
Promising Targets for Cancer Immunotherapy: TLRs, RLRs, and STING-Mediated Innate Immune Pathways

Abstract
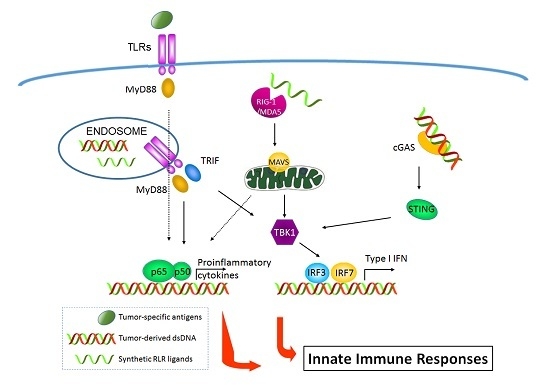
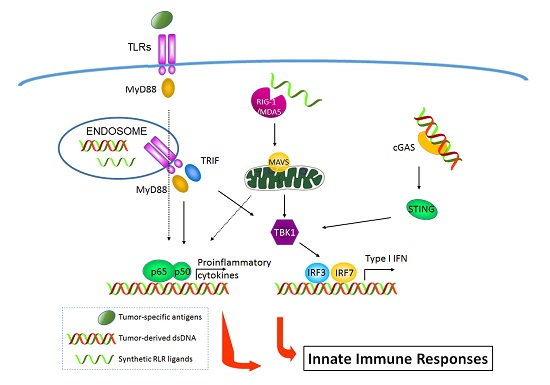
:
1. Introduction
2. Targeting Toll-Like Receptors in Cancer Immunotherapy
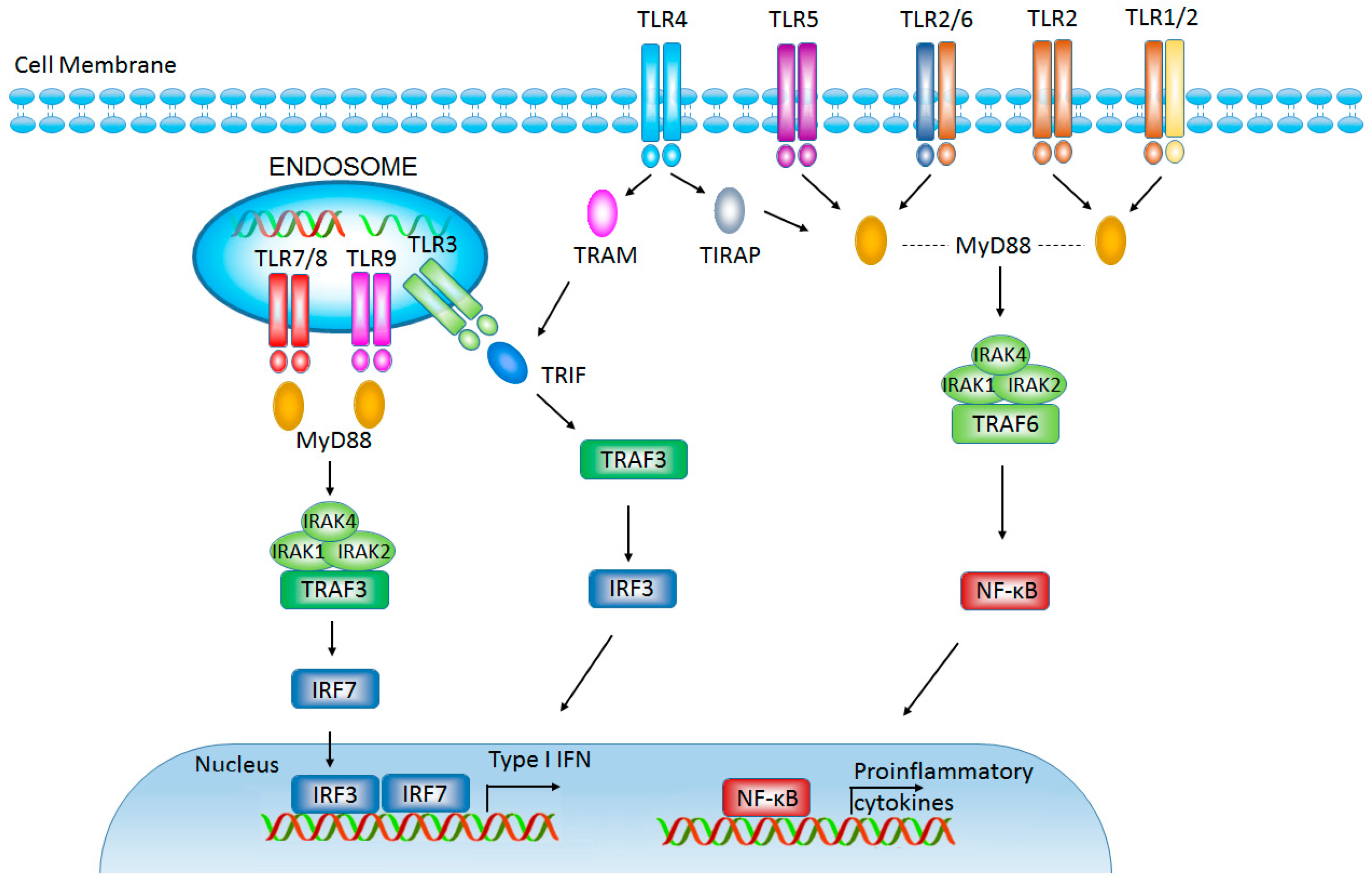
2.1. Toll-Like Receptors
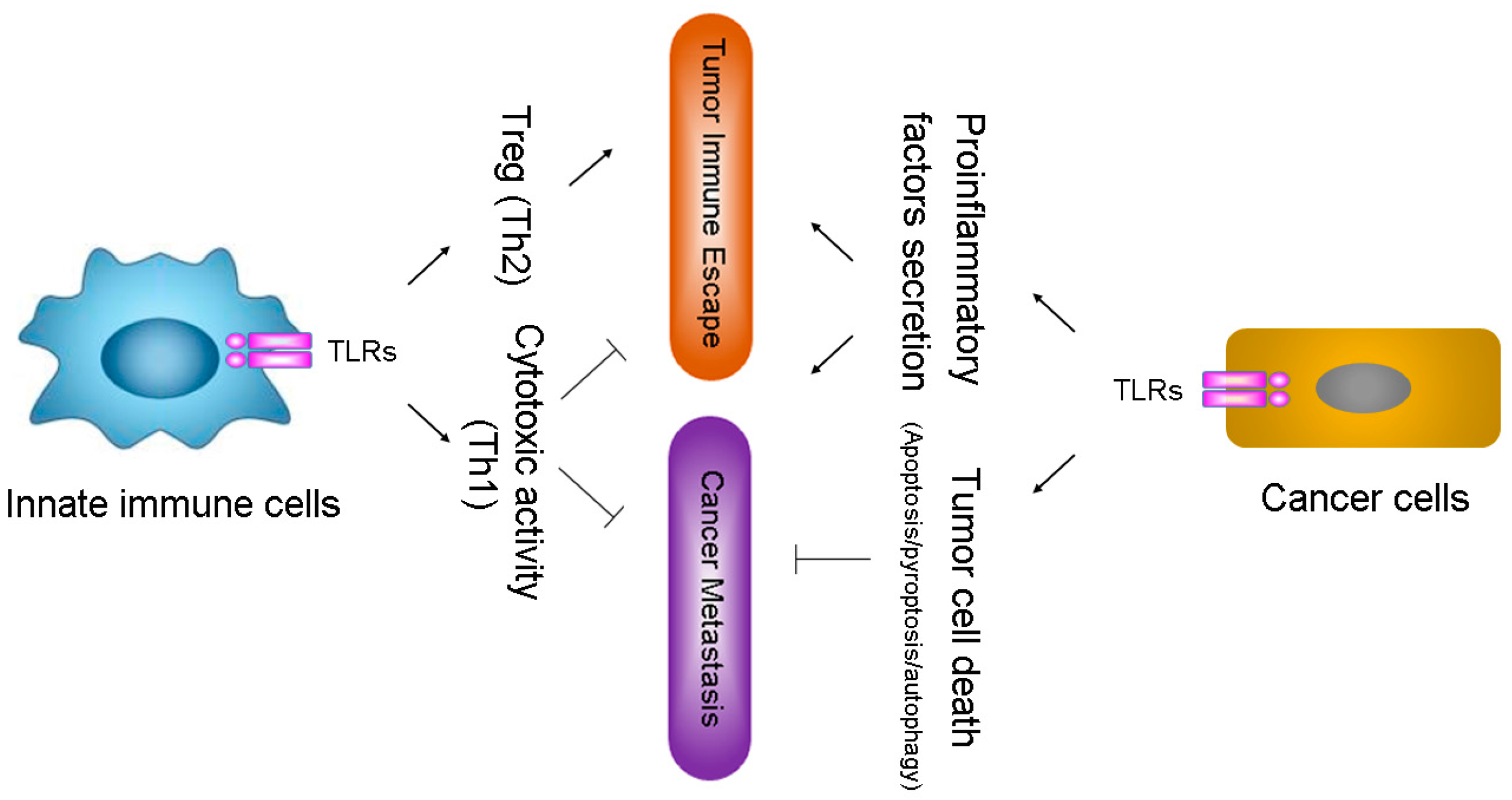
2.2. TLRs Influence Tumor Evasion from Immune Surveillance
2.3. TLRs Facilitate Inflaming Metastasis of Cancer
2.4. Triggering of TLRs Induces Strong Immune Response against Multiple Cancers
3. Antitumor Properties of RIG-I-Like Receptors (RLR) Signaling
3.1. RIG-I-Like Receptors (RLRs)
3.2. Activation of RLRs Directly Triggers Cancer Cell Death
3.3. Triggering of RLRs Signaling for Cancer Immunotherapy
4. Targeting Stimulator of Interferon Genes (STING) Pathway for Cancer Immunotherapy
4.1. STING Signaling Pathway
4.2. Activation of STING Pathway Directly Triggers Cancer Cell Death
4.3. The Role of STING in Cancer Immunotherapy
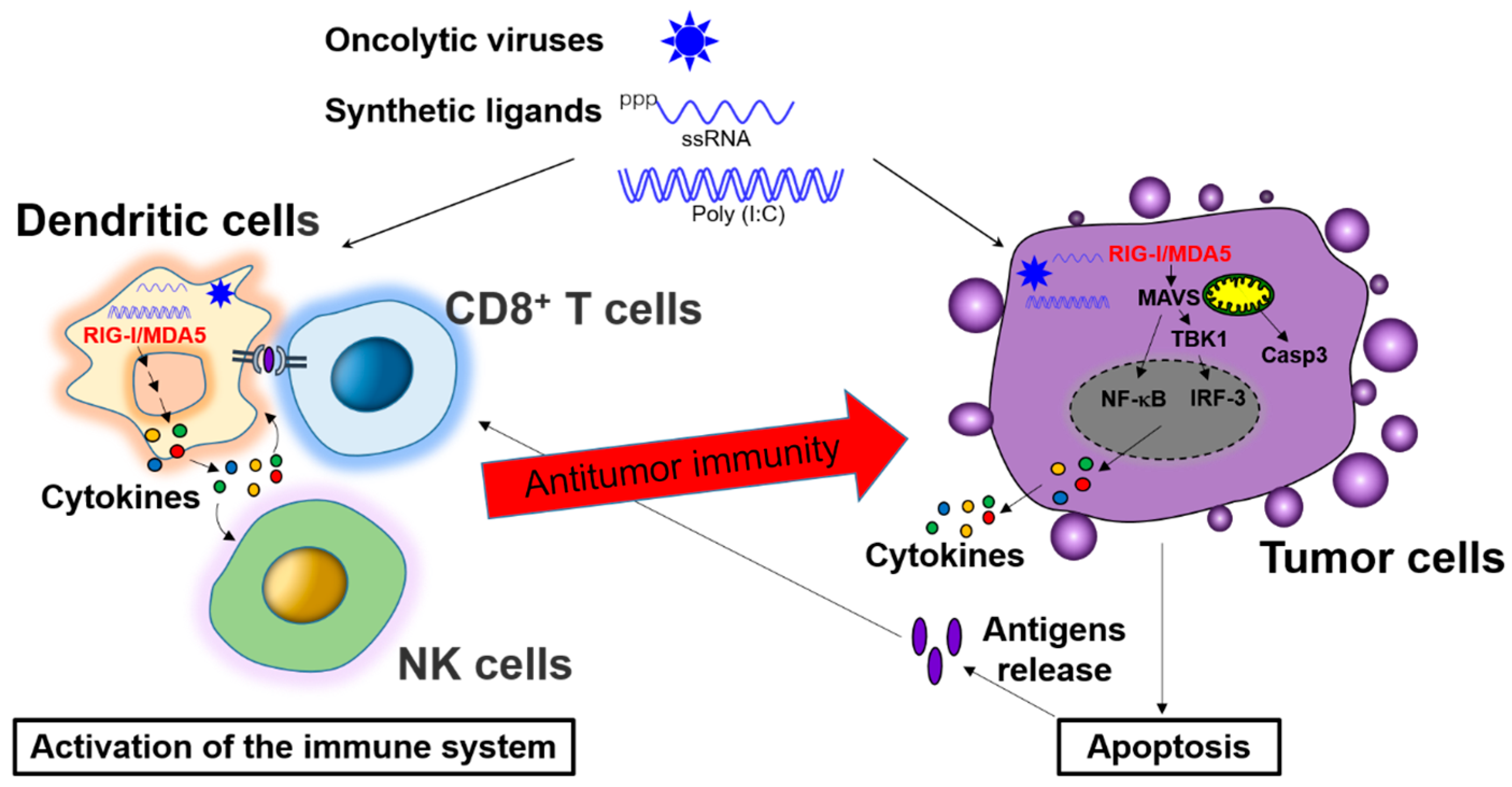
5. Combined Cancer Immunotherapies with Synergistic Activation of Innate and Adaptive Immune Responses
5.1. Activation of Adaptive Immune Responses by Innate Immunity in Cancer Therapy
5.2. Enhancement of Innate Immune Effectors Activation by Adaptive Immunity
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Travis, J. Origins. On the origin of the immune system. Science 2009, 324, 580–582. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Pattern recognition theory and the launch of modern innate immunity. J. Immunol. 2013, 191, 4473–4474. [Google Scholar] [CrossRef] [PubMed]
- Matzinger, P. The danger model: A renewed sense of self. Science 2002, 296, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Marcus, A.; Gowen, B.G.; Thompson, T.W.; Iannello, A.; Ardolino, M.; Deng, W.; Wang, L.; Shifrin, N.; Raulet, D.H. Recognition of tumors by the innate immune system and natural killer cells. Adv. Immunol. 2014, 122, 91–128. [Google Scholar] [PubMed]
- Mocellin, S.; Nitti, D. Therapeutics targeting tumor immune escape: Towards the development of new generation anticancer vaccines. Med. Res. Rev. 2008, 28, 413–444. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.R.; Corrales, L.; Gajewski, T.F. Innate immune recognition of cancer. Annu. Rev. Immunol. 2015, 33, 445–474. [Google Scholar] [CrossRef] [PubMed]
- Rakoff-Nahoum, S.; Medzhitov, R. Toll-like receptors and cancer. Nat. Rev. Cancer 2009, 9, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Poltorak, A.; Kurmyshkina, O.; Volkova, T. Stimulator of interferon genes (STING): A “new chapter” in virus-associated cancer research. Lessons from wild-derived mouse models of innate immunity. Cytokine Growth Factor Rev. 2016, 29, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.S.; Xu, F. Anticancer function of polyinosinic-polycytidylic acid. Cancer Biol. Ther. 2010, 10, 1219–1223. [Google Scholar] [CrossRef] [PubMed]
- Kottke, T.; Boisgerault, N.; Diaz, R.M.; Donnelly, O.; Rommelfanger-Konkol, D.; Pulido, J.; Thompson, J.; Mukhopadhyay, D.; Kaspar, R.; Coffey, M.; et al. Detecting and targeting tumor relapse by its resistance to innate effectors at early recurrence. Nat. Med. 2013, 19, 1625–1631. [Google Scholar] [CrossRef] [PubMed]
- Berraondo, P.; Nouze, C.; Preville, X.; Ladant, D.; Leclerc, C. Eradication of large tumors in mice by a tritherapy targeting the innate, adaptive, and regulatory components of the immune system. Cancer Res. 2007, 67, 8847–8855. [Google Scholar] [CrossRef] [PubMed]
- Moynihan, K.D.; Opel, C.F.; Szeto, G.L.; Tzeng, A.; Zhu, E.F.; Engreitz, J.M.; Williams, R.T.; Rakhra, K.; Zhang, M.H.; Rothschilds, A.M.; et al. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat. Med. 2016, 22, 1402–1410. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, Y.; Zhang, Q.; Wang, F.; Zhang, D. Toll-like receptors and prostate cancer. Front. Immunol. 2014, 5, 352. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhang, G.; Hayden, M.S.; Greenblatt, M.B.; Bussey, C.; Flavell, R.A.; Ghosh, S. A Toll-like receptor that prevents infection by uropathogenic bacteria. Science 2004, 303, 1522–1526. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Zhang, Y.; Liu, L.; Zhang, T.; Han, F.; Cleveland, J.; Wang, F.; McKeehan, W.L.; Li, Y.; Zhang, D. MAP1S protein regulates the phagocytosis of bacteria and Toll-like receptor (TLR) Signaling. J. Biol. Chem. 2016, 291, 1243–1250. [Google Scholar] [CrossRef]
