
Reduced SHARPIN and LUBAC Formation May Contribute to CCl4- or Acetaminophen-Induced Liver Cirrhosis in Mice

 ,
,
Abstract
:1. Introduction
2. Results
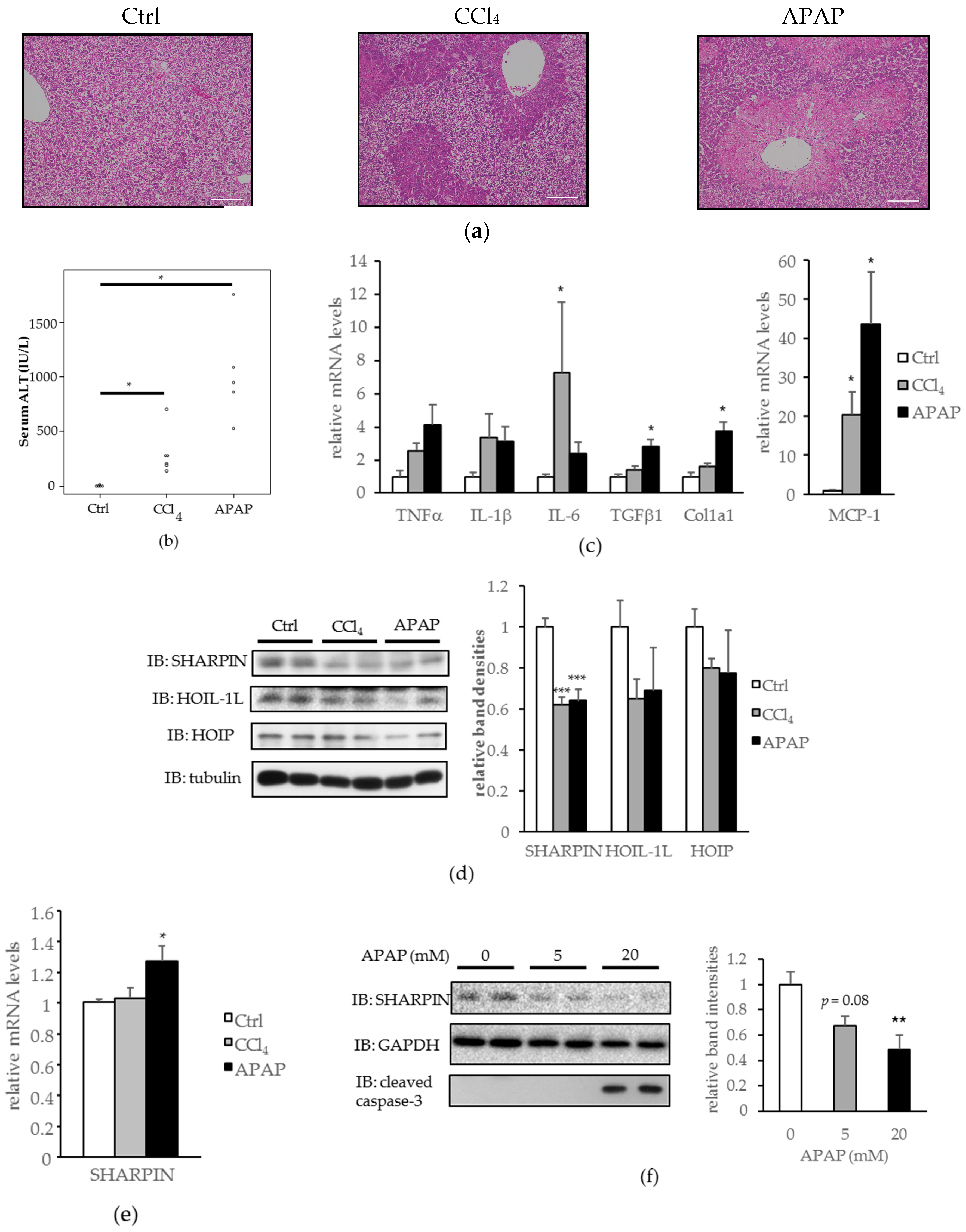
2.1. SHANK-Associated RH Domain-Interacting Protein (SHARPIN) Expression Was Downregulated in the Livers of Carbon Tetrachloride (CCl4) and Acetaminophen (APAP) Induced Acute Liver Injury Models, and in Hepa1-6 Cells Treated with APAP
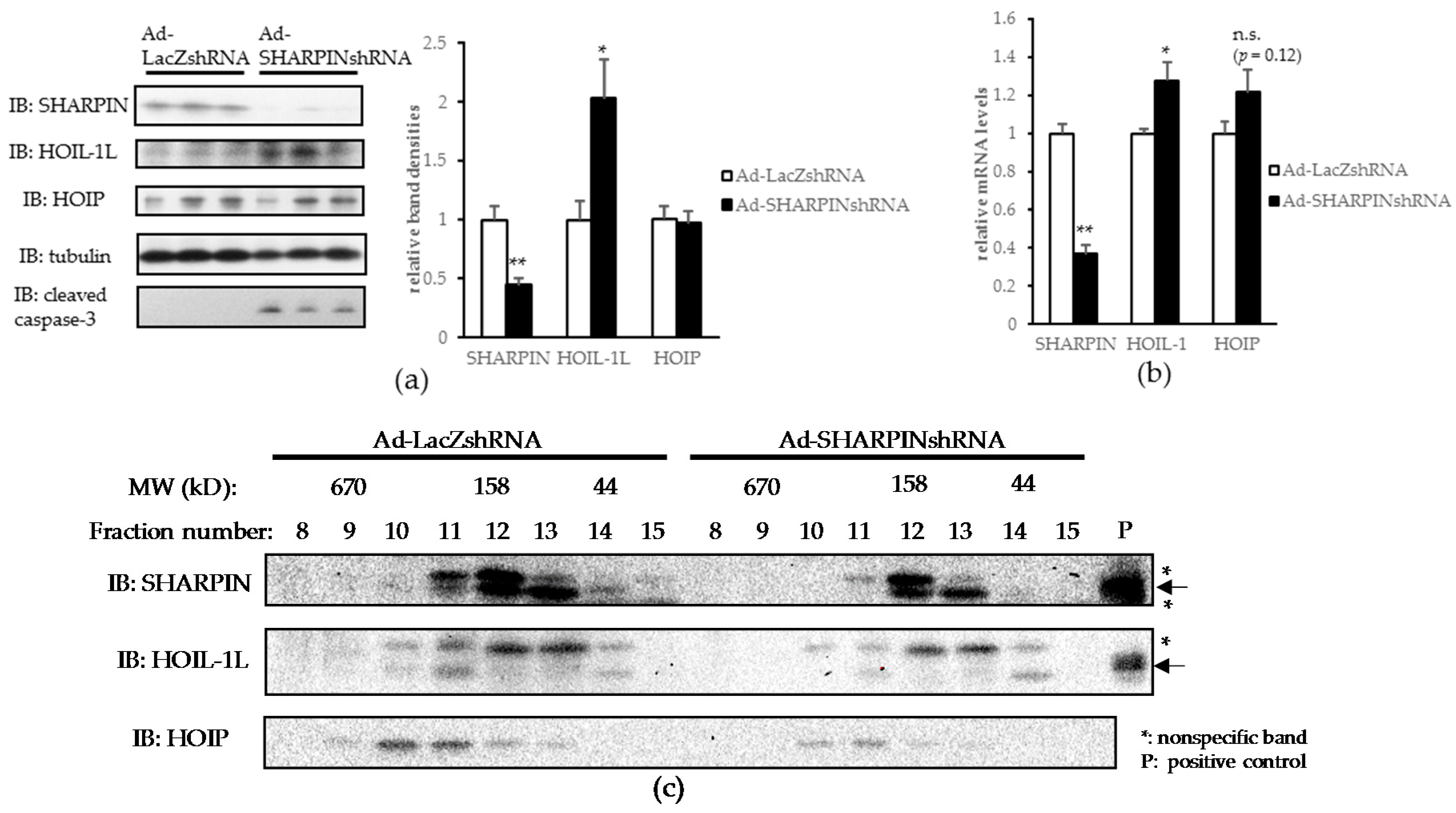
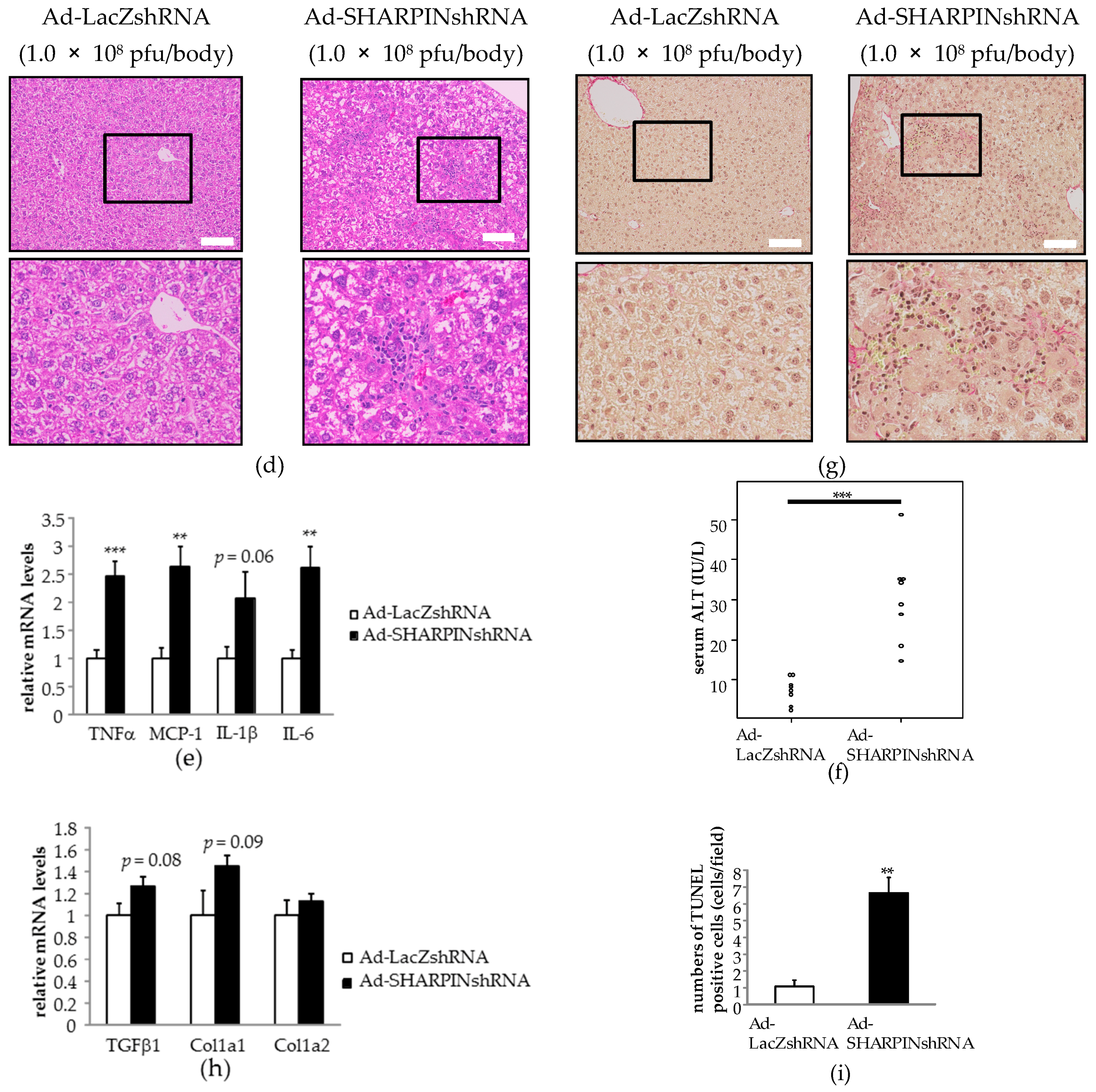
2.2. Hepatic SHARPIN Knockdown Induces Apoptosis in Hepatocytes and Leads to Inflammation and Fibrosis in the Liver
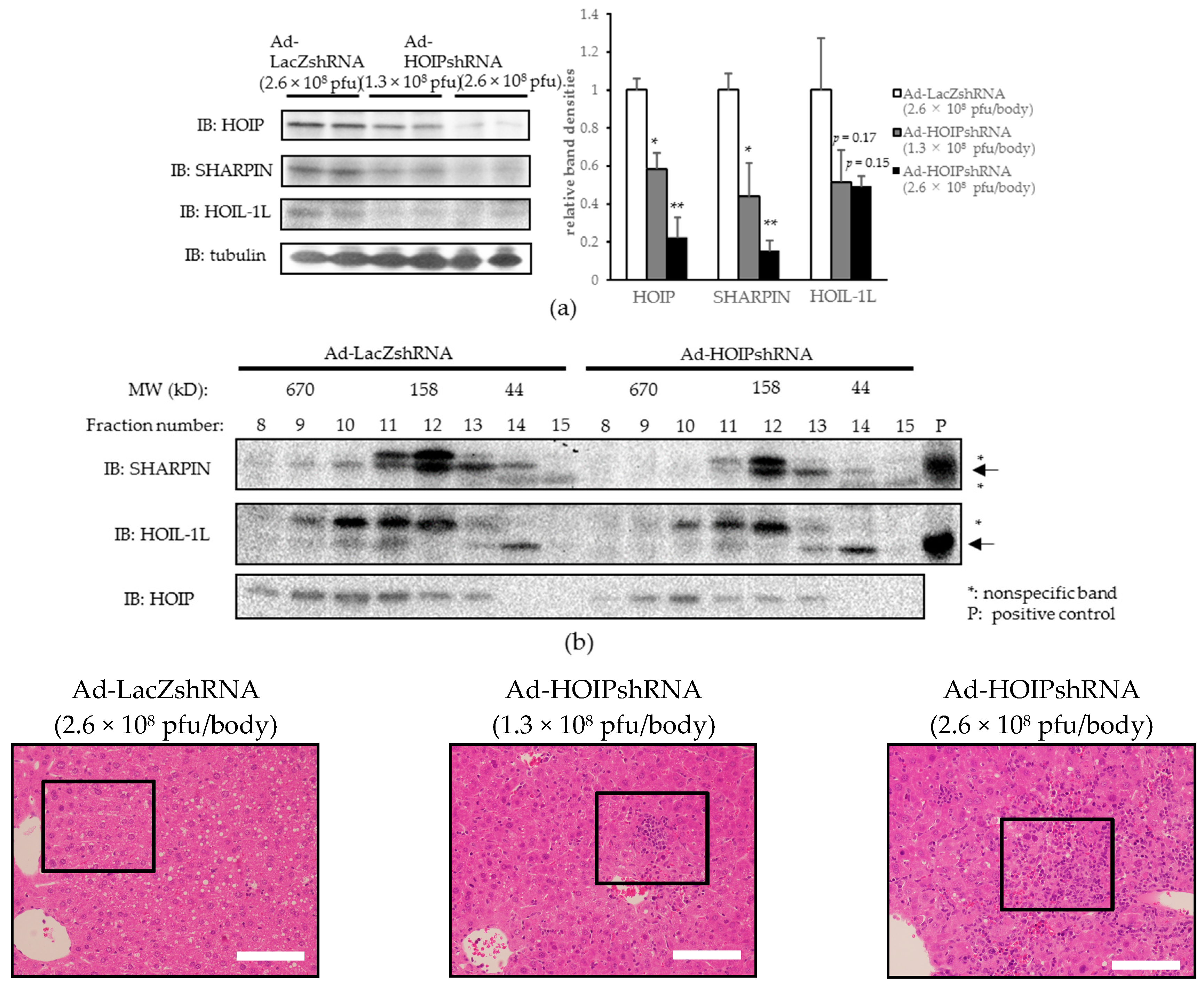
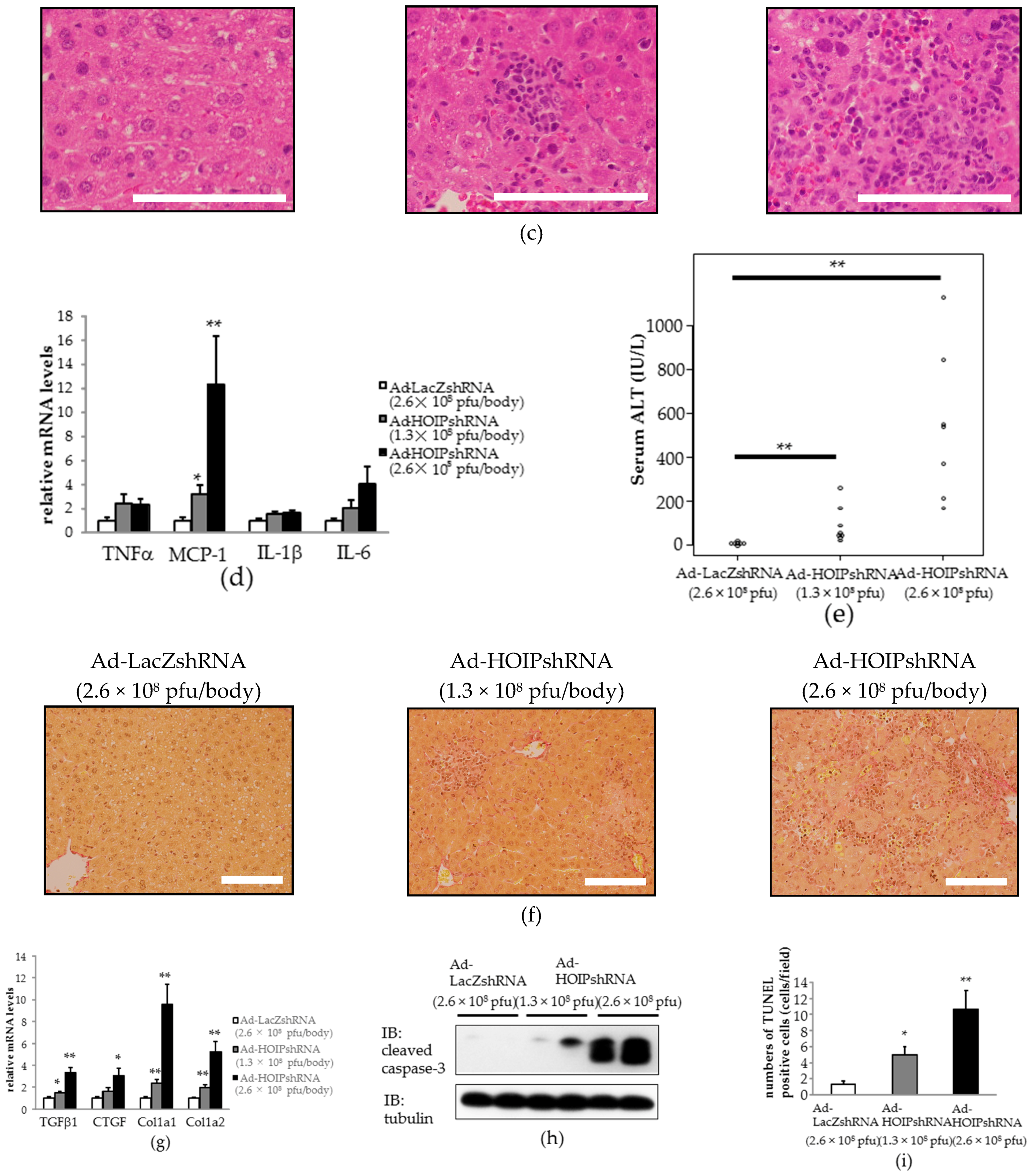
2.3. Hepatic HOIL-1L Interacting Protein (HOIP) Knockdown Also Induces Hepatocyte Apoptosis, Severe Inflammation, and Fibrosis in the Liver
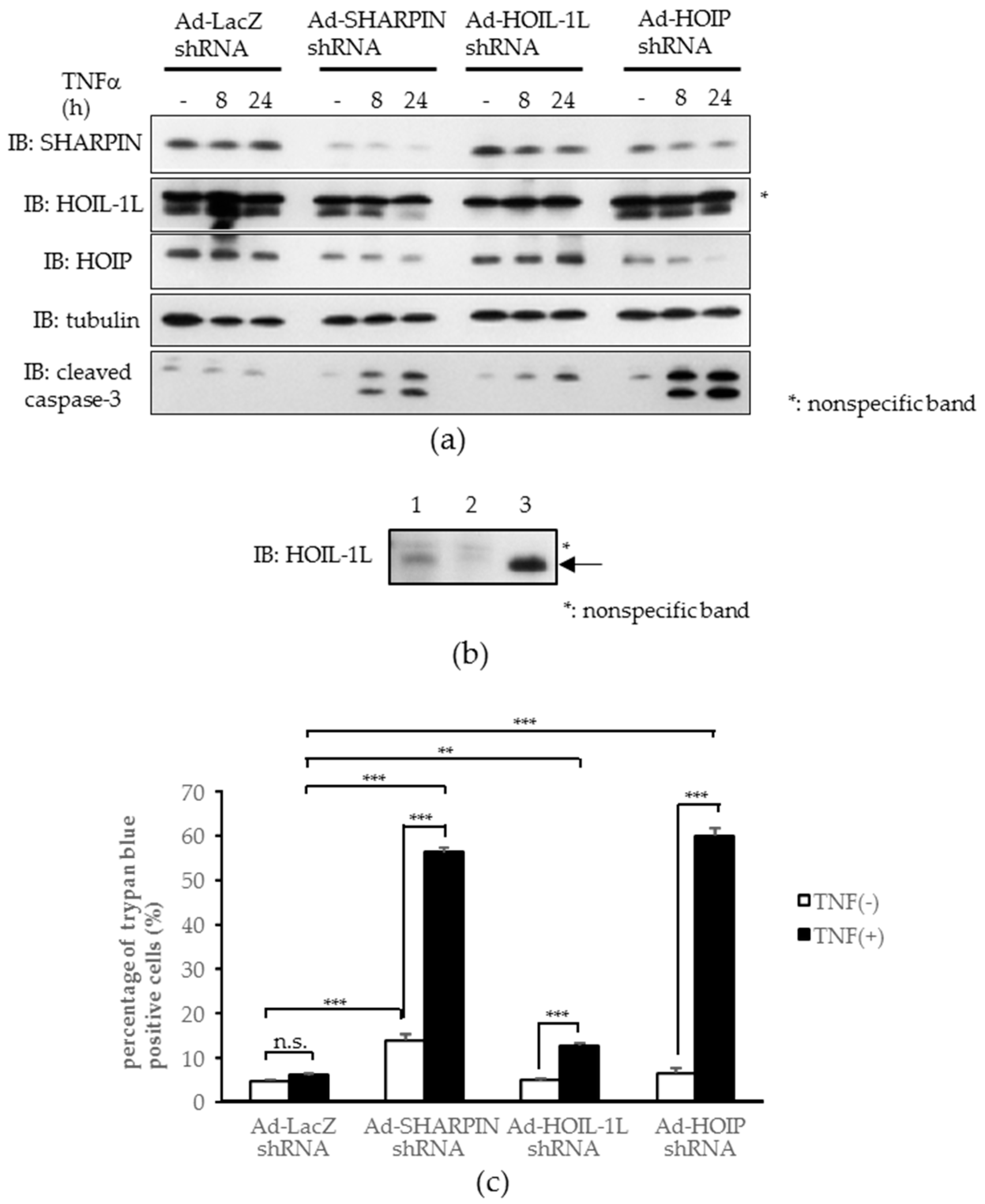
2.4. Knockdown of Each Component of Linear Ubiquitin Chain Assembly Complex (LUBAC) Leads to Increased Tumor Necrosis Factor-α (TNFα)-induced Apoptosis in Hepatoma Cells In Vitro
3. Discussion
4. Materials and Methods
4.1. Animals and Experimental Protocols
4.2. Histological Studies
4.3. Serum Investigations
4.4. Cell Culture and Cell Death Assay
4.5. Antibodies
4.6. Western Blotting
4.7. RNAi Interference
4.8. Real-Time PCR (Polymerase Chain Reaction)
4.9. Gel Filtration
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| LUBAC | Linear ubiquitin chain assembly complex |
| SHARPIN | SHANK-associated RH domain-interacting protein |
