J. Biol. Chem. 2003, 278, 6323–6329. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, G.; Udasin, R.G.; Livneh, I.; Ciechanover, A. Identification of UBact, a ubiquitin-like protein, along with other homologous components of a conjugation system and the proteasome in different gram-negative Bacteria. Biochem. Biophys. Res. Commun. 2017, 483, 946–950. [Google Scholar] [CrossRef] [PubMed]
- Delley, C.L.; Muller, A.U.; Ziemski, M.; Weber-Ban, E. Prokaryotic ubiquitin-like protein and its ligase/deligase enyzmes. J. Mol. Biol. 2017, 429, 3486–3499. [Google Scholar] [CrossRef] [PubMed]
- Hicke, L. Protein regulation by monoubiquitin. Nat. Rev. Mol. Cell Biol. 2001, 2, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Pickart, C.M. Ubiquitin enters the new millennium. Mol. Cell 2001, 8, 499–504. [Google Scholar] [CrossRef]
- Pornillos, O.; Garrus, J.E.; Sundquist, W.I. Mechanisms of enveloped RNA virus budding. Trends Cell Biol. 2002, 12, 569–579. [Google Scholar] [CrossRef]
- Muratani, M.; Tansey, W.P. How the ubiquitin-proteasome system controls transcription. Nat. Rev. Mol. Cell Biol. 2003, 4, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Terrell, J.; Shih, S.; Dunn, R.; Hicke, L. A function for monoubiquitination in the internalization of a G protein-coupled receptor. Mol. Cell 1998, 1, 193–202. [Google Scholar] [CrossRef]
- Carmichael, R.E.; Henley, J.M. Transcriptional and post-translational regulation of Arc in synaptic plasticity. Semin. Cell Dev. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Torrecilla, I.; Oehler, J.; Ramadan, K. The role of ubiquitin-dependent segregase p97 (VCP or Cdc48) in chromatin dynamics after DNA double strand breaks. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef] [PubMed]
- Rome, S.; Meugnier, E.; Vidal, H. The ubiquitin-proteasome pathway is a new partner for the control of insulin signaling. Curr. Opin. Clin. Nutr. Metab. Care 2004, 7, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Izzi, L.; Attisano, L. Regulation of the TGFbeta signalling pathway by ubiquitin-mediated degradation. Oncogene 2004, 23, 2071–2078. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; He, C.; Wang, L.; Ge, B. Post-translational regulation of antiviral innate signaling. Eur. J. Immunol. 2017, 47, 1414–1426. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Best, R.B.; Depristo, M.A.; Dobson, C.M.; Vendruscolo, M. Simultaneous determination of protein structure and dynamics. Nature 2005, 433, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Solomon, W.C.; Kang, Y.; Cerda-Maira, F.; Darwin, K.H.; Walters, K.J. Prokaryotic ubiquitin-like protein pup is intrinsically disordered. J. Mol. Biol. 2009, 392, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Shang, Q.; Zhang, X.; Zhang, J.; Xu, C.; Tu, X. Pup, a prokaryotic ubiquitin-like protein, is an intrinsically disordered protein. Biochem. J. 2009, 422, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Vidal, M.; Goudreau, N.; Cornille, F.; Cussac, D.; Gincel, E.; Garbay, C. Molecular and cellular analysis of GRB2 SH3 domain mutants: Interaction with Sos and dynamin. J. Mol. Biol. 1999, 290, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Ettmayer, P.; France, D.; Gounarides, J.; Jarosinski, M.; Martin, M.S.; Rondeau, J.M.; Sabio, M.; Topiol, S.; Weidmann, B.; Zurini, M.; et al. Structural and conformational requirements for high-affinity binding to the SH2 domain of GRB2(1). J. Med. Chem. 1999, 42, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Rice, C.; Shastrula, P.K.; Kossenkov, A.V.; Hills, R.; Baird, D.M.; Showe, L.C.; Doukov, T.; Janicki, S.; Skordalakes, E. Structural and functional analysis of the human POT1-TPP1 telomeric complex. Nat. Commun. 2017, 8, 14928. [Google Scholar] [CrossRef] [PubMed]