- Kim, S.; Takahashi, H.; Lin, W.W.; Descargues, P.; Grivennikov, S.; Kim, Y.; Luo, J.L.; Karin, M. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 2009, 457, 102–106. [Google Scholar] [CrossRef]
- Dybdahl, B.; Wahba, A.; Lien, E.; Flo, T.H.; Waage, A.; Qureshi, N.; Sellevold, O.F.; Espevik, T.; Sundan, A. Inflammatory response after open heart surgery: Release of heat-shock protein 70 and signaling through Toll-like receptor-4. Circulation 2002, 105, 685–690. [Google Scholar] [CrossRef]
- Herr, H.W.; Morales, A. History of bacillus Calmette-Guerin and bladder cancer: An immunotherapy success story. J. Urol. 2008, 179, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Luckett, R.; Feldman, S. Impact of 2-, 4- and 9-valent HPV vaccines on morbidity and mortality from cervical cancer. Hum. Vaccin. Immunother. 2016, 12, 1332–1342. [Google Scholar] [CrossRef]
- Shi, M.; Chen, X.; Ye, K.; Yao, Y.; Li, Y. Application potential of Toll-like receptors in cancer immunotherapy: Systematic review. Medicine (Baltimore) 2016, 95, e3951. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.K.; Dinc, G.; Sharma, R.K.; Yolcu, E.S.; Zhao, H.; Shirwan, H. SA-4-1BBL and monophosphoryl lipid A constitute an efficacious combination adjuvant for cancer vaccines. Cancer Res. 2014, 74, 6441–6451. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Kozhaya, L.; Martiniuk, F.; Meng, T.C.; Chiriboga, L.; Liebes, L.; Hochman, T.; Shuman, N.; Axelrod, D.; Speyer, J.; et al. Topical TLR7 agonist imiquimod can induce immune-mediated rejection of skin metastases in patients with breast cancer. Clin. Cancer Res. 2012, 18, 6748–6757. [Google Scholar] [CrossRef] [PubMed]
- Burdelya, L.G.; Brackett, C.M.; Kojouharov, B.; Gitlin, I.I.; Leonova, K.I.; Gleiberman, A.S.; Aygun-Sunar, S.; Veith, J.; Johnson, C.; Haderski, G.J.; et al. Central role of liver in anticancer and radioprotective activities of Toll-like receptor 5 agonist. Proc. Natl. Acad. Sci. USA 2013, 110, E1857–E1866. [Google Scholar] [CrossRef] [PubMed]
- Weigel, B.J.; Cooley, S.; DeFor, T.; Weisdorf, D.J.; Panoskaltsis-Mortari, A.; Chen, W.; Blazar, B.R.; Miller, J.S. Prolonged subcutaneous administration of 852A, a novel systemic Toll-like receptor 7 agonist, to activate innate immune responses in patients with advanced hematologic malignancies. Am. J. Hematol. 2012, 87, 953–956. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, A.; Metellus, P.; Ursu, R.; Zohar, S.; Lafitte, F.; Barrie, M.; Meng, Y.; Richard, M.; Parizot, C.; Laigle-Donadey, F.; et al. Intracerebral administration of CpG oligonucleotide for patients with recurrent glioblastoma: A phase II study. Neuro Oncol. 2010, 12, 401–408. [Google Scholar] [CrossRef]
- Ammi, R.; De Waele, J.; Willemen, Y.; Van Brussel, I.; Schrijvers, D.M.; Lion, E.; Smits, E.L. Poly(I:C) as cancer vaccine adjuvant: Knocking on the door of medical breakthroughs. Pharmacol. Ther. 2015, 146, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Poeck, H.; Besch, R.; Maihoefer, C.; Renn, M.; Tormo, D.; Morskaya, S.S.; Kirschnek, S.; Gaffal, E.; Landsberg, J.; Hellmuth, J.; et al. 5′-Triphosphate-siRNA: Turning gene silencing and Rig-I activation against melanoma. Nat. Med. 2008, 14, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Ellermeier, J.; Wei, J.; Duewell, P.; Hoves, S.; Stieg, M.R.; Adunka, T.; Noerenberg, D.; Anders, H.J.; Mayr, D.; Poeck, H.; et al. Therapeutic efficacy of bifunctional siRNA combining TGF-beta1 silencing with RIG-I activation in pancreatic cancer. Cancer Res. 2013, 73, 1709–1720. [Google Scholar] [CrossRef] [PubMed]
- Matsushima-Miyagi, T.; Hatano, K.; Nomura, M.; Li-Wen, L.; Nishikawa, T.; Saga, K.; Shimbo, T.; Kaneda, Y. TRAIL and Noxa are selectively upregulated in prostate cancer cells downstream of the RIG-I/MAVS signaling pathway by nonreplicating Sendai virus particles. Clin. Cancer Res. 2012, 18, 6271–6283. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, M.; Nimura, K.; Shimbo, T.; Hamasaki, T.; Yamamoto, T.; Matsumura, A.; Kaneda, Y. Immunogene therapy using immunomodulating HVJ-E vector augments anti-tumor effects in murine malignant glioma. J. Neurooncol. 2011, 103, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Kubler, K.; tho Pesch, C.; Gehrke, N.; Riemann, S.; Dassler, J.; Coch, C.; Landsberg, J.; Wimmenauer, V.; Polcher, M.; Rudlowski, C.; et al. Immunogenic cell death of human ovarian cancer cells induced by cytosolic poly(I:C) leads to myeloid cell maturation and activates NK cells. Eur. J. Immunol. 2011, 41, 3028–3039. [Google Scholar] [CrossRef] [PubMed]