| HOIL-1L | Longer isoform of heme-oxidized iron-regulatory protein 2 ubiquitin ligase-1 |
| HOIP | HOIL-1L interacting protein |
| NASH | Non-alcoholic steatohepatitis |
| APAP | Acetaminophen |
References
- Kirisako, T.; Kamei, K.; Murata, S.; Kato, M.; Fukumoto, H.; Kanie, M.; Sano, S.; Tokunaga, F.; Tanaka, K.; Iwai, K. A ubiquitin ligase complex assembles linear polyubiquitin chains. Embo. J. 2006, 25, 4877–4887. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, F.; Sakata, S.; Saeki, Y.; Satomi, Y.; Kirisako, T.; Kamei, K.; Nakagawa, T.; Kato, M.; Murata, S.; Yamaoka, S.; et al. Involvement of linear polyubiquitylation of NEMO in NF-κB activation. Nat. Cell. Biol. 2009, 11, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, F.; Nakagawa, T.; Nakahara, M.; Saeki, Y.; Taniguchi, M.; Sakata, S.; Tanaka, K.; Nakano, H.; Iwai, K. SHARPIN is a component of the NF-κB-activating linear ubiquitin chain assembly complex. Nature 2011, 471, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, F.; Deribe, Y.L.; Skanland, S.S.; Stieglitz, B.; Grabbe, C.; Franz-Wachtel, M.; van Wijk, S.J.; Goswami, P.; Nagy, V.; Terzic, J.; et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-κB activity and apoptosis. Nature 2011, 471, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, B.; Cordier, S.M.; Schmukle, A.C.; Emmerich, C.H.; Rieser, E.; Haas, T.L.; Webb, A.I.; Rickard, J.A.; Anderton, H.; Wong, W.W.; et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 2011, 471, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Tosello-Trampont, A.C.; Landes, S.G.; Nguyen, V.; Novobrantseva, T.I.; Hahn, Y.S. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-α production. J. Biol. Chem. 2012, 287, 40161–40172. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Yang, L.; van Rooijen, N.; Brenner, D.A.; Ohnishi, H.; Seki, E. Toll-like receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation in mice. Hepatology 2013, 57, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Jarrar, M.H.; Baranova, A.; Collantes, R.; Ranard, B.; Stepanova, M.; Bennett, C.; Fang, Y.; Elariny, H.; Goodman, Z.; Chandhoke, V.; et al. Adipokines and cytokines in non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2008, 27, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Martius, G.; Alwahsh, S.M.; Rave-Frank, M.; Hess, C.F.; Christiansen, H.; Ramadori, G.; Malik, I.A. Hepatic fat accumulation and regulation of FAT/CD36: An effect of hepatic irradiation. Int. J. Clin. Exp. Pathol. 2014, 7, 5379–5392. [Google Scholar] [PubMed]
- Schoemaker, M.H.; Ros, J.E.; Homan, M.; Trautwein, C.; Liston, P.; Poelstra, K.; van Goor, H.; Jansen, P.L.; Moshage, H. Cytokine regulation of pro- and anti-apoptotic genes in rat hepatocytes: NF-κB-regulated inhibitor of apoptosis protein 2 (cIAP2) prevents apoptosis. J. Hepatol. 2002, 36, 742–750. [Google Scholar] [CrossRef]
