1. Introduction
Although the DNA sequence of the human genome, which carries genetic information, is almost invariant in various human cells, the epigenetic features of each cell show great differences, leading to distinguishable gene expression patterns and cell-type specificities [
1]. Among these features (such as histone modifications mediating changes in chromatin conformation), DNA methylation is the best studied epigenetic modification [
2,
3]. In mammals, DNA methylation mainly occurs at CpG dinucleotide sites with an added methyl group to the fifth carbon of the cytosine residue [
2,
4,
5,
6,
7,
8]. In general, the patterns of DNA methylation are mediated by the main three enzymes, DNMT3 (for establishment), DNMT1 (for maintenance) and MBD4 (for demethylation) [
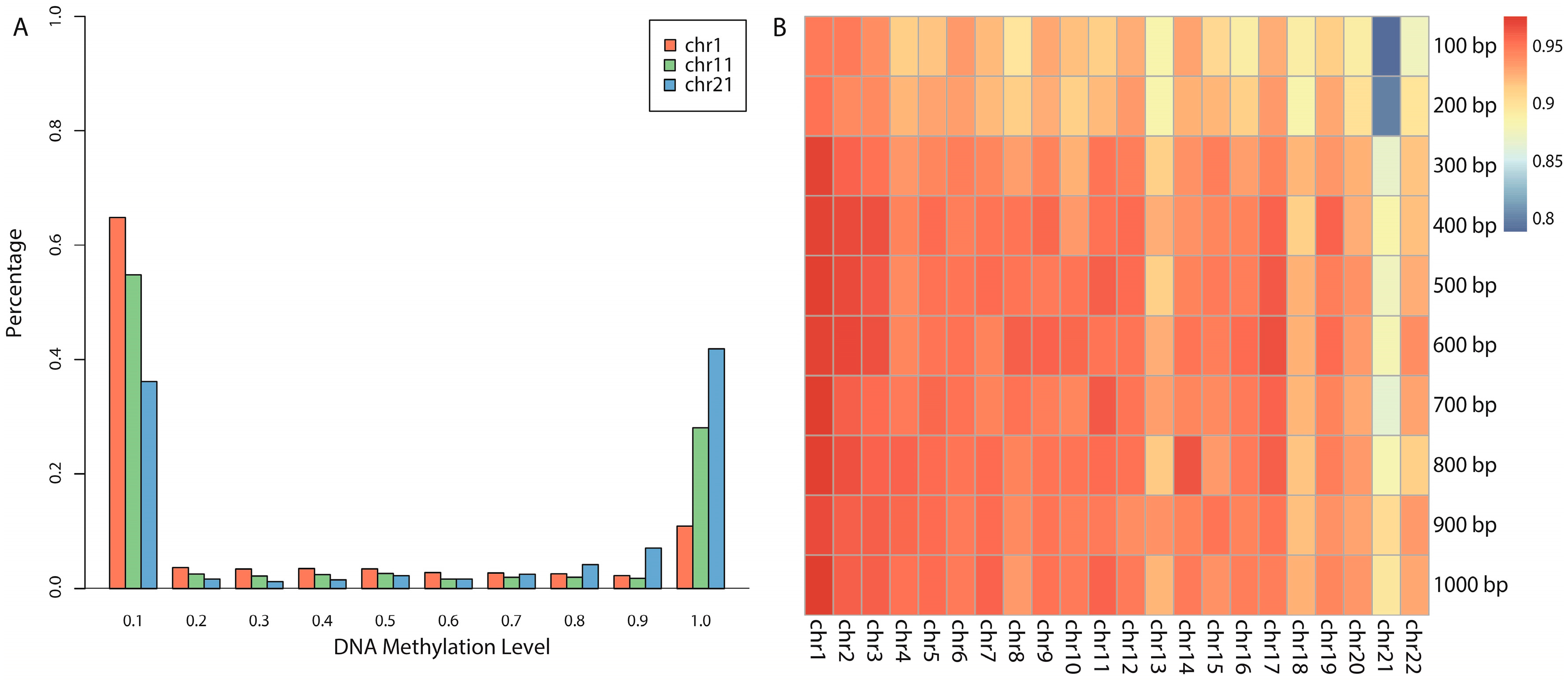
3]. The human genome contains approximately 28 million CpG sites, 60%–80% of which are generally methylated [
2], and the remaining unmethylated CpG sites are mostly located near promoter or exonic regions where GC content is much greater than 50%, which are usually called
CpG islands (CGIs) [
3,
9].