- Bhoopathi, P.; Quinn, B.A.; Gui, Q.; Shen, X.N.; Grossman, S.R.; Das, S.K.; Sarkar, D.; Fisher, P.B.; Emdad, L. Pancreatic cancer-specific cell death induced in vivo by cytoplasmic-delivered polyinosine-polycytidylic acid. Cancer Res. 2014, 74, 6224–6235. [Google Scholar] [CrossRef]
- Li, T.; Cheng, H.; Yuan, H.; Xu, Q.; Shu, C.; Zhang, Y.; Xu, P.; Tan, J.; Rui, Y.; Li, P.; et al. Antitumor Activity of cGAMP via Stimulation of cGAS-cGAMP-STING-IRF3 mediated innate immune response. Sci. Rep. 2016, 6, 19049. [Google Scholar] [CrossRef]
- Nakamura, T.; Miyabe, H.; Hyodo, M.; Sato, Y.; Hayakawa, Y.; Harashima, H. Liposomes loaded with a STING pathway ligand, cyclic di-GMP, enhance cancer immunotherapy against metastatic melanoma. J. Control. Release 2015, 216, 149–157. [Google Scholar] [CrossRef]
- Fu, J.; Kanne, D.B.; Leong, M.; Glickman, L.H.; McWhirter, S.M.; Lemmens, E.; Mechette, K.; Leong, J.J.; Lauer, P.; Liu, W.; et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci. Transl. Med. 2015, 7, 283ra252. [Google Scholar] [CrossRef] [PubMed]
- Shatz, M.; Menendez, D.; Resnick, M.A. The human TLR innate immune gene family is differentially influenced by DNA stress and p53 status in cancer cells. Cancer Res. 2012, 72, 3948–3957. [Google Scholar] [CrossRef] [PubMed]
- Ridnour, L.A.; Cheng, R.Y.; Switzer, C.H.; Heinecke, J.L.; Ambs, S.; Glynn, S.; Young, H.A.; Trinchieri, G.; Wink, D.A. Molecular pathways: Toll-like receptors in the tumor microenvironment-poor prognosis or new therapeutic opportunity. Clin. Cancer Res. 2013, 19, 1340–1346. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Yao, Y.; Han, F.; Li, Y. MAP1S controls breast cancer cell TLR5 signaling pathway and promotes TLR5 signaling-based tumor suppression. PLoS ONE 2014, 9, e86839. [Google Scholar] [CrossRef] [PubMed]
- DeCarlo, C.A.; Rosa, B.; Jackson, R.; Niccoli, S.; Escott, N.G.; Zehbe, I. Toll-like receptor transcriptome in the HPV-positive cervical cancer microenvironment. Clin. Dev. Immunol. 2012, 2012, 785825. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Sanchez, A.; Shi, Z.; Zhang, T.; Liu, M.; Zhang, D. Activation of Toll-like receptor 5 on breast cancer cells by flagellin suppresses cell proliferation and tumor growth. Cancer Res. 2011, 71, 2466–2475. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Amakawa, R.; Fukuhara, S. Roles of Toll-like receptors in natural interferon-producing cells as sensors in immune surveillance. Hum. Immunol. 2002, 63, 1120–1125. [Google Scholar] [CrossRef]
- Kalb, M.L.; Glaser, A.; Stary, G.; Koszik, F.; Stingl, G. TRAIL(+) human plasmacytoid dendritic cells kill tumor cells in vitro: Mechanisms of imiquimod- and IFN-alpha-mediated antitumor reactivity. J. Immunol. 2012, 188, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Pisetsky, D.S.; Gauley, J.; Ullal, A.J. HMGB1 and microparticles as mediators of the immune response to cell death. Antioxid. Redox Sign. 2011, 15, 2209–2219. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Zhao, J.; Li, H.; He, K.L.; Chen, Y.; Chen, S.H.; Mayer, L.; Unkeless, J.C.; Xiong, H. Toll-like receptors on tumor cells facilitate evasion of immune surveillance. Cancer Res. 2005, 65, 5009–5014. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Alvero, A.B.; Silasi, D.A.; Steffensen, K.D.; Mor, G. Cancers take their Toll—The function and regulation of Toll-like receptors in cancer cells. Oncogene 2008, 27, 225–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A. Cancer: Inflaming metastasis. Nature 2009, 457, 36–37. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yan, W.; Tohme, S.; Chen, M.; Fu, Y.; Tian, D.; Lotze, M.; Tang, D.; Tsung, A. Hypoxia induced HMGB1 and mitochondrial DNA interactions mediate tumor growth in hepatocellular carcinoma through Toll-like receptor 9. J. Hepatol. 2015, 63, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Monlish, D.A.; Bhatt, S.T.; Schuettpelz, L.G. The role of Toll-Like Receptors in hematopoietic malignancies. Front. Immunol. 2016, 7, 390. [Google Scholar] [CrossRef] [PubMed]