- Wang, K.; Lin, B. Inhibitor of apoptosis proteins (IAPs) as regulatory factors of hepatic apoptosis. Cell. Signal. 2013, 25, 1970–1980. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, Y.; Nakatsu, Y.; Fukushima, T.; Okubo, H.; Iwashita, M.; Sakoda, H.; Fujishiro, M.; Yamamotoya, T.; Kushiyama, A.; Takahashi, S.; et al. LUBAC formation is impaired in the livers of mice with MCD-dependent nonalcoholic steatohepatitis. Mediators Inflamm. 2015, 2015, 125380. [Google Scholar] [CrossRef] [PubMed]
- Alwahsh, S.M.; Xu, M.; Seyhan, H.A.; Ahmad, S.; Mihm, S.; Ramadori, G.; Schultze, F.C. Diet high in fructose leads to an overexpression of lipocalin-2 in rat fatty liver. World J. Gastroenterol. 2014, 20, 1807–1821. [Google Scholar] [CrossRef] [PubMed]
- Jegatheesan, P.; Beutheu, S.; Freese, K.; Waligora-Dupriet, A.J.; Nubret, E.; Butel, M.J.; Bergheim, I.; de Bandt, J.P. Preventive effects of citrulline on western diet-induced non-alcoholic fatty liver disease in rats. Br. J. Nutr. 2016, 116, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Maeda, S.; Hikiba, Y.; Ohmae, T.; Shibata, W.; Yanai, A.; Sakamoto, K.; Ogura, K.; Noguchi, T.; Karin, M.; et al. Deletion of apoptosis signal-regulating kinase 1 attenuates acetaminophen-induced liver injury by inhibiting c-jun N-terminal kinase activation. Gastroenterology 2008, 135, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology 2014, 147, 765.e4–783.e4. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Sano, S.; Nakahara, M.; Murata, S.; Kometani, K.; Aiba, Y.; Sakamoto, S.; Watanabe, Y.; Tanaka, K.; Kurosaki, T.; et al. Defective immune responses in mice lacking LUBAC-mediated linear ubiquitination in B cells. Embo. J. 2013, 32, 2463–2476. [Google Scholar] [CrossRef] [PubMed]