- Bono, F.; Ebert, J.; Lorentzen, E.; Conti, E. The crystal structure of the exon junction complex reveals how it maintains a stable grip on mRNA. Cell 2006, 126, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Tanase, C.A. Histidine domain-protein tyrosine phosphatase interacts with GRB2 and GRPL. PLoS ONE 2010, 5, e14339. [Google Scholar] [CrossRef] [PubMed]
- Belov, A.A.; Mohammadi, M. GRB2, a double-edged sword of receptor tyrosine kinase signaling. Sci. Signal. 2012, 5, pe49. [Google Scholar] [CrossRef] [PubMed]
- Jang, I.K.; Zhang, J.; Gu, H. GRB2, a simple adapter with complex roles in lymphocyte development, function, and signaling. Immunol. Rev. 2009, 232, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Giubellino, A.; Burke, T.R., Jr.; Bottaro, D.P. GRB2 signaling in cell motility and cancer. Expert Opin. Ther. Targets 2008, 12, 1021–1033. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Kharbanda, S.; Kufe, D. Association of the DF3/MUC1 breast cancer antigen with GRB2 and the SOS/RAS exchange protein. Cancer Res. 1995, 55, 4000–4003. [Google Scholar] [PubMed]
- Chardin, P.; Camonis, J.H.; Gale, N.W.; van Aelst, L.; Schlessinger, J.; Wigler, M.H.; Bar-Sagi, D. Human SOS1: A guanine nucleotide exchange factor for RAS that binds to GRB2. Science 1993, 260, 1338–1343. [Google Scholar] [CrossRef] [PubMed]
- Tarcic, G.; Boguslavsky, S.K.; Wakim, J.; Kiuchi, T.; Liu, A.; Reinitz, F.; Nathanson, D.; Takahashi, T.; Mischel, P.S.; Ng, T.; et al. An unbiased screen identifies dep-1 tumor suppressor as a phosphatase controlling egfr endocytosis. Curr. Biol. 2009, 19, 1788–1798. [Google Scholar] [CrossRef] [PubMed]
- Braverman, L.E.; Quilliam, L.A. Identification of GRB4/Nckbeta, a src homology 2 and 3 domain-containing adapter protein having similar binding and biological properties to Nck. J. Biol. Chem. 1999, 274, 5542–5549. [Google Scholar] [CrossRef] [PubMed]
- Pfrepper, K.I.; Marie-Cardine, A.; Simeoni, L.; Kuramitsu, Y.; Leo, A.; Spicka, J.; Hilgert, I.; Scherer, J.; Schraven, B. Structural and functional dissection of the cytoplasmic domain of the transmembrane adaptor protein SIT (SHP2-interacting transmembrane adaptor protein). Eur. J. Immunol. 2001, 31, 1825–1836. [Google Scholar] [CrossRef]
- Fantin, V.R.; Sparling, J.D.; Slot, J.W.; Keller, S.R.; Lienhard, G.E.; Lavan, B.E. Characterization of insulin receptor substrate 4 in human embryonic kidney 293 cells. J. Biol. Chem. 1998, 273, 10726–10732. [Google Scholar] [CrossRef] [PubMed]
- Tobe, K.; Matuoka, K.; Tamemoto, H.; Ueki, K.; Kaburagi, Y.; Asai, S.; Noguchi, T.; Matsuda, M.; Tanaka, S.; Hattori, S.; et al. Insulin stimulates association of insulin receptor substrate-1 with the protein abundant src homology/growth factor receptor-bound protein 2. J. Biol. Chem. 1993, 268, 11167–11171. [Google Scholar] [PubMed]
- Skolnik, E.Y.; Lee, C.H.; Batzer, A.; Vicentini, L.M.; Zhou, M.; Daly, R.; Myers, M.J., Jr.; Backer, J.M.; Ullrich, A.; White, M.F.; et al. The SH2/SH3 domain-containing protein GRB2 interacts with tyrosine-phosphorylated IRS1 and SHC: Implications for insulin control of RAS signalling. EMBO J. 1993, 12, 1929–1936. [Google Scholar] [PubMed]