- Tye, H.; Kennedy, C.L.; Najdovska, M.; McLeod, L.; McCormack, W.; Hughes, N.; Dev, A.; Sievert, W.; Ooi, C.H.; Ishikawa, T.O.; et al. STAT3-driven upregulation of TLR2 promotes gastric tumorigenesis independent of tumor inflammation. Cancer Cell 2012, 22, 466–478. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, J.L.; Sondel, P.M. Enhancing cancer immunotherapy via activation of innate immunity. Semin. Oncol. 2015, 42, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vacchelli, E.; Eggermont, A.; Fridman, W.H.; Galon, J.; Sautes-Fridman, C.; Tartour, E.; Zitvogel, L.; Kroemer, G. Trial Watch: Experimental Toll-like receptor agonists for cancer therapy. Oncoimmunology 2012, 1, 699–716. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, S.; Masuko, K.; Wakita, D.; Tanaka, S.; Mitamura, R.; Kato, Y.; Tabata, H.; Nakahara, M.; Kitamura, H.; Nishimura, T. Extracts of Larix Leptolepis effectively augments the generation of tumor antigen-specific cytotoxic T lymphocytes via activation of dendritic cells in TLR-2 and TLR-4-dependent manner. Cell. Immunol. 2012, 276, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Dewan, M.Z.; Vanpouille-Box, C.; Kawashima, N.; DiNapoli, S.; Babb, J.S.; Formenti, S.C.; Adams, S.; Demaria, S. Synergy of topical Toll-like receptor 7 agonist with radiation and low-dose cyclophosphamide in a mouse model of cutaneous breast cancer. Clin. Cancer Res. 2012, 18, 6668–6678. [Google Scholar] [CrossRef] [PubMed]
- Jeung, H.C.; Moon, Y.W.; Rha, S.Y.; Yoo, N.C.; Roh, J.K.; Noh, S.H.; Min, J.S.; Kim, B.S.; Chung, H.C. Phase III trial of adjuvant 5-fluorouracil and adriamycin versus 5-fluorouracil, adriamycin, and polyadenylic-polyuridylic acid (poly A:U) for locally advanced gastric cancer after curative surgery: Final results of 15-year follow-up. Ann. Oncol. 2008, 19, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Van De Voort, T.J.; Felder, M.A.; Yang, R.K.; Sondel, P.M.; Rakhmilevich, A.L. Intratumoral delivery of low doses of anti-CD40 mAb combined with monophosphoryl lipid a induces local and systemic antitumor effects in immunocompetent and T cell-deficient mice. J. Immunother. 2013, 36, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Scholch, S.; Rauber, C.; Weitz, J.; Koch, M.; Huber, P.E. TLR activation and ionizing radiation induce strong immune responses against multiple tumor entities. Oncoimmunology 2015, 4, e1042201. [Google Scholar] [CrossRef] [PubMed]
- Tel, J.; Sittig, S.P.; Blom, R.A.; Cruz, L.J.; Schreibelt, G.; Figdor, C.G.; de Vries, I.J. Targeting uptake receptors on human plasmacytoid dendritic cells triggers antigen cross-presentation and robust type I IFN secretion. J. Immunol. 2013, 191, 5005–5012. [Google Scholar] [CrossRef] [PubMed]
- Le Bon, A.; Etchart, N.; Rossmann, C.; Ashton, M.; Hou, S.; Gewert, D.; Borrow, P.; Tough, D.F. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat. Immunol. 2003, 4, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Onomoto, K.; Jogi, M.; Akaboshi, T.; Fujita, T. Viral RNA detection by RIG-I-like receptors. Curr. Opin. Immunol. 2015, 32, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Schlee, M. Master sensors of pathogenic RNA—RIG-I like receptors. Immunobiology 2013, 218, 1322–1335. [Google Scholar] [CrossRef] [PubMed]
- Kolakofsky, D.; Kowalinski, E.; Cusack, S. A structure-based model of RIG-I activation. RNA 2012, 18, 2118–2127. [Google Scholar] [CrossRef] [PubMed]
- Peisley, A.; Wu, B.; Xu, H.; Chen, Z.J.; Hur, S. Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I. Nature 2014, 509, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Kinch, L.N.; Brautigam, C.A.; Chen, X.; Du, F.; Grishin, N.V.; Chen, Z.J. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity 2012, 36, 959–973. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Zhang, X.; Wang, G.; Zheng, H. The laboratory of genetics and physiology 2: Emerging insights into the controversial functions of this RIG-I-like receptor. BioMed Res. Int. 2014, 2014, 960190. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wang, H.; Li, X.; Guo, C.; Yuan, F.; Fisher, P.B.; Wang, X.Y. Activation of the MDA-5-IPS-1 viral sensing pathway induces cancer cell death and type I IFN-dependent antitumor immunity. Cancer Res. 2016, 76, 2166–2176. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, S.; Roberts, P.C.; Kipperman, T.; Bhalla, K.N.; Compans, R.W.; Archer, D.R.; Barber, G.N. Alpha/beta interferons potentiate virus-induced apoptosis through activation of the FADD/Caspase-8 death signaling pathway. J. Virol. 2000, 74, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.; Alcami, J.; Esteban, M. Induction of apoptosis by double-stranded-RNA-dependent protein kinase (PKR) involves the alpha subunit of eukaryotic translation initiation factor 2 and NF-kappaB. Mol. Cell. Biol. 1999, 19, 4653–4663. [Google Scholar] [CrossRef] [PubMed]