- Peltzer, N.; Rieser, E.; Taraborrelli, L.; Draber, P.; Darding, M.; Pernaute, B.; Shimizu, Y.; Sarr, A.; Draberova, H.; Montinaro, A.; et al. HOIP deficiency causes embryonic lethality by aberrant TNFR1-mediated endothelial cell death. Cell Rep. 2014, 9, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, A.E.; Gores, G.J. Apoptosis in alcoholic and nonalcoholic steatohepatitis. Front. Biosci. 2005, 10, 3093–3099. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Williams, C.D.; Ramachandran, A.; Bajt, M.L. Acetaminophen hepatotoxicity and repair: The role of sterile inflammation and innate immunity. Liver Int. 2012, 32, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Ingawale, D.K.; Mandlik, S.K.; Naik, S.R. Models of hepatotoxicity and the underlying cellular, biochemical and immunological mechanism(s): A critical discussion. Environ. Toxicol. Pharmacol. 2014, 37, 118–133. [Google Scholar] [CrossRef] [PubMed]
- Kudo, H.; Takahara, T.; Yata, Y.; Kawai, K.; Zhang, W.; Sugiyama, T. Lipopolysaccharide triggered TNF-α-induced hepatocyte apoptosis in a murine non-alcoholic steatohepatitis model. J. Hepatol. 2009, 51, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Imajo, K.; Fujita, K.; Yoneda, M.; Nozaki, Y.; Ogawa, Y.; Shinohara, Y.; Kato, S.; Mawatari, H.; Shibata, W.; Kitani, H.; et al. Hyperresponsivity to low-dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin-mediated signaling. Cell Metab. 