
mRNA Quantification of NIPBL Isoforms A and B in Adult and Fetal Human Tissues, and a Potentially Pathological Variant Affecting Only Isoform A in Two Patients with Cornelia de Lange Syndrome

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Results
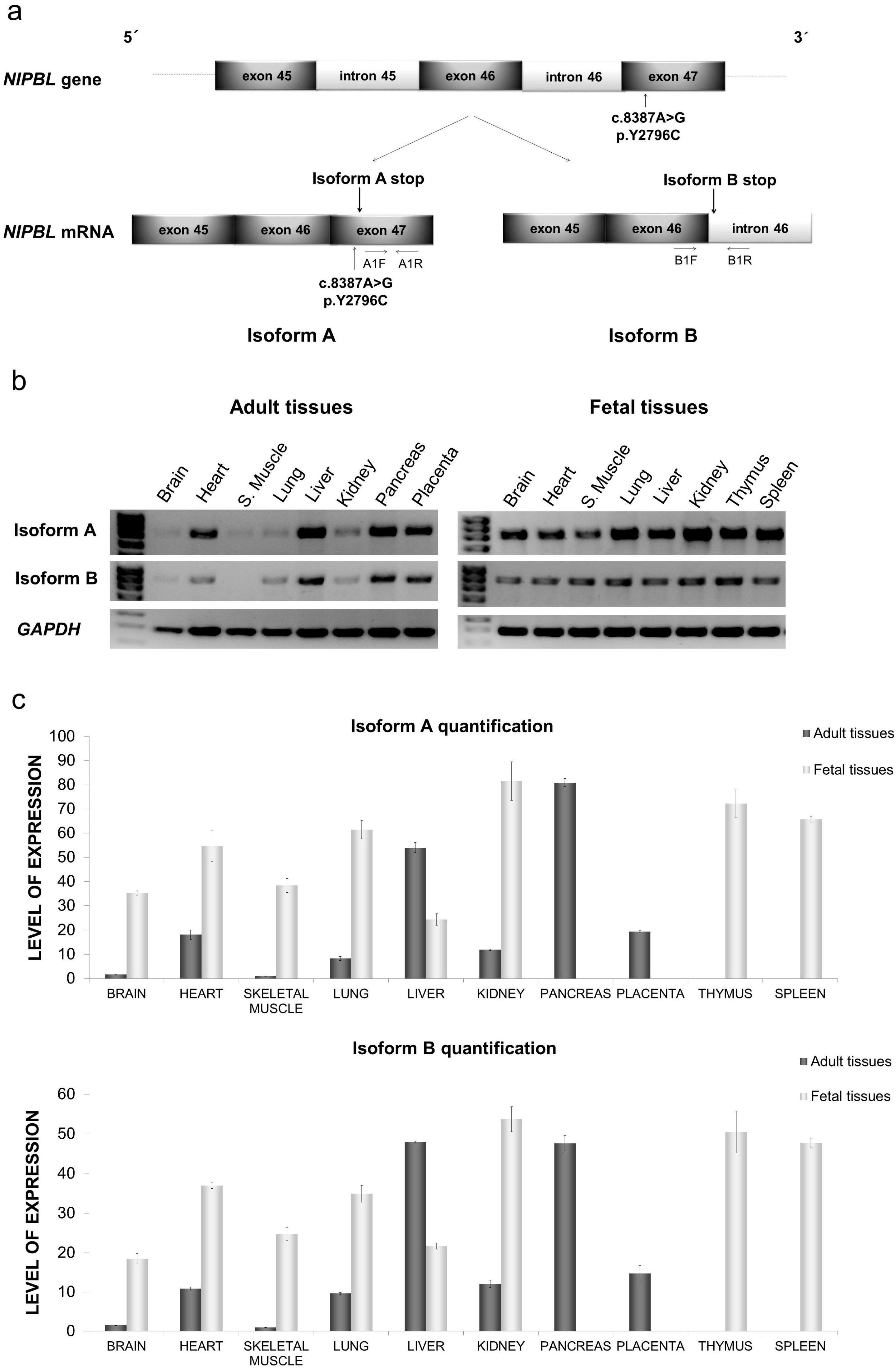
2.1. Distribution of mRNA NIPBL Isoforms A and B in Foetal and Adult Human Tissues
2.2. Quantitative mRNA Analysis of Isoforms A and B in Foetal and Adult Human Tissues
2.3. Structure Prediction of the C-Terminal Segment of the Isoforms A and B of the NIPBL Gene
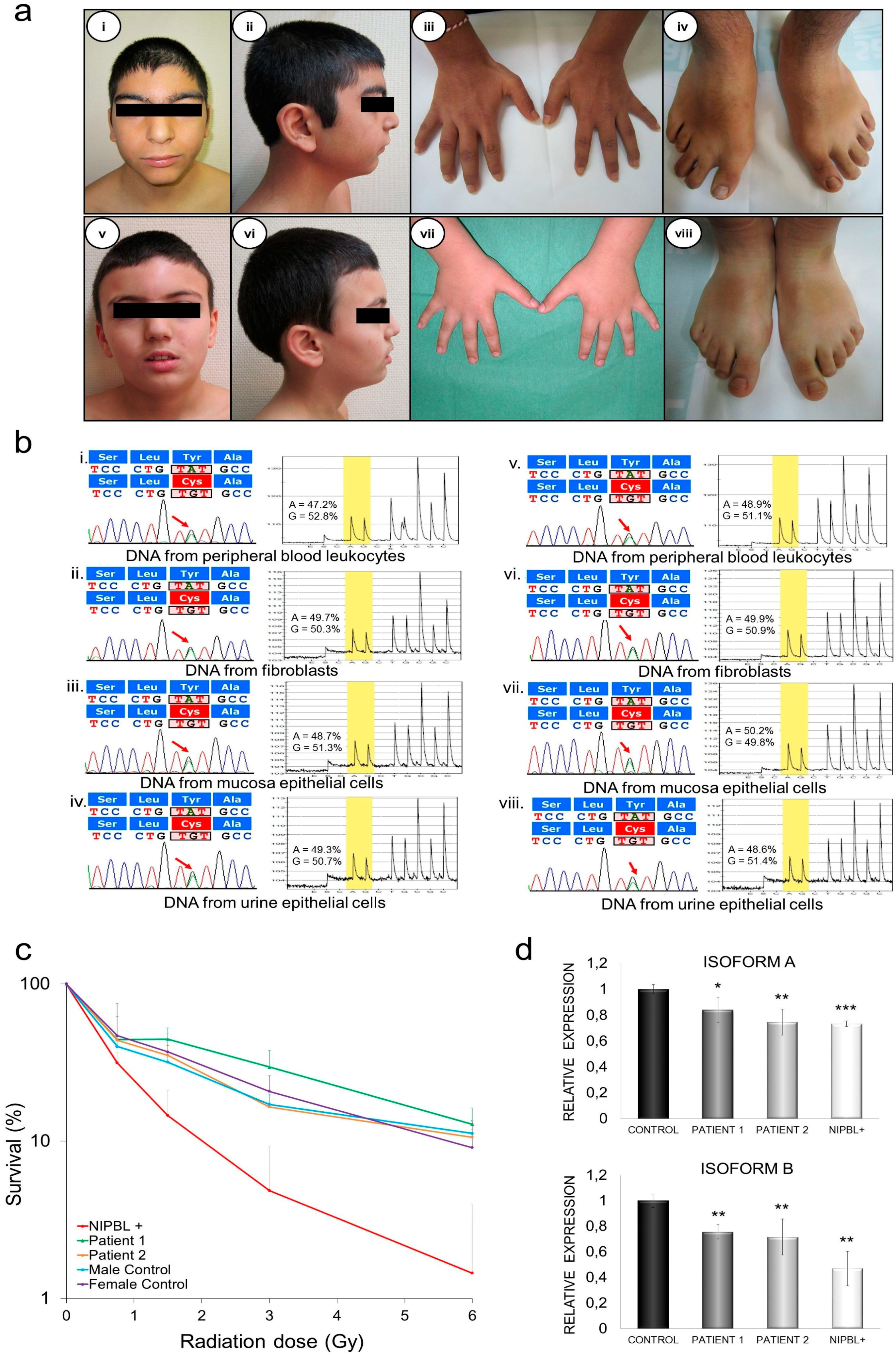
2.4. Clinical Description
2.4.1. Patient 1
2.4.2. Patient 2
2.5. Genetic Diagnosis and Pyrosequencing Analysis in Different Tissues
2.6. DNA Damage Sensibility in Patients’ Fibroblasts
2.7. Quantification of mRNA NIPBL Isoforms A and B in Patients’ Fibroblasts
3. Discussion
4. Materials and Methods
4.1. Identification of NIPBL Isoforms A and B in Foetal and Adult Human Tissues
4.2. Quantification of mRNA NIPBL Isoforms A and B by qPCR in Human Tissues
4.3. Patients
4.4. Genetic Diagnosis and Pyrosequencing Analysis
4.5. Structural In Silico Modeling of C-Terminal Domain of NIPBL Isoforms A and B and Bioinformatic Analysis of the Missense Pathological Variant in Isoform A
4.6. Cell Culture and Colony Formation Assay
4.7. Quantification of mRNA from NIPBL Isoforms A and B in Patients’ Fibroblasts
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Opitz, J.M. The Brachmann-de Lange syndrome. Am. J. Med. Genet. 1985, 22, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Kline, A.D.; Krantz, I.D.; Sommer, A.; Kliewer, M.; Jackson, L.G.; FitzPatrick, D.R.; Levin, A.V.; Selicorni, A. Cornelia de Lange syndrome: Clinical review, diagnostic and scoring systems, and anticipatory guidance. Am. J. Med. Genet. A 2007, 143A, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Krantz, I.D.; McCallum, J.; DeScipio, C.; Kaur, M.; Gillis, L.A.; Yaeger, D.; Jukofsky, L.; Wasserman, N.; Bottani, A.; Morris, C.A.; et al. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat. Genet. 2004, 36, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Tonkin, E.T.; Wang, T.J.; Lisgo, S.; Bamshad, M.J.; Strachan, T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat. Genet. 