- Castelli, J.C.; Hassel, B.A.; Wood, K.A.; Li, X.L.; Amemiya, K.; Dalakas, M.C.; Torrence, P.F.; Youle, R.J. A study of the interferon antiviral mechanism: Apoptosis activation by the 2–5A system. J. Exp. Med. 1997, 186, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Maitra, R.K.; Silverman, R.H. Regulation of human immunodeficiency virus replication by 2′,5′-oligoadenylate-dependent RNase L. J. Virol. 1998, 72, 1146–1152. [Google Scholar] [PubMed]
- Chawla-Sarkar, M.; Lindner, D.J.; Liu, Y.F.; Williams, B.R.; Sen, G.C.; Silverman, R.H.; Borden, E.C. Apoptosis and interferons: Role of interferon-stimulated genes as mediators of apoptosis. Apoptosis 2003, 8, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.Y.; Chiang, R.L.; Chang, T.H.; Liao, C.L.; Lin, Y.L. The interferon stimulator mitochondrial antiviral signaling protein facilitates cell death by disrupting the mitochondrial membrane potential and by activating caspases. J. Virol. 2010, 84, 2421–2431. [Google Scholar] [CrossRef] [PubMed]
- Guan, K.; Zheng, Z.; Song, T.; He, X.; Xu, C.; Zhang, Y.; Ma, S.; Wang, Y.; Xu, Q.; Cao, Y.; et al. MAVS regulates apoptotic cell death by decreasing K48-linked ubiquitination of voltage-dependent anion channel 1. Mol. Cell. Biol. 2013, 33, 3137–3149. [Google Scholar] [CrossRef] [PubMed]
- El Maadidi, S.; Faletti, L.; Berg, B.; Wenzl, C.; Wieland, K.; Chen, Z.J.; Maurer, U.; Borner, C. A novel mitochondrial MAVS/Caspase-8 platform links RNA virus-induced innate antiviral signaling to Bax/Bak-independent apoptosis. J. Immunol. 2014, 192, 1171–1183. [Google Scholar] [CrossRef] [PubMed]
- Besch, R.; Poeck, H.; Hohenauer, T.; Senft, D.; Hacker, G.; Berking, C.; Hornung, V.; Endres, S.; Ruzicka, T.; Rothenfusser, S.; et al. Proapoptotic signaling induced by RIG-I and MDA-5 results in type I interferon-independent apoptosis in human melanoma cells. J. Clin. Investig. 2009, 119, 2399–2411. [Google Scholar] [CrossRef] [PubMed]
- Eitz Ferrer, P.; Potthoff, S.; Kirschnek, S.; Gasteiger, G.; Kastenmuller, W.; Ludwig, H.; Paschen, S.A.; Villunger, A.; Sutter, G.; Drexler, I.; et al. Induction of Noxa-mediated apoptosis by modified vaccinia virus Ankara depends on viral recognition by cytosolic helicases, leading to IRF-3/IFN-beta-dependent induction of pro-apoptotic Noxa. PLoS Pathog. 2011, 7, e1002083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Y.; Moore, C.B.; Liesman, R.M.; O’Connor, B.P.; Bergstralh, D.T.; Chen, Z.J.; Pickles, R.J.; Ting, J.P. MAVS-mediated apoptosis and its inhibition by viral proteins. PLoS ONE 2009, 4, e5466. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, S.; Marques, J.T.; Yamashita, M.; Peters, K.L.; Smith, K.; Desai, A.; Williams, B.R.; Sen, G.C. Viral apoptosis is induced by IRF-3-mediated activation of Bax. EMBO J. 2010, 29, 1762–1773. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, S.; Kuzmanovic, T.; Zhang, Y.; Wetzel, J.L.; Sen, G.C. Ubiquitination of the transcription factor IRF-3 Activates RIPA, the apoptotic pathway that protects mice from viral pathogenesis. Immunity 2016, 44, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Kirshner, J.R.; Karpova, A.Y.; Kops, M.; Howley, P.M. Identification of TRAIL as an interferon regulatory factor 3 transcriptional target. J. Virol. 2005, 79, 9320–9324. [Google Scholar] [CrossRef] [PubMed]
- Knowlton, J.J.; Dermody, T.S.; Holm, G.H. Apoptosis induced by mammalian reovirus is beta interferon (IFN) independent and enhanced by IFN regulatory factor 3- and NF-kappaB-dependent expression of Noxa. J. Virol. 2012, 86, 1650–1660. [Google Scholar] [CrossRef] [PubMed]
- Ranoa, D.R.; Parekh, A.D.; Pitroda, S.P.; Huang, X.; Darga, T.; Wong, A.C.; Huang, L.; Andrade, J.; Staley, J.P.; Satoh, T.; et al. Cancer therapies activate RIG-I-like receptor pathway through endogenous non-coding RNAs. Oncotarget 2016, 7, 26496–26515. [Google Scholar] [CrossRef] [PubMed]