The biological role of DNA methylation is to repress transcriptional activity. Different cell types show distinct methylation distributions across the genome, particularly in regulatory regions of cell-specific genes [
2,
3]. Notably, the average methylation levels in specific regions are consistent with other signals and modifications that are related to transcriptional regulation, such as transcription factor binding sites (TFBSs), DNase I hypersensitive sites (DHSs) and various histone modifications [
1,
9]. Recent studies have shown that hypomethylated regions were associated with promoters near transcriptional start sites (TSSs), whereas hypermethylated regions were considered as silenced regions. Interestingly, the low-methylated regions, in which average methylation levels are approximately 0.3, an intermediate status between hypomethylation and hypermethylation, were reported to be associated with distal regulatory regions, such as enhancers [
9]. Therefore, DNA methylation is a key biomarker and plays a critical role in transcriptional regulation.
Considering the significance of DNA methylation, change of its level in specific regions is usually regarded as an important factor affecting the expressions of target genes and downstream phenotypes, such as embryonic development and tumorigenesis [
10]. When compared to normal cells, aberrant average levels of DNA methylation in important regulatory regions (such as promoters) were linked with the altered expression profiles of cancer cells [
11,
12,
13,
14]. Interestingly, recent findings revealed that abnormal DNA methylation levels at distal regulatory regions (such as enhancers and super-enhancers) were closely related to gene dysregulation in cancer [
14,
15]. In summary, the average DNA methylation level in specific regions is an ideal biomarker of tumorigenesis, and detecting aberrant methylations in these regions is a promising approach for early diagnosis and classification of cancer [
10].
Traditional methods for identifying DNA methylation mainly include whole-genome bisulfite sequencing (WGBS) [
16], reduced representation bisulfite sequencing (RRBS) [
17] and the methylation-microarray-based Illumina 450K BeadChip [
18]. The 450K BeadChip uses microarray-based technology to assay approximately 0.4–0.6 million preselected CpG sites. In contrast, WGBS is a standard method in whole-genome sequencing at base resolution with approximately 1–18 million CpG sites captured [
19]. Additionally, RRBS is an efficient technique designed for enriching the regions with a high CpG content, leading to high-efficiency and saving compared with WGBS. The total coverage of RRBS is roughly equivalent to that of WGBS, and the number of CpGs captured per sample genome by RRBS ranged from 0.5 million to 13 million [
19]. However, there are 28 million CpG sites in the whole human genome, only 10%–50% of which are covered by WGBS or RRBS. Why it is difficult to assay the majority of CpGs in the whole genome even by the high-throughput techniques? Generally, there are many experimental difficulties hampering the analysis of the methylation status within this “hidden” fraction of CpG sites, such as copy-number variation bias, incomplete bisulfite conversion bias, bisulfite PCR bias, GC content bias and CpG density bias [
19]. For RRBS or WGBS experiments, still two coverage limitations (incomplete bisulfite conversion bias and bisulfite PCR bias) could happen [
19,
20,
21], which are the reasons for their low coverages. Therefore, more quantitative methods are urgently demanded to predict the methylation status of the remaining unassayed CpG sites of the human genome.
Computational approaches are alternative methods to identify DNA methylation status. Actually, a number of computational methods had been developed to predict DNA methylation status with the rapid developments of bioinformatics and machine learning approaches, which usually contain three vital steps—data collection, feature extraction and a classification algorithm [
22]. Similarly, many efforts have been made for predicting the functional sites in proteins (such as cysteine S-nitrosylation sites [
23,
24,
25,
26,
27], protein methylation sites [
28], hydroxyproline and hydroxylysine in proteins [
29], lysine ubiquitination sites [
30], lysine succinylation sites [
31]. For more details, please refer to two recent reviews [
32,
33].
Due to the restrictions of experimental data (such as microarray), some early methods limited their attentions on specific genomic regions (such as CGIs) and obtained satisfactory prediction accuracies (with accuracy >90%) [
34,
35,
36,
37]. In contrast, when turning to more common regions predicted using RRBS and WGBS data, the prediction accuracies clearly decreased to 75%–89% [
38]. This is because when we focused on the prediction in CGI regions, GC contents of positive samples are high, whereas GC contents of negative samples are lower. Thus, it is easy to distinguish the methylated CpGs and unmethylated ones only by GC content feature. However, when turning to more common regions, the predictive role of GC content will not be as significant as that of CGI regions and the corresponding prediction accuracies will decrease. In these situations, more complex features are needed for better predictions.
Regarding feature extraction, various methods have been employed to formulate methylation status, including: DNA composition [
34,
36,
39,
40], pseudo trinucleotide composition (PseTNC) [
41,
42,
43,
44,
45], predicted DNA structure [
34,
46], single nucleotide polymorphisms (SNPs) [
34], TFBSs [
34,
46], histone modifications [
36,
46], neighboring CpG site methylation status and distance [
46]. It is worth noting that a powerful web-server called “Pse-in-One” has been used to extract various features from DNA or protein sequences [
47].