2012, 16, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Alwahsh, S.M.; Gebhardt, R. Dietary fructose as a risk factor for non-alcoholic fatty liver disease (NAFLD). Arch. Toxicol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kovalovich, K.; DeAngelis, R.A.; Li, W.; Furth, E.E.; Ciliberto, G.; Taub, R. Increased toxin-induced liver injury and fibrosis in interleukin-6-deficient mice. Hepatology 2000, 31, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Cressman, D.E.; Greenbaum, L.E.; DeAngelis, R.A.; Ciliberto, G.; Furth, E.E.; Poli, V.; Taub, R. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science 1996, 274, 1379–1383. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Alwahsh, S.M.; Ramadori, G.; Kollmar, O.; Slotta, J.E. Upregulation of hepatic melanocortin 4 receptor during rat liver regeneration. J. Surg. Res. 2016, 203, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Starkey Lewis, P.J.; Dear, J.; Platt, V.; Simpson, K.J.; Craig, D.G.; Antoine, D.J.; French, N.S.; Dhaun, N.; Webb, D.J.; Costello, E.M.; et al. Circulating microRNAs as potential markers of human drug-induced liver injury. Hepatology 2011, 54, 1767–1776. [Google Scholar] [CrossRef] [PubMed]
- Kia, R.; Kelly, L.; Sison-Young, R.L.; Zhang, F.; Pridgeon, C.S.; Heslop, J.A.; Metcalfe, P.; Kitteringham, N.R.; Baxter, M.; Harrison, S.; et al. MicroRNA-122: A novel hepatocyte-enriched in vitro marker of drug-induced cellular toxicity. Toxicol. Sci. 