- Tchelebi, L.; Ashamalla, H.; Graves, P.R. Mutant p53 and the response to chemotherapy and radiation. Subcell. Biochem. 2014, 85, 133–159. [Google Scholar] [PubMed]
- Ablasser, A.; Hertrich, C.; Wassermann, R.; Hornung, V. Nucleic acid driven sterile inflammation. Clin. Immunol. 2013, 147, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.H.; Macmillan, J.B.; Chen, Z.J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Burckstummer, T.; Baumann, C.; Bluml, S.; Dixit, E.; Durnberger, G.; Jahn, H.; Planyavsky, M.; Bilban, M.; Colinge, J.; Bennett, K.L.; et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 2009, 10, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Pazhoor, S.; Bao, M.; Zhang, Z.; Hanabuchi, S.; Facchinetti, V.; Bover, L.; Plumas, J.; Chaperot, L.; Qin, J.; et al. Aspartate-glutamate-alanine-histidine box motif (DEAH)/RNA helicase A helicases sense microbial DNA in human plasmacytoid dendritic cells. Proc. Natl. Acad. Sci. USA 2010, 107, 15181–15186. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; An, H.; Liu, X.; Wen, M.; Zheng, Y.; Rui, Y.; Cao, X. The cytosolic nucleic acid sensor LRRFIP1 mediates the production of type I interferon via a beta-catenin-dependent pathway. Nat. Immunol. 2010, 11, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yuan, B.; Bao, M.; Lu, N.; Kim, T.; Liu, Y.J. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat. Immunol. 2011, 12, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Brann, T.W.; Zhou, M.; Yang, J.; Oguariri, R.M.; Lidie, K.B.; Imamichi, H.; Huang, D.W.; Lempicki, R.A.; Baseler, M.W.; et al. Cutting edge: Ku70 is a novel cytosolic DNA sensor that induces type III rather than type I IFN. J. Immunol. 2011, 186, 4541–4545. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, B.J.; Mansur, D.S.; Peters, N.E.; Ren, H.; Smith, G.L. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. eLife 2012, 1, e00047. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Kobayashi, J.; Saitoh, T.; Maruyama, K.; Ishii, K.J.; Barber, G.N.; Komatsu, K.; Akira, S.; Kawai, T. DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. Proc. Natl. Acad. Sci. USA 2013, 110, 2969–2974. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Yang, Y.; Li, S.; Wang, Y.Y.; Li, Y.; Diao, F.; Lei, C.; He, X.; Zhang, L.; Tien, P.; et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 2008, 29, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Burdette, D.L.; Monroe, K.M.; Sotelo-Troha, K.; Iwig, J.S.; Eckert, B.; Hyodo, M.; Hayakawa, Y.; Vance, R.E. STING is a direct innate immune sensor of cyclic di-GMP. Nature 2011, 478, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Hill, K.K.; Filak, H.; Mogan, J.; Knowles, H.; Zhang, B.; Perraud, A.L.; Cambier, J.C.; Lenz, L.L. MPYS is required for IFN response factor 3 activation and type I IFN production in the response of cultured phagocytes to bacterial second messengers cyclic-di-AMP and cyclic-di-GMP. J. Immunol. 2011, 187, 2595–2601. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Rohl, I.; Hopfner, K.P.; Ludwig, J.; Hornung, V. cGAS produces a 2′–5′-linked cyclic dinucleotide second messenger that activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shi, H.; Wu, J.; Sun, L.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol. Cell 2013, 51, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Barber, G.N. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-kappaB activation through TBK1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.H.; Zundell, J.A.; Ranatunga, S.; Lin, C.; Nefedova, Y.; Del Valle, J.R.; Hu, C.C. Agonist-mediated activation of STING induces apoptosis in malignant B cells. Cancer Res. 2016, 76, 2137–2152. [Google Scholar] [CrossRef] [PubMed]
- Sze, A.; Belgnaoui, S.M.; Olagnier, D.; Lin, R.; Hiscott, J.; van Grevenynghe, J. Host restriction factor SAMHD1 limits human T cell leukemia virus type I infection of monocytes via STING-mediated apoptosis. Cell Host Microbe 2013, 14, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Petrasek, J.; Iracheta-Vellve, A.; Csak, T.; Satishchandran, A.; Kodys, K.; Kurt-Jones, E.A.; Fitzgerald, K.A.; Szabo, G. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc. Natl. Acad. Sci. USA 2013, 110, 16544–16549. [Google Scholar] [CrossRef] [PubMed]