Particularly, Das et al. analyzed GC content and Alu elements features for 800 bp regions centered on CpG sites using DNA methylation data of human brain, and developed classifiers with accuracy of 86% [
40]. Moreover, Bock et al. employed 1184 DNA attributes for discriminating between CpG islands that are prone to methylation from those that remain unmethylated using CpG island methylation data on human Chromosome 21, and they found that certain sequence patterns, specific DNA repeats and a particular DNA structure played significant roles in the prediction of DNA methylation status [
34]. In the same year, Fang et al. [
39] used nucleotide sequence features including GC content, CpG ratio, TpG content and Alu distribution, as well as TFBSs features to predict DNA methylation status on human brain dataset. They tested four different window sizes (200 bp, 300 bp, 400 bp, and 500 bp) and found 400 bp is a better choice for prediction, with accuracy of 85%. Recently, Liu et al. invented a novel method called “pseudo trinucleotide composition (PseTNC)” which can both capture the local or short-range sequence order effects and the global or long-range effects of DNA sequences for predicting the methylation status of DNA fragments using 41-bp window centered on CpG sites [
41]. Notably, Zhang et al. [
46] integrated four groups of features, including neighboring features, genomic position, DNA sequence properties and
cis-regulatory elements, to predict DNA methylation status using the whole blood sample dataset. As a result, 400-bp window was also found to be the best size for prediction and DHSs as well as GC content were found to be the most predictive features. In addition, Wang et al. combined “PseTNC” and chromatin interaction features (Hi-C features) to predict methylation states using the methylation datasets taken from two cell lines (GM12878 and K562). A series of window sizes (500–1000) were tested to search the best one, and 600 bp was found to be the appropriate choice [
48].
These methods were mainly developed based on some common machine-learning algorithms: support vector machine (SVM) [
34,
35,
36,
38,
40,
49], random forest (RF) [
37,
46], naive Bayes (NB) and stacked denoising autoencoders (SDA) [
48]. One can refer to the textbook written by James et al. [
50] for more details of these algorithms and their implementations in R. The majority of these studies used SVM as the classifier due to its powerful classification ability and universality for various data types. Notably, some research showed the superiority of RF compared with SVM [
46], and the SDA method from deep learning field emerged for prediction of DNA methylation [
48].
The methods above achieved remarkable results, but there were still some defects among them: (a) most of them could not perform predictions at the whole genome-wide level because the data they used were specific fragments of the genome (such as the 450K BeadChip); (b) although some of them developed their classifiers based on RRBS and WGBS and achieved satisfactory results [
46], obtaining important features for prediction (such as neighboring CpG site methylation status and DHSs) is difficult and would require comprehensive and expensive epigenetic experiments; and (c) most of them did not test the validities of their predicting models on unassayed CpG sites.
In this work, we introduced a novel computational algorithm called “sequence complexity”, together with DNA composition (72 features in total) to predict the DNA methylation status of CpG sites in the whole human genome. Our prediction method has the following advantages compared with current classifiers: (a) a group of novel features called “sequence complexity” were developed, and subsequent analysis confirmed that these new features played significant roles for predictions; (b) by integrating the fundamental features (DNA composition), the prediction model achieved satisfactory results; (c) all the features we used were only extracted from the primary DNA sequence of the human genome without additional experiments, and comparisons with previous works showed the superiority of our method; and (d) a statistical test of the experimental data and the predicted data on functional regions annotated by ChromHMM found that six out of 10 regions were consistent, which implies reliable prediction of unassayed CpG sites. Thus, we believe that our novel model will be useful and reliable in predicting DNA methylation.
As illustrated by many recently published papers [
51,
52,
53,
54,
55,
56,
57], to establish a powerful predictor for a biological problem, one should obey the following five steps: (a) build or choose a benchmark dataset to train and test the predictor; (b) transform the raw biological sequences into mathematical feature vectors that can truly extract their intrinsic features from the target to be predicted; (c) employ or create a powerful algorithm to operate the prediction; (d) accurately use cross-validation tests to impersonally evaluate the predicting ability of the predictor; and (e) build a user-friendly web-server to make their dataset and predictor publicly available. Next, the current study will be organized following these steps one-by-one.