2015, 144, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Eakins, R.; Walsh, J.; Randle, L.; Jenkins, R.E.; Schuppe-Koistinen, I.; Rowe, C.; Starkey Lewis, P.; Vasieva, O.; Prats, N.; Brillant, N.; et al. Adaptation to acetaminophen exposure elicits major changes in expression and distribution of the hepatic proteome. Sci. Rep. 2015, 5, 16423. [Google Scholar] [CrossRef] [PubMed]
- Ilavenil, S.; Al-Dhabi, N.A.; Srigopalram, S.; Ock Kim, Y.; Agastian, P.; Baru, R.; Choi, K.C.; Valan Arasu, M. Acetaminophen induced hepatotoxicity in wistar rats—A proteomic approach. Molecules 2016, 21, 161. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Sets for Real-Time PCR |
|---|
| mSHARPIN-F: CCTGTGTATGCCTGAACGAA |
| mSHARPIN-R: AGAGGATCCCAAGCACAGG |
| mHOIL-1-F: TCTCCCCAACACAGGACATC |
| mHOIL-1-R: AAATGGTGACGGTGTGCAT |
| mHOIP-F: CCCAGTGTCACCAGACCTTC |
| mHOIP-R: CCTCACAACTCCGTCCTCTG |
| mTNFα-F: GAACTGGCAGAAGAGGCACT |
| mTNFα-R: AGGGTCTGGGCCATAGAACT |
| mMCP-1-F: AGGTCCCTGTCATGCTTCTG |
| mMCP-1-R: TCTGGACCCATTCCTTCTTG |
| mIL-1β-F: TGACGGACCCCAAAAGATG |
| mIL-1β-R: TGGACAGCCCAGGTCAAAG |
| mIL-6-F: TCGTGGAAATGAGAAAAGAGTTG |
| mIL-6-R: AGTGCATCATCGTTGTTCATACA |
| mTGF-β1-F: AAGTTGGCATGGTAGCCCTT |
| mTGF-β1-R: GCCCTGGATACCAACTATTGC |
| mCTGF-F: AGGGCCTCTTCTGCGATTTC |
| mCTGF-R: CATTTCCCAGGCAGCTTGAC |
| mCollagen1a1-F: TCATCGTGGCTTCTCTGGT |
| mCollagen1a1-R: GACCGTTGAGTCCGTCTTTG |
| mCollagen1a2-F: CCGTGCTTCTCAGAACATCA |