- Yokouchi, M.; Suzuki, R.; Masuhara, M.; Komiya, S.; Inoue, A.; Yoshimura, A. Cloning and characterization of APS, an adaptor molecule containing PH and SH2 domains that is tyrosine phosphorylated upon B-cell receptor stimulation. Oncogene 1997, 15, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Janssen, E.; Zhu, M.; Zhang, W.; Koonpaew, S.; Zhang, W. Lab: A new membrane-associated adaptor molecule in B cell activation. Nat. Immunol. 2003, 4, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Brdicka, T.; Imrich, M.; Angelisova, P.; Brdickova, N.; Horvath, O.; Spicka, J.; Hilgert, I.; Luskova, P.; Draber, P.; Novak, P.; et al. Non-T cell activation linker (NTAL): A transmembrane adaptor protein involved in immunoreceptor signaling. J. Exp. Med. 2002, 196, 1617–1626. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Janssen, E.; Leung, K.; Zhang, W. Molecular cloning of a novel gene encoding a membrane-associated adaptor protein (LAX) in lymphocyte signaling. J. Biol. Chem. 2002, 277, 46151–46158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Sloan-Lancaster, J.; Kitchen, J.; Trible, R.P.; Samelson, L.E. LAT: The ZAP-70 tyrosine kinase substrate that links T cell receptor to cellular activation. Cell 1998, 92, 83–92. [Google Scholar] [CrossRef]
- Kharitonenkov, A.; Chen, Z.; Sures, I.; Wang, H.; Schilling, J.; Ullrich, A. A family of proteins that inhibit signalling through tyrosine kinase receptors. Nature 1997, 386, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Sano, H.; Liu, S.C.; Lane, W.S.; Piletz, J.E.; Lienhard, G.E. Insulin receptor substrate 4 associates with the protein IRAS. J. Biol. Chem. 2002, 277, 19439–19447. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Ishida, O.; Hinoi, T.; Kishida, S.; Kikuchi, A. Identification and characterization of a novel protein interacting with Ral-binding protein 1, a putative effector protein of Ral. J. Biol. Chem. 1998, 273, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, M.; Domin, J. Recruitment of the class ii phosphoinositide 3-kinase c2beta to the epidermal growth factor receptor: Role of GRB2. Mol. Cell. Biol. 2001, 21, 6660–6667. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D.; Blanchoin, L.; Mullins, R.D. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 545–576. [Google Scholar] [CrossRef] [PubMed]
- Povarova, O.I.; Uversky, V.N.; Kuznetsova, I.M.; Turoverov, K.K. Actinous enigma or enigmatic actin: Folding, structure, and functions of the most abundant eukaryotic protein. Intrinsically Disord. Proteins 2014, 2, e34500. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, I.M.; Povarova, O.I.; Uversky, V.N.; Turoverov, K.K. Native globular actin has a thermodynamically unstable quasi-stationary structure with elements of intrinsic disorder. FEBS J. 2016, 283, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Barua, B.; Winkelmann, D.A.; White, H.D.; Hitchcock-DeGregori, S.E. Regulation of actin-myosin interaction by conserved periodic sites of tropomyosin. Proc. Natl. Acad. Sci. USA 2012, 109, 18425–18430. [Google Scholar] [CrossRef] [PubMed]
- Carlier, M.F.; Valentin-Ranc, C.; Combeau, C.; Fievez, S.; Pantoloni, D. Actin polymerization: Regulation by divalent metal ion and nucleotide binding, atp hydrolysis and binding of myosin. Adv. Exp. Med. Biol. 1994, 358, 71–81. [Google Scholar] [PubMed]
- Guharoy, M.; Szabo, B.; Contreras Martos, S.; Kosol, S.; Tompa, P. Intrinsic structural disorder in cytoskeletal proteins. Cytoskeleton 2013, 70, 550–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Remedios, C.G.; Chhabra, D.; Kekic, M.; Dedova, I.V.; Tsubakihara, M.; Berry, D.A.; Nosworthy, N.J. Actin binding proteins: Regulation of cytoskeletal microfilaments. Physiol. Rev. 2003, 83, 433–473. [Google Scholar] [CrossRef] [PubMed]
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; O’Connor, M.S.; Qin, J.; Songyang, Z. Telosome, a mammalian telomere-associated complex formed by multiple telomeric proteins. J. Biol. Chem. 2004, 279, 51338–51342. [Google Scholar] [CrossRef] [PubMed]