3. Discussion
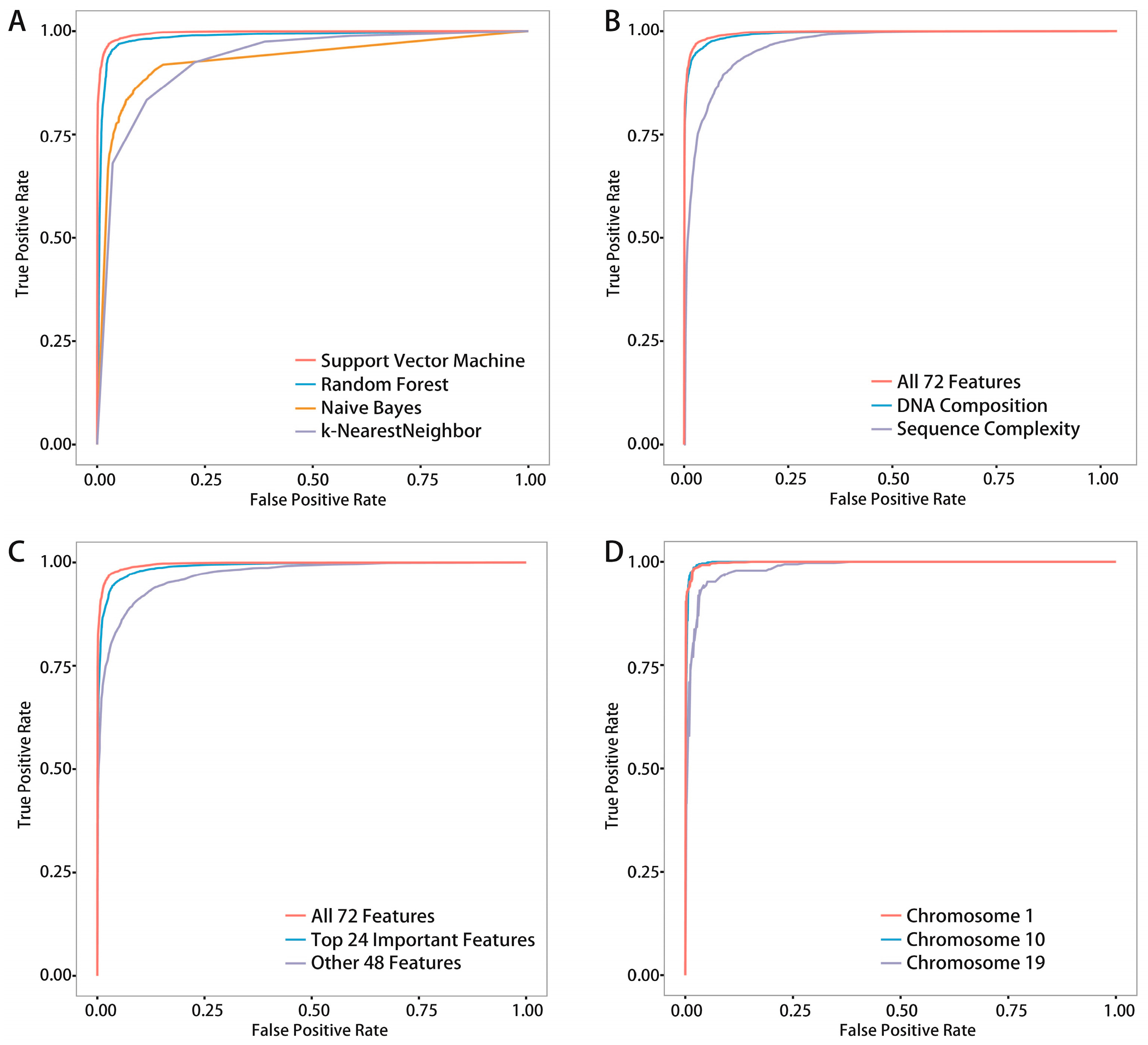
We proposed a novel algorithm that could accurately extract the sequence complexity features of DNA methylation status, and developed a SVM-based prediction model by integrating DNA composition features based on human embryonic stem cell methylation profiles. Different window sizes (100–1000 bp) and different chromosomes were combined pairwise to display the overall prediction results, and 600-bp windows whose center are methylated CpG sites were found to achieve the best accuracies.