- White, M.J.; McArthur, K.; Metcalf, D.; Lane, R.M.; Cambier, J.C.; Herold, M.J.; van Delft, M.F.; Bedoui, S.; Lessene, G.; Ritchie, M.E.; et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 2014, 159, 1549–1562. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. Weichselbaum RR7. STING-Dependent Cytosolic DNA Sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Pu, Y.; Cron, K.; Deng, L.; Kline, J.; Frazier, W.A.; Xu, H.; Peng, H.; Fu, Y.X.; Xu, M.M. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat. Med. 2015, 21, 1209–1215. [Google Scholar] [CrossRef] [PubMed]
- Perera, P.Y.; Barber, S.A.; Ching, L.M.; Vogel, S.N. Activation of LPS-inducible genes by the antitumor agent 5,6-dimethylxanthenone-4-acetic acid in primary murine macrophages. Dissection of signaling pathways leading to gene induction and tyrosine phosphorylation. J. Immunol. 1994, 153, 4684–4693. [Google Scholar] [PubMed]
- Conlon, J.; Burdette, D.L.; Sharma, S.; Bhat, N.; Thompson, M.; Jiang, Z.; Rathinam, V.A.; Monks, B.; Jin, T.; Xiao, T.S.; et al. Mouse, but not human STING, binds and signals in response to the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid. J. Immunol. 2013, 190, 5216–5225. [Google Scholar] [CrossRef] [PubMed]
- Downey, C.M.; Aghaei, M.; Schwendener, R.A.; Jirik, F.R. DMXAA causes tumor site-specific vascular disruption in murine non-small cell lung cancer, and like the endogenous non-canonical cyclic dinucleotide STING agonist, 2′3′-cGAMP, induces M2 macrophage repolarization. PLoS ONE 2014, 9, e99988. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Ascano, M.; Zillinger, T.; Wang, W.; Dai, P.; Serganov, A.A.; Gaffney, B.L.; Shuman, S.; Jones, R.A.; Deng, L.; et al. Structure-function analysis of STING activation by c[G(2′,5′)pA(3′,5′)p] and targeting by antiviral DMXAA. Cell 2013, 154, 748–762. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Monie, A.; Pang, X.; Hung, C.F.; Wu, T.C. Vascular disrupting agent DMXAA enhances the antitumor effects generated by therapeutic HPV DNA vaccines. J. Biomed. Sci. 2011, 18, 21. [Google Scholar] [CrossRef] [PubMed]
- Corrales, L.; Glickman, L.H.; McWhirter, S.M.; Kanne, D.B.; Sivick, K.E.; Katibah, G.E.; Woo, S.R.; Lemmens, E.; Banda, T.; Leong, J.J.; et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 2015, 11, 1018–1030. [Google Scholar] [CrossRef] [PubMed]
- Demaria, O.; De Gassart, A.; Coso, S.; Gestermann, N.; Di Domizio, J.; Flatz, L.; Gaide, O.; Michielin, O.; Hwu, P.; Petrova, T.V.; et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc. Natl. Acad. Sci. USA 2015, 112, 15408–15413. [Google Scholar] [CrossRef] [PubMed]
- Ohkuri, T.; Ghosh, A.; Kosaka, A.; Zhu, J.; Ikeura, M.; David, M.; Watkins, S.C.; Sarkar, S.N.; Okada, H. STING contributes to antiglioma immunity via triggering type I IFN signals in the tumor microenvironment. Cancer Immunol. Res. 2014, 2, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Celis, E. STING activator c-di-GMP enhances the anti-tumor effects of peptide vaccines in melanoma-bearing mice. Cancer Immunol. Immunother. 2015, 64, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Li, L.; Lemos, H.; Chandler, P.R.; Pacholczyk, G.; Baban, B.; Barber, G.N.; Hayakawa, Y.; McGaha, T.L.; Ravishankar, B.; et al. Cutting edge: DNA sensing via the STING adaptor in myeloid dendritic cells induces potent tolerogenic responses. J. Immunol. 2013, 191, 3509–3513. [Google Scholar] [CrossRef] [PubMed]
- Lemos, H.; Mohamed, E.; Huang, L.; Ou, R.; Pacholczyk, G.; Arbab, A.S.; Munn, D.; Mellor, A.L. STING promotes the growth of tumors characterized by low antigenicity via IDO activation. Cancer Res. 2016, 76, 2076–2081. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; Jacob, L.S.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016, 533, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Inhibiting the inhibitors: Evaluating agents targeting cancer immunosuppression. Expert Opin. Biol. Ther. 2010, 10, 1019–1035. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.Q.; Mills, K.H. Immunosuppressive networks and checkpoints controlling antitumor immunity and their blockade in the development of cancer immunotherapeutics and vaccines. Oncogene 2014, 33, 4623–4631. [Google Scholar] [CrossRef] [PubMed]