- Venteicher, A.S.; Abreu, E.B.; Meng, Z.; McCann, K.E.; Terns, R.M.; Veenstra, T.D.; Terns, M.P.; Artandi, S.E. A human telomerase holoenzyme protein required for cajal body localization and telomere synthesis. Science 2009, 323, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, U.; Im, K.B.; Benzing, C.; Janjetovic, S.; Rippe, K.; Lichter, P.; Wachsmuth, M. Assembly and mobility of exon-exon junction complexes in living cells. RNA 2009, 15, 862–876. [Google Scholar] [CrossRef] [PubMed]
- Jurica, M.S.; Licklider, L.J.; Gygi, S.R.; Grigorieff, N.; Moore, M.J. Purification and characterization of native spliceosomes suitable for three-dimensional structural analysis. RNA 2002, 8, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Michelle, L.; Cloutier, A.; Toutant, J.; Shkreta, L.; Thibault, P.; Durand, M.; Garneau, D.; Gendron, D.; Lapointe, E.; Couture, S.; et al. Proteins associated with the exon junction complex also control the alternative splicing of apoptotic regulators. Mol. Cell. Biol. 2012, 32, 954–967. [Google Scholar] [CrossRef] [PubMed]
- Tange, T.O.; Nott, A.; Moore, M.J. The ever-increasing complexities of the exon junction complex. Curr. Opin. Cell Biol. 2004, 16, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Licata, L.; Briganti, L.; Peluso, D.; Perfetto, L.; Iannuccelli, M.; Galeota, E.; Sacco, F.; Palma, A.; Nardozza, A.P.; Santonico, E.; et al. Mint, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012, 40, D857–D861. [Google Scholar] [CrossRef] [PubMed]
- Licata, L.; Orchard, S. The mintact project and molecular interaction databases. Methods Mol. Biol. 2016, 1415, 55–69. [Google Scholar] [PubMed]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; del-Toro, N.; et al. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef] [PubMed]
- Salwinski, L.; Miller, C.S.; Smith, A.J.; Pettit, F.K.; Bowie, J.U.; Eisenberg, D. The database of interacting proteins: 2004 update. Nucleic Acids Res. 2004, 32, D449–D451. [Google Scholar] [CrossRef] [PubMed]
- Launay, G.; Salza, R.; Multedo, D.; Thierry-Mieg, N.; Ricard-Blum, S. Matrixdb, the extracellular matrix interaction database: Updated content, a new navigator and expanded functionalities. Nucleic Acids Res. 2015, 43, D321–D327. [Google Scholar] [CrossRef] [PubMed]
- Chatr-Aryamontri, A.; Oughtred, R.; Boucher, L.; Rust, J.; Chang, C.; Kolas, N.K.; O’Donnell, L.; Oster, S.; Theesfeld, C.; Sellam, A.; et al. The biogrid interaction database: 2017 update. Nucleic Acids Res. 2017, 45, D369–D379. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. Uniprot: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar]
- Kiran, M.; Nagarajaram, H.A. Global versus local hubs in human protein-protein interaction network. J. Proteome Res. 2013, 12, 5436–5446. [Google Scholar] [CrossRef] [PubMed]
- Bertin, N.; Simonis, N.; Dupuy, D.; Cusick, M.E.; Han, J.D.; Fraser, H.B.; Roth, F.P.; Vidal, M. Confirmation of organized modularity in the yeast interactome. PLoS Biol. 2007, 5, e153. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Kim, P.M.; Sprecher, E.; Trifonov, V.; Gerstein, M. The importance of bottlenecks in protein networks: Correlation with gene essentiality and expression dynamics. PLoS Comput. Biol. 2007, 3, e59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batada, N.N.; Reguly, T.; Breitkreutz, A.; Boucher, L.; Breitkreutz, B.J.; Hurst, L.D.; Tyers, M. Still stratus not altocumulus: Further evidence against the date/party hub distinction. PLoS Biol. 2007, 5, 1202–1206. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Deane, C.M.; Porter, M.A.; Jones, N.S. Revisiting date and party hubs: Novel approaches to role assignment in protein interaction networks. PLoS Comput. Biol. 2010, 6, e1000817. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.L.; Kurgan, L. Comprehensive comparative assessment of in-silico predictors of disordered regions. Curr. Protein Pept. Sci. 2012, 13, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Walsh, I.; Giollo, M.; Di Domenico, T.; Ferrari, C.; Zimmermann, O.; Tosatto, S.C. Comprehensive large-scale assessment of intrinsic protein disorder. Bioinformatics 2015, 31, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Uversky, V.N.; Kurgan, L. Comprehensive review of methods for prediction of intrinsic disorder and its molecular functions. Cell. Mol. Life Sci. 2017, 74, 3069–3090. [Google Scholar] [CrossRef] [PubMed]
- Piovesan, D.; Tabaro, F.; Micetic, I.; Necci, M.; Quaglia, F.; Oldfield, C.J.; Aspromonte, M.C.; Davey, N.E.; Davidovic, R.; Dosztanyi, Z.; et al. Disprot 7.0: A major update of the database of disordered proteins. Nucleic Acids Res. 2016, D1, D219–D227. [Google Scholar] [CrossRef] [PubMed]
- Sickmeier, M.; Hamilton, J.A.; LeGall, T.; Vacic, V.; Cortese, M.S.; Tantos, A.; Szabo, B.; Tompa, P.; Chen, J.; Uversky, V.N.; et al. Disprot: The database of disordered proteins. Nucleic Acids Res. 2007, 35, D786–D793. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Kurgan, L. On the complementarity of the consensus-based disorder prediction. Pac. Symp. Biocomput. 2012, 8, 176–187. [Google Scholar]
- Fan, X.; Kurgan, L. Accurate prediction of disorder in protein chains with a comprehensive and empirically designed consensus. J. Biomol. Struct. Dyn. 2014, 32, 448–464. [Google Scholar] [CrossRef] [PubMed]
- Na, I.; Meng, F.; Kurgan, L.; Uversky, V.N. Autophagy-related intrinsically disordered proteins in intra-nuclear compartments. Mol. Biosyst. 2016, 12, 2798–2817. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Oldfield, C.J.; Xue, B.; Mizianty, M.J.; Dunker, A.K.; Kurgan, L.; Uversky, V.N. A creature with a hundred waggly tails: Intrinsically disordered proteins in the ribosome. Cell. Mol. Life Sci. 2014, 71, 1477–1504. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Wu, Z.; Wang, K.; Uversky, V.N.; Kurgan, L. Untapped potential of disordered proteins in current druggable human proteome. Curr. Drug Targets 2016, 17, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Uversky, V.N.; Kurgan, L. Genes encoding intrinsic disorder in Eukaryota have high gc content. Intrinsically Disord. Proteins 2016, 4, e1262225. [Google Scholar] [CrossRef] [PubMed]
- Oates, M.E.; Romero, P.; Ishida, T.; Ghalwash, M.; Mizianty, M.J.; Xue, B.; Dosztanyi, Z.; Uversky, V.N.; Obradovic, Z.; Kurgan, L.; et al. D(2)p(2): Database of disordered protein predictions. Nucleic Acids Res. 2013, 41, D508–D516. [Google Scholar] [CrossRef] [PubMed]
- Monastyrskyy, B.; Kryshtafovych, A.; Moult, J.; Tramontano, A.; Fidelis, K. Assessment of protein disorder region predictions in casp10. Proteins 2014, 82 (Suppl. 2), 127–137. [Google Scholar] [CrossRef] [PubMed]
- Pentony, M.M.; Jones, D.T. Modularity of intrinsic disorder in the human proteome. Proteins 2010, 78, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P.; Fuxreiter, M.; Oldfield, C.J.; Simon, I.; Dunker, A.K.; Uversky, V.N. Close encounters of the third kind: Disordered domains and the interactions of proteins. Bioessays 2009, 31, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Disfani, F.M.; Hsu, W.L.; Mizianty, M.J.; Oldfield, C.J.; Xue, B.; Dunker, A.K.; Uversky, V.N.; Kurgan, L. Morfpred, a computational tool for sequence-based prediction and characterization of short disorder-to-order transitioning binding regions in proteins. Bioinformatics 2012, 28, i75–i83. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Cozzetto, D. Disopred3: Precise disordered region predictions with annotated protein-binding activity. Bioinformatics 2015, 31, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Samudrala, R. Incorporating background frequency improves entropy-based residue conservation measures. BMC Bioinform. 2006, 7, 385. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped blast and psi-blast: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Van Roey, K.; Uyar, B.; Weatheritt, R.J.; Dinkel, H.; Seiler, M.; Budd, A.; Gibson, T.J.; Davey, N.E. Short linear motifs: Ubiquitous and functionally diverse protein interaction modules directing cell regulation. Chem. Rev. 2014, 114, 6733–6778. [Google Scholar] [CrossRef] [PubMed]
- Dinkel, H.; Van Roey, K.; Michael, S.; Kumar, M.; Uyar, B.; Altenberg, B.; Milchevskaya, V.; Schneider, M.; Kuhn, H.; Behrendt, A.; et al. Elm 2016—Data update and new functionality of the eukaryotic linear motif resource. Nucleic Acids Res. 2016, 44, D294–D300. [Google Scholar] [CrossRef] [PubMed]
- Van Roey, K.; Dinkel, H.; Weatheritt, R.J.; Gibson, T.J.; Davey, N.E. The switches.Elm resource: A compendium of conditional regulatory interaction interfaces. Sci. Signal. 2013, 6, rs7. [Google Scholar] [CrossRef] [PubMed]
- Davey, N.E.; Van Roey, K.; Weatheritt, R.J.; Toedt, G.; Uyar, B.; Altenberg, B.; Budd, A.; Diella, F.; Dinkel, H.; Gibson, T.J. Attributes of short linear motifs. Mol. Biosyst. 2012, 8, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M.; Tompa, P.; Simon, I. Local structural disorder imparts plasticity on linear motifs. Bioinformatics 2007, 23, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Dinkel, H.; Van Roey, K.; Michael, S.; Davey, N.E.; Weatheritt, R.J.; Born, D.; Speck, T.; Kruger, D.; Grebnev, G.; Kuban, M.; et al. The eukaryotic linear motif resource elm: 10 years and counting. Nucleic Acids Res. 2014, 42, D259–D266. [Google Scholar] [CrossRef] [PubMed]
- Gould, C.M.; Diella, F.; Via, A.; Puntervoll, P.; Gemund, C.; Chabanis-Davidson, S.; Michael, S.; Sayadi, A.; Bryne, J.C.; Chica, C.; et al. Elm: The status of the 2010 eukaryotic linear motif resource. Nucleic Acids Res. 2010, 38, D167–D180. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Functional/Structural Characteristic 1 | Measure 1 | Results for Hubs Involved in Hub PPIs | Results for all Hub Interactors Involved in Hub PPIs | Results for Hub Interactors Involved in Hub PPIs that Exclude Hubs | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Avg for Disorder-Enriched Proteins | Avg for Remaining Proteins | p-Value | Relative Difference | Avg for Disorder-Enriched Proteins | Avg for Remaining Proteins | p-Value | Relative Difference | Avg for Disorder-Enriched Proteins | Avg for Remaining Proteins | p-Value | Relative Difference | ||
| Structural properties | Disorder content | 0.542 | 0.224 | <0.001 | 141% | 0.540 | 0.227 | <0.001 | 138% | 0.527 | 0.213 | <0.001 | 148% |
| Number of LIDPRs (per 1000 AAs) | 4.428 | 1.900 | <0.001 | 133% | 4.436 | 1.910 | <0.001 | 132% | 4.863 | 1.954 | <0.001 | 149% | |
| Number of disulfide links (per 1000 AAs) | 0.765 | 1.240 | <0.001 | −62% | 0.764 | 1.249 | <0.001 | −63% | 0.973 | 1.828 | <0.001 | −47% | |
| Sequence | Average protein chain length | 736.8 | 637.6 | <0.001 | 16% | 734.5 | 653.2 | <0.001 | 12% | 791.5 | 654.2 | <0.001 | 21% |
| Evolutionary conservation | Average conservation per protein | 0.920 | 1.069 | <0.001 | −16% | 0.930 | 1.053 | <0.001 | −13% | 0.965 | 1.048 | <0.001 | −8% |