2004, 36, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Musio, A.; Selicorni, A.; Focarelli, M.L.; Gervasini, C.; Milani, D.; Russo, S.; Vezzoni, P.; Larizza, L. X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat. Genet. 2006, 38, 528–530. [Google Scholar] [CrossRef] [PubMed]
- Deardorff, M.A.; Kaur, M.; Yaeger, D.; Rampuria, A.; Korolev, S.; Pie, J.; Gil-Rodríguez, C.; Arnedo, M.; Loeys, B.; Kline, A.D.; et al. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of Cornelia de Lange syndrome with predominant mental retardation. Am. J. Hum. Genet. 2007, 80, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Deardorff, M.A.; Wilde, J.J.; Albrecht, M.; Dickinson, E.; Tennstedt, S.; Braunholz, D.; Mönnich, M.; Yan, Y.; Xu, W.; Gil-Rodríguez, M.C.; et al. RAD21 mutations cause a human cohesinopathy. Am. J. Hum. Genet. 2012, 90, 1014–1027. [Google Scholar] [CrossRef] [PubMed]
- Deardorff, M.A.; Bando, M.; Nakato, R.; Watrin, E.; Itoh, T.; Minamino, M.; Saitoh, K.; Komata, M.; Katou, Y.; Clark, D.; et al. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 2012, 489, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Remeseiro, S.; Losada, A. Cohesin, a chromatin engagement ring. Curr. Opin. Cell. Biol. 2013, 25, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Mannini, L.; Cucco, F.; Quarantotti, V.; Krantz, I.D.; Musio, A. Mutation spectrum and genotype–phenotype correlation in Cornelia de Lange syndrome. Hum. Mutat. 2013, 34, 1589–1596. [Google Scholar] [CrossRef] [PubMed]
- Teresa-Rodrigo, M.E.; Eckhold, J.; Puisac, B.; Dalski, A.; Gil-Rodríguez, M.C.; Braunholz, D.; Baquero, C.; Hernández-Marcos, M.; de Karam, J.C.; Ciero, M.; et al. Functional characterization of NIPBL physiological splice variants and eight splicing mutations in patients with Cornelia de Lange syndrome. Int. J. Mol. Sci. 2014, 15, 10350–10364. [Google Scholar] [PubMed]
- Källberg, M.; Haipeng, W.; Sheng, W.; Jian, P.; Zhiyong, W.; Hui, L.; Jinbo, X. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 2012, 7, 1511–1522. [Google Scholar] [CrossRef] [PubMed]