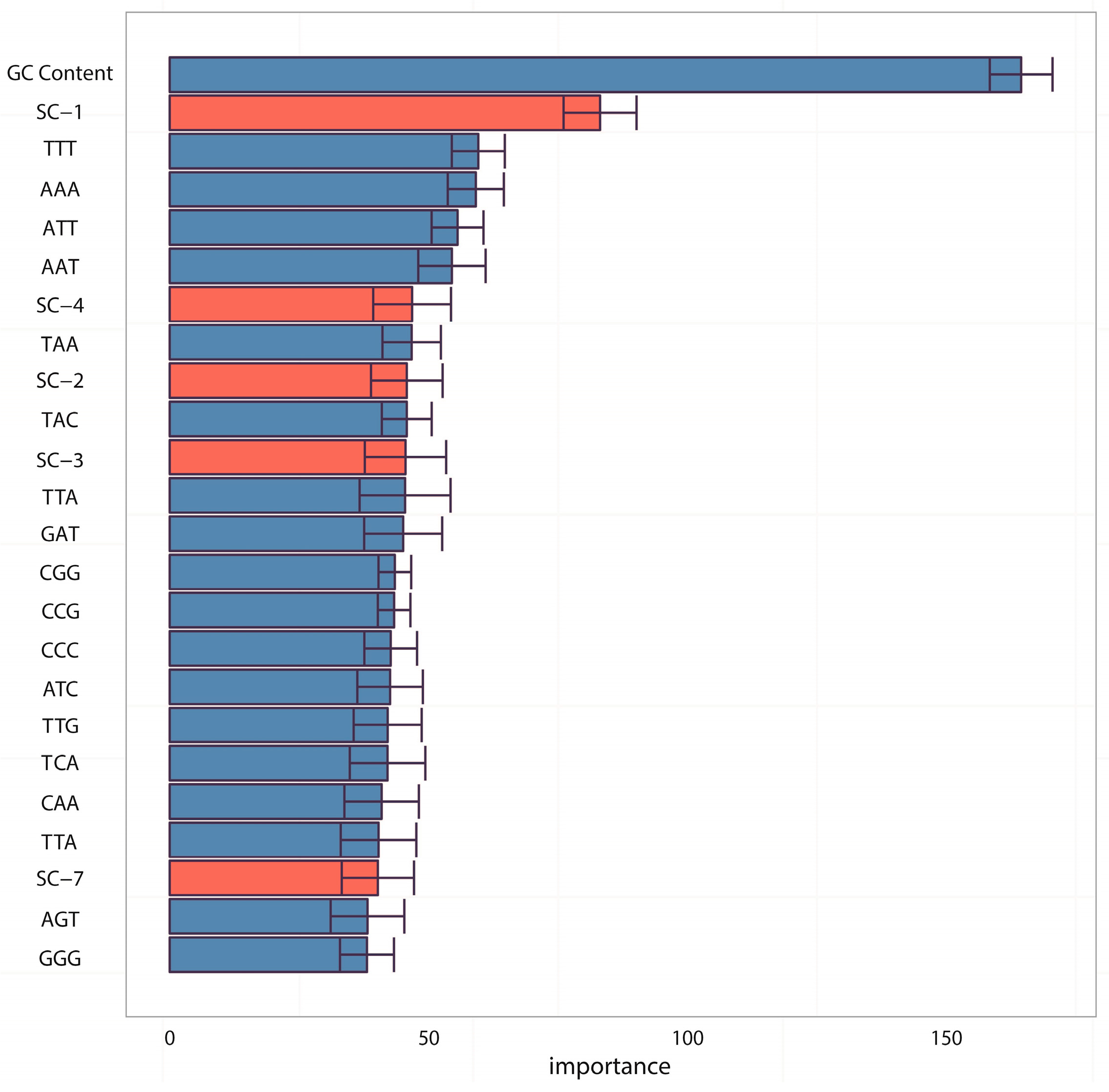
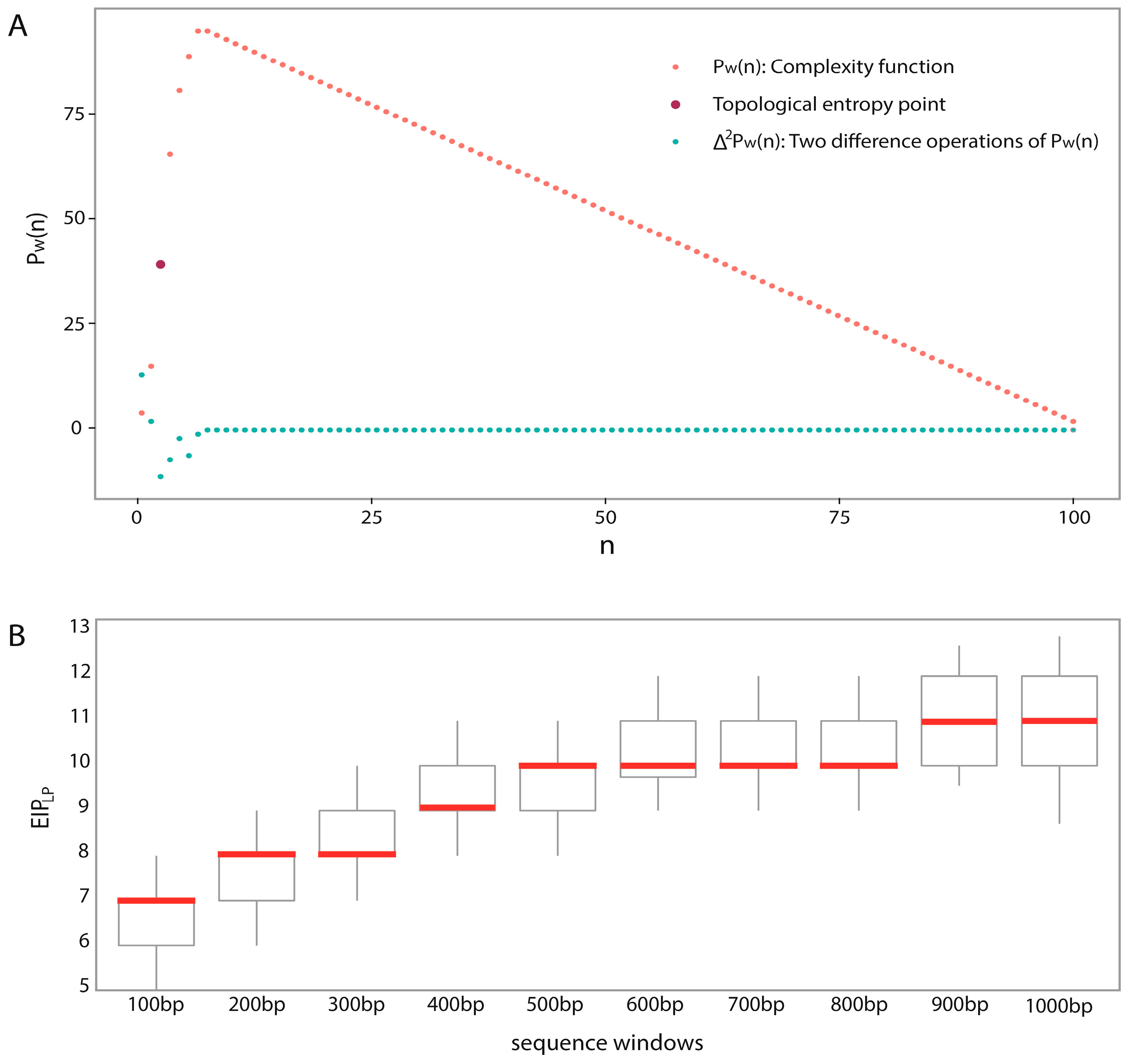
In the analyses of feature importance, the feature group of DNA composition was found to be the fundamental features and the feature group of sequence complexity was found to be the important complementary features. From the ranking of
Figure 3, it is worth noting that some components of sequence complexity feature group, such as SC-1 and SC-2, are important for prediction. Let us recall the definitions of SC-1 and SC-2 features (see Materials and Methods, 600-bp window): SC-1 feature is exactly the fourth point of complexity function and SC-2 feature is exactly the fifth point of complexity function. That means that methylated samples and unmethylated samples probably have different complexities of four-nucleotide and five-nucleotide usages of DNA sequence.
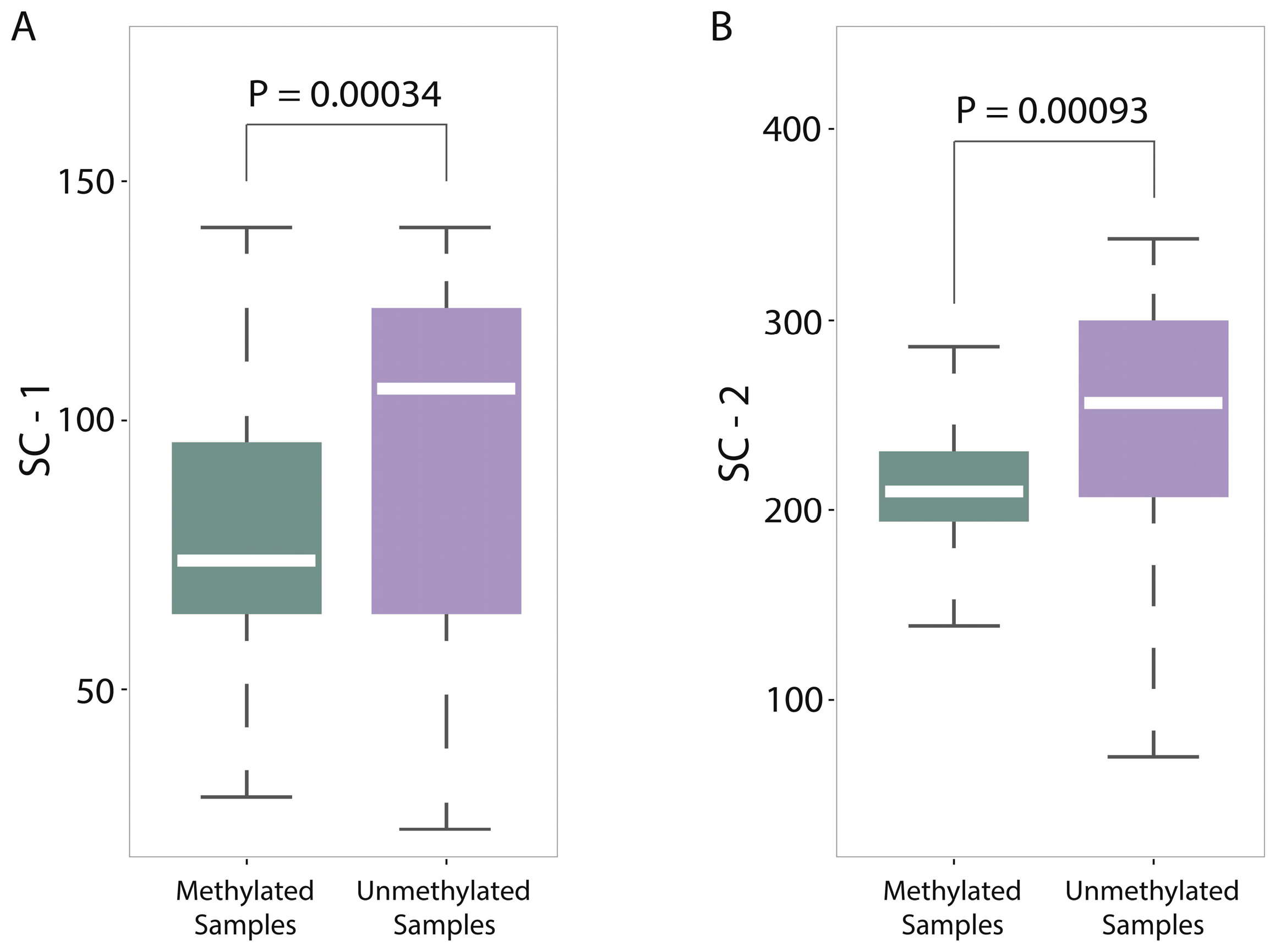
For a deep discussion, we further analyzed the distribution differences between methylated samples and unmethylated samples on SC-1 and SC-2 features (600-bp window) and then performed the corresponding statistical tests (
T test). Both results were shown in
Figure 5A,B, from which we found two interesting things: (1) The
p-value on SC-1 feature is 0.00034 and the