| Average conservation of LIDPRs | 0.735 | 0.742 | 0.113 | −1% | 0.762 | 0.763 | 0.811 | 0% | 0.790 | 0.455 | <0.001 | 74% | |
| Functional regions | MoRF content | 0.014 | 0.008 | <0.001 | 68% | 0.014 | 0.008 | <0.001 | 67% | 0.018 | 0.012 | <0.001 | 53% |
| Number of MoRF regions (per 1000 AAs) | 1.843 | 1.090 | <0.001 | 69% | 1.834 | 1.096 | <0.001 | 67% | 2.398 | 1.577 | <0.001 | 52% | |
| Disordered protein binding (DPB) content | 0.230 | 0.092 | <0.001 | 149% | 0.231 | 0.093 | <0.001 | 148% | 0.201 | 0.074 | <0.001 | 171% | |
| Number of DPB regions (per 1000 AAs) | 8.764 | 3.665 | <0.001 | 139% | 8.784 | 3.663 | <0.001 | 140% | 8.402 | 3.270 | <0.001 | 157% | |
| Functional motifs (ELMs) | Proteolytic cleavage sites (per 1000 AAs) | 0.023 | 0.033 | 0.017 | −43% | 0.022 | 0.032 | 0.039 | −46% | 0.000 | 0.003 | 0.044 | −89% |
| Degradation sites (per 1000 AAs) | 0.014 | 0.019 | 0.051 | −31% | 0.014 | 0.018 | 0.016 | −32% | 0.003 | 0.004 | 0.342 | −40% | |
| Docking sites for catalysis (per 1000 AAs) | 0.137 | 0.074 | <0.001 | 85% | 0.137 | 0.074 | <0.001 | 85% | 0.010 | 0.005 | 0.031 | 88% | |
| Non-catalytic ligand binding sites (per 1000 AAs) | 0.278 | 0.240 | 0.026 | 15% | 0.257 | 0.234 | 0.061 | 10% | 0.119 | 0.043 | <0.001 | 174% | |
| PTM sites (per 1000 AAs) | 0.180 | 0.084 | <0.001 | 114% | 0.183 | 0.087 | <0.001 | 110% | 0.028 | 0.013 | 0.022 | 121% | |
| Targeting sites for localization (per 1000 AAs) | 0.149 | 0.053 | <0.001 | 182% | 0.146 | 0.058 | <0.001 | 151% | 0.000 | 0.008 | <0.001 | −100% | |
| PTMs | Number of PTMs (per 1000 AAs) | 413.3 | 275.3 | <0.001 | 50% | 412.5 | 275.8 | <0.001 | 50% | 410.4 | 277.9 | <0.001 | 48% |
| Number of residues with PTMs (per 1000 AAs) | 325.9 | 226.5 | <0.001 | 44% | 325.7 | 227.1 | <0.001 | 43% | 327.3 | 230.0 | <0.001 | 42% | |
| Acetylation sites (per 1000 AAs) | 9.603 | 7.954 | <0.001 | 21% | 9.481 | 7.919 | <0.001 | 20% | 8.951 | 7.759 | <0.001 | 15% | |
| ADP-ribosylation sites (per 1000 AAs) | 32.02 | 18.52 | <0.001 | 73% | 31.69 | 18.39 | <0.001 | 72% | 30.97 | 17.50 | <0.001 | 77% | |
| Amidation sites (per 1000 AAs) | 48.67 | 36.64 | <0.001 | 33% | 49.05 | 36.59 | <0.001 | 34% | 49.62 | 39.75 | <0.001 | 25% | |
| Carboxylation sites (per 1000 AAs) | 39.24 | 23.42 | <0.001 | 68% | 39.22 | 23.43 | <0.001 | 67% | 38.22 | 22.28 | <0.001 | 72% | |
| C-linked_glycosylation sites (per 1000 AAs) | 0.200 | 0.261 | <0.001 | −31% | 0.212 | 0.287 | <0.001 | −35% | 0.262 | 0.331 | 0.004 | −21% | |
| Farnesylation sites (per 1000 AAs) | 0.007 | 0.008 | 0.303 | −21% | 0.006 | 0.008 | 0.059 | −30% | 0.008 | 0.021 | 0.083 | −61% | |
| Geranylgeranylation sites (per 1000 AAs) | 0.008 | 0.010 | 0.059 | −32% | 0.007 | 0.009 | 0.045 | −40% | 0.011 | 0.031 | 0.226 | −63% | |
| GPI anchor amidation sites (per 1000 AAs) | 1.695 | 1.621 | 0.050 | 5% | 1.689 | 1.626 | 0.056 | 4% | 1.917 | 1.865 | 0.500 | 3% | |
| Hydroxylation sites (per 1000 AAs) | 26.75 | 12.56 | <0.001 | 113% | 26.98 | 13.16 | <0.001 | 105% | 26.18 | 12.99 | <0.001 | 101% | |
| Methylation sites (per 1000 AAs) | 15.41 | 10.27 | <0.001 | 50% | 15.33 | 10.27 | <0.001 | 49% | 13.98 | 9.97 | <0.001 | 40% | |
| Myristoylation sites (per 1000 AAs) | 0.012 | 0.017 | 0.002 | −39% | 0.014 | 0.013 | 0.908 | 1% | 0.021 | 0.015 | 0.222 | 41% | |
| N-linked glycosylation sites (per 1000 AAs) | 3.754 | 3.772 | 0.577 | 0% | 3.748 | 3.781 | 0.356 | −1% | 3.474 | 3.660 | 0.020 | −5% | |
| N-terminal acetylation sites (per 1000 AAs) | 0.525 | 0.517 | 0.345 | 2% | 0.542 | 0.515 | 0.035 | 5% | 0.769 | 0.797 | 0.321 | −3% | |
| O-linked glycosylation sites (per 1000 AAs) | 14.22 | 8.18 | <0.001 | 74% | 14.71 | 8.13 | <0.001 | 81% | 14.21 | 7.81 | <0.001 | 82% | |