- PolyPhen-2: Prediction of Functional Effects of Human nsSNPs. Available online: http://genetics.bwh.harvard.edu/pph/ (accessed on 21 December 2016).
- PredictSNP: Consensus Classifiers for Prediction of Disease-Related Mutations. Available online: http://loschmidt.chemi.muni.cz/predictsnp/ (accessed on 21 December 2016).
- Pié, J.; Gil-Rodríguez, M.C.; Ciero, M.; López-Viñas, E.; Ribate, M.P.; Arnedo, M.; Deardorff, M.A.; Puisac, B.; Legarreta, J.; de Karam, J.C.; et al. Mutations and variants in the cohesion factor genes NIPBL, SMC1A, and SMC3 in a cohort of 30 unrelated patients with Cornelia de Lange syndrome. Am. J. Med. Gene A 2010, 152A, 924–929. [Google Scholar] [CrossRef] [PubMed]
- Enervald, E.; Du, L.; Visnes, T.; Björkman, A.; Lindgren, E.; Wincent, J.; Borck, G.; Colleaux, L.; Cormier-Daire, V.; van Gent, D.C. A regulatory role for the cohesin loader NIPBL in nonhomologous end joining during immunoglobulin class switch recombination. J. Exp. Med. 2013, 210, 2503–2513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorsett, D. Cohesin, gene expression and development: Lessons from Drosophila. Chromosom. Res. 2009, 17, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Muto, A.; Ikeda, S.; Lopez-Burks, M.E.; Kikuchi, Y.; Calof, A.L.; Lander, A.D.; Schilling, T.F. NIPBL and mediator cooperatively regulate gene expression to control limb development. PLoS Genet. 2014, 10, e1004671. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, T.W.; Graveley, B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Saifi, G.M.; Wierzba, T.H.; Withers, M.; Bien-Willner, G.A.; Limon, J.; Stankiewicz, P.; Lupski, J.R.; Wierzba, J. Mutational and genotype-phenotype correlation analyses in 28 Polish patients with Cornelia de Lange syndrome. Am. J. Med. Genet. A 2006, 140, 1531–1541. [Google Scholar] [CrossRef] [PubMed]
- Holmberg, J.; Clarke, D.L.; Frisén, J. Regulation of repulsion versus adhesion by different splice forms of an EPH receptor. Nature 2000, 408, 203–206. [Google Scholar] [PubMed]
- Chen, T.; Tang, Z.; Yan, A.; Li, W.; Lin, H. Molecular cloning and mRNA expression analysis of two GH secretagogue receptor transcripts in orange-spotted grouper (Epinephelus coioides