p-value on SC-2 feature is 0.00093, which shows that there are statistically significant differences between methylated samples and unmethylated ones on these two features. Therefore, it is not difficult to understand why the group of sequence complexity is an important group of features; (2) The variations of methylated samples are obviously lower than those of unmethylated samples for both two features (for SC-1 feature, variation of methylated samples equals to 60.62 and variation of unmethylated samples equals to 307.94; for SC-2 feature, variation of methylated samples equals to 90.47 and variation of unmethylated samples equals to 980.82), which implies that methylated samples tend to be more conservative with having more consistent complexity of four-nucleotide and five-nucleotide usages, whereas unmethylated samples tend to use them more randomly. That leads us to conjecture that DNA sequence motifs with length four or five are influential factors for DNA methylation, which should be validated by more future studies.
Moreover, we compared four common classifiers, and SVM resulted in the best records. We also compared two existing methods,
iDNA-Methyl [
41] and
DeepMethyl [
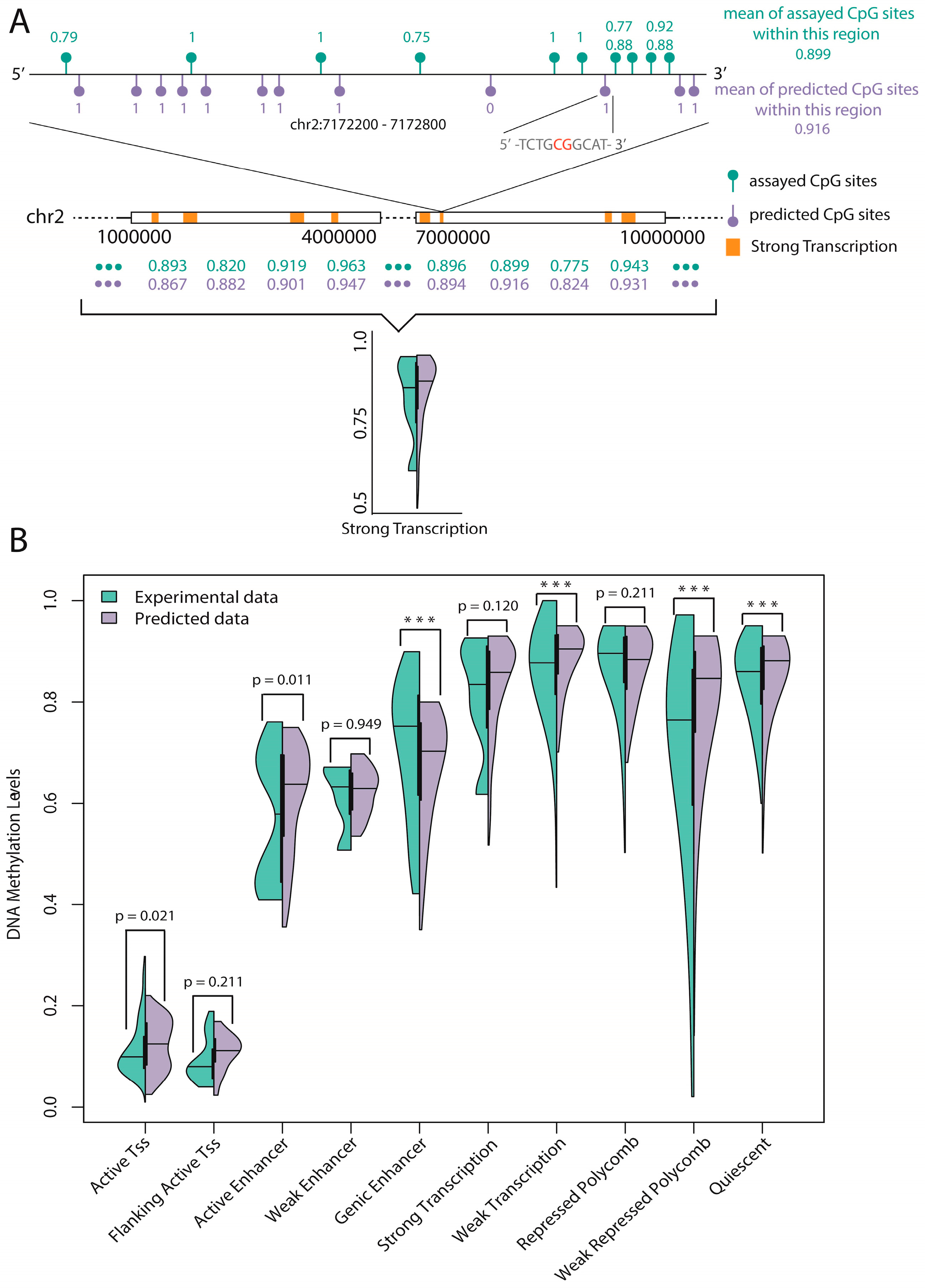
48] to show the advantages of our method. Additionally, we tested our model on methylation data of mouse genome for demonstrating that our model has certain generalization ability. Finally, a statistical test of experimental data and predicted data on 10 functional regions annotated by ChromHMM found that six regions were consistent, which implies reliable prediction of unassayed CpG sites.