| Palmitoylation sites (per 1000 AAs) | 1.334 | 1.734 | <0.001 | −30% | 1.342 | 1.786 | <0.001 | −33% | 1.783 | 2.751 | <0.001 | −35% | |
| Phosphorylation sites (per 1000 AAs) | 44.52 | 21.63 | <0.001 | 106% | 43.75 | 21.71 | <0.001 | 102% | 42.60 | 18.45 | <0.001 | 131% | |
| PUPylation sites (per 1000 AAs) | 1.949 | 2.603 | <0.001 | −34% | 1.920 | 2.633 | <0.001 | −37% | 1.838 | 2.436 | <0.001 | −25% | |
| Pyrrolidone carboxylic acid sites (per 1000 AAs) | 4.803 | 4.357 | <0.001 | 10% | 4.813 | 4.366 | <0.001 | 10% | 4.998 | 4.883 | 0.197 | 2% | |
| Sulfation sites (per 1000 AAs) | 3.589 | 2.291 | <0.001 | 57% | 3.603 | 2.305 | <0.001 | 56% | 3.148 | 2.026 | <0.001 | 55% | |
| SUMOylation sites (per 1000 AAs) | 6.844 | 6.484 | 0.003 | 6% | 6.876 | 6.467 | <0.001 | 6% | 6.457 | 6.682 | 0.035 | −3% | |
| Ubiquitination sites (per 1000 AAs) | 9.377 | 6.924 | <0.001 | 35% | 9.378 | 6.984 | <0.001 | 34% | 9.194 | 6.931 | <0.001 | 33% | |
| Disease Class (ID at the Second MeSH Level) | Average for Disorder-Enriched Proteins | Average for all Human Proteins | p-Value | Relative Difference | |
|---|---|---|---|---|---|
| ALL Diseases | 0.519 | 0.433 | <0.001 | 20% | |
| Specific disease classes | Neoplasms, including cancers (C04) | 3.150 | 0.436 | <0.001 | 622% |
| Stomatognathic Diseases (C07) | 1.833 | 0.437 | <0.001 | 320% | |
| Endocrine System Diseases (C19) | 1.594 | 0.432 | <0.001 | 269% | |
| Digestive System Diseases (C06) | 1.440 | 0.436 | <0.001 | 230% | |
| Respiratory Tract Diseases (C08) | 1.060 | 0.437 | <0.001 | 143% | |
| Female Urogenital Diseases and Pregnancy Complications (C13) | 0.855 | 0.437 | <0.001 | 96% | |
| Nervous System Diseases (C10) | 0.775 | 0.432 | <0.001 | 79% | |
| Musculoskeletal Diseases (C05) | 0.654 | 0.433 | <0.001 | 51% | |
| Hemic and Lymphatic Diseases (C15) | 0.543 | 0.434 | <0.001 | 25% | |
| Pathological Conditions, Signs and Symptoms (C23) | 0.467 | 0.434 | <0.001 | 8% | |
| Congenital, Hereditary, and Neonatal Diseases and Abnormalities (C16) | 0.456 | 0.433 | <0.001 | 5% | |
| Male Urogenital Diseases (C12) | 0.459 | 0.436 | 0.009 | 5% | |
| Immune System Diseases (C20) | 0.409 | 0.435 | 0.81 | −6% | |
| Eye Diseases (C11) | 0.373 | 0.437 | <0.001 | −17% | |
| Cardiovascular Diseases (C14) | 0.347 | 0.437 | <0.001 | −26% | |
| Nutritional and Metabolic Diseases (C18) | 0.248 | 0.433 | <0.001 | −75% | |
| Skin and Connective Tissue Diseases (C17) | 0.247 | 0.435 | <0.001 | −76% | |
| Otorhinolaryngologic Diseases (C09) | 0.162 | 0.431 | <0.001 | −165% | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, G.; Wu, Z.; Uversky, V.N.; Kurgan, L. Functional Analysis of Human Hub Proteins and Their Interactors Involved in the Intrinsic Disorder-Enriched Interactions. Int. J. Mol. Sci. 2017, 18, 2761. https://doi.org/10.3390/ijms18122761
Hu G, Wu Z, Uversky VN, Kurgan L. Functional Analysis of Human Hub Proteins and Their Interactors Involved in the Intrinsic Disorder-Enriched Interactions. International Journal of Molecular Sciences. 2017; 18(12):2761. https://doi.org/10.3390/ijms18122761
Chicago/Turabian StyleHu, Gang, Zhonghua Wu, Vladimir N. Uversky, and Lukasz Kurgan. 2017. "Functional Analysis of Human Hub Proteins and Their Interactors Involved in the Intrinsic Disorder-Enriched Interactions" International Journal of Molecular Sciences 18, no. 12: 2761. https://doi.org/10.3390/ijms18122761
APA StyleHu, G., Wu, Z., Uversky, V. N., & Kurgan, L. (2017). Functional Analysis of Human Hub Proteins and Their Interactors Involved in the Intrinsic Disorder-Enriched Interactions. International Journal of Molecular Sciences, 18(12), 2761. https://doi.org/10.3390/ijms18122761