- Levy, L.; Mishalian, I.; Bayuch, R.; Zolotarov, L.; Michaeli, J.; Fridlender, Z.G. Splenectomy inhibits non-small cell lung cancer growth by modulating anti-tumor adaptive and innate immune response. Oncoimmunology 2015, 4, e998469. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Jassar, A.; Mishalian, I.; Wang, L.C.; Kapoor, V.; Cheng, G.; Sun, J.; Singhal, S.; Levy, L.; Albelda, S.M. Using macrophage activation to augment immunotherapy of established tumours. Br. J. Ccancer 2013, 108, 1288–1297. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Honjo, T. PD-1 and PD-1 ligands: From discovery to clinical application. Int. Immunol. 2007, 19, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Ribas, A.; Wolchok, J.D.; Hodi, F.S.; Hamid, O.; Kefford, R.; Weber, J.S.; Joshua, A.M.; Hwu, W.J.; Gangadhar, T.C.; et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: A randomised dose-comparison cohort of a phase 1 trial. Lancet 2014, 384, 1109–1117. [Google Scholar] [CrossRef]
- Buferne, M.; Chasson, L.; Grange, M.; Mas, A.; Arnoux, F.; Bertuzzi, M.; Naquet, P.; Leserman, L.; Schmitt-Verhulst, A.M.; Auphan-Anezin, N. IFNgamma producing CD8+ T cells modified to resist major immune checkpoints induce regression of MHC class I-deficient melanomas. Oncoimmunology 2015, 4, e974959. [Google Scholar] [CrossRef] [PubMed]
- Sim, G.C.; Radvanyi, L. The IL-2 cytokine family in cancer immunotherapy. Cytokine Growth Factor Rev. 2014, 25, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A. IL-2: The first effective immunotherapy for human cancer. J. Immunol. 2014, 192, 5451–5458. [Google Scholar] [CrossRef] [PubMed]
- Conlon, K.C.; Lugli, E.; Welles, H.C.; Rosenberg, S.A.; Fojo, A.T.; Morris, J.C.; Fleisher, T.A.; Dubois, S.P.; Perera, L.P.; Stewart, D.M.; et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J. Clin. Oncol. 2015, 33, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Locati, M. Macrophage polarization comes of age. Immunity 2005, 23, 344–346. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Liu, L.; Che, G.; Yu, N.; Dai, F.; You, Z. The M1 form of tumor-associated macrophages in non-small cell lung cancer is positively associated with survival time. BMC Cancer 2010, 10, 112. [Google Scholar] [CrossRef] [PubMed]
- Alderson, K.L.; Luangrath, M.; Elsenheimer, M.M.; Gillies, S.D.; Navid, F.; Rakhmilevich, A.L.; Sondel, P.M. Enhancement of the anti-melanoma response of Hu14.18K322A by alphaCD40 + CpG. Cancer Immunol. Immunother. 2013, 62, 665–675. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Promising Agents | Receptors | Cancer Types | References |
|---|---|---|---|
| Bacillus Calmette-Guérin (BCG) | TLR2/4 | Bladder cancer | [19] |
| monophosphoryl lipid A(MPL) | TLR4 | Cervical cancer | [22] |
| Imiquimod | TLR7 | Breast cancer | [23] |
| Flagellin-derived CBLB502 (Entolimod) | TLR5 | Hepatoma | [24] |
| 852A | TLR7 | Hematologic malignancy | [25] |
| CpG ODN | TLR9 | Glioblastoma | [26] |
| poly(I:C)/poly-ICLC | TLR3 | Multiple cancer types | [27] |
| 5′ ppp-siRNA for Bcl-2 | RIG-I | Melanoma | [28] |
| 5′ ppp-siRNA for TGF-β | RIG-I | Pancreatic cancer | [29] |
| HVJ-E | RIG-I | Prostate cancer, gliomas | [30,31] |
| poly(I:C) | MDA5 | Ovarian cancer, Pancreatic cancer | [32,33] |
| cGAMP | STING | Colon cancer | [34] |
| c-di-GMP | STING | Melanoma | [35] |
| STINGVAX | STING | Melanoma | [36] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, K.; Qu, S.; Chen, X.; Wu, Q.; Shi, M. Promising Targets for Cancer Immunotherapy: TLRs, RLRs, and STING-Mediated Innate Immune Pathways. Int. J. Mol. Sci. 2017, 18, 404. https://doi.org/10.3390/ijms18020404
Li K, Qu S, Chen X, Wu Q, Shi M. Promising Targets for Cancer Immunotherapy: TLRs, RLRs, and STING-Mediated Innate Immune Pathways. International Journal of Molecular Sciences. 2017; 18(2):404. https://doi.org/10.3390/ijms18020404
Chicago/Turabian StyleLi, Kai, Shuai Qu, Xi Chen, Qiong Wu, and Ming Shi. 2017. "Promising Targets for Cancer Immunotherapy: TLRs, RLRs, and STING-Mediated Innate Immune Pathways" International Journal of Molecular Sciences 18, no. 2: 404. https://doi.org/10.3390/ijms18020404
APA StyleLi, K., Qu, S., Chen, X., Wu, Q., & Shi, M. (2017). Promising Targets for Cancer Immunotherapy: TLRs, RLRs, and STING-Mediated Innate Immune Pathways. International Journal of Molecular Sciences, 18(2), 404. https://doi.org/10.3390/ijms18020404