Based on the above efforts, we summarize that our predictor brings some new benefits to the area of DNA methylation prediction:
Methodology: The biggest novelty of this work is the successful utilization of sequence complexity features for characterizing DNA methylation patterns. Earlier methods for selecting rational points when estimating topological entropy were not accurate and left out useful information. We provide a simple way to detect intrinsic features of sequence complexities, which were successfully used to predict DNA methylation status. Moreover, the feature importance analyses show that sequence complexities are the important complementary features.
Predicting window size: Previous works used different window sizes for predictions, such as 41 bp [
41], 400 bp [
46], and 100–1000 bp [
48]. We found that 600 bp is the most appropriate by considering pairwise combinations of 22 chromosomes and different window sizes (from 100 to 1000 bp) based on a large dataset.
Prediction of unassayed CpG sites: A statistical test of the average methylation level of experimental CpG sites and predicted CpG sites (unassayed CpG sites, predicted by our trained model) on 10 functional regions annotated by ChromHMM found six regions were consistent. Based on this, we believe that the average methylation level of specific functional regulatory regions (such as promoters and enhancers) can be reliably predicted by our model.
The above in-depth analyses demonstrated the advantages of our computational model from different perspectives, however, there are still some limitations. For example, our predicting model only depends on the primary DNA sequence, which results in the same features extracted from different cells or cell lines. This might lead to the predicting deviation when predicting the DNA methylation level of different cells which show great changes in DNA methylation, especially between normal cells and cancer cells.
Moreover, important future work includes the investigation of the regulatory roles of DNA methylation, including the relationships between DNA methylation and transcription factor binding, especially in important regions of DNA regulatory elements, such as promoters and enhancers. Another future work is to establish a web-server to make our predictor publicly available just as shown in many recent publications (see, e.g., [
52,
53,
55,
57,
63,
64,
65,
66,
67,
68,
69]) and to significantly enhance the influence of our predictor.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}