| mCollagen1a2-R: GAGCAGCCATCGACTAGGAC |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamamotoya, T.; Nakatsu, Y.; Matsunaga, Y.; Fukushima, T.; Yamazaki, H.; Kaneko, S.; Fujishiro, M.; Kikuchi, T.; Kushiyama, A.; Tokunaga, F.; et al. Reduced SHARPIN and LUBAC Formation May Contribute to CCl4- or Acetaminophen-Induced Liver Cirrhosis in Mice. Int. J. Mol. Sci. 2017, 18, 326. https://doi.org/10.3390/ijms18020326
Yamamotoya T, Nakatsu Y, Matsunaga Y, Fukushima T, Yamazaki H, Kaneko S, Fujishiro M, Kikuchi T, Kushiyama A, Tokunaga F, et al. Reduced SHARPIN and LUBAC Formation May Contribute to CCl4- or Acetaminophen-Induced Liver Cirrhosis in Mice. International Journal of Molecular Sciences. 2017; 18(2):326. https://doi.org/10.3390/ijms18020326
Chicago/Turabian StyleYamamotoya, Takeshi, Yusuke Nakatsu, Yasuka Matsunaga, Toshiaki Fukushima, Hiroki Yamazaki, Sunao Kaneko, Midori Fujishiro, Takako Kikuchi, Akifumi Kushiyama, Fuminori Tokunaga, and et al. 2017. "Reduced SHARPIN and LUBAC Formation May Contribute to CCl4- or Acetaminophen-Induced Liver Cirrhosis in Mice" International Journal of Molecular Sciences 18, no. 2: 326. https://doi.org/10.3390/ijms18020326
APA StyleYamamotoya, T., Nakatsu, Y., Matsunaga, Y., Fukushima, T., Yamazaki, H., Kaneko, S., Fujishiro, M., Kikuchi, T., Kushiyama, A., Tokunaga, F., Asano, T., & Sakoda, H. (2017). Reduced SHARPIN and LUBAC Formation May Contribute to CCl4- or Acetaminophen-Induced Liver Cirrhosis in Mice. International Journal of Molecular Sciences, 18(2), 326. https://doi.org/10.3390/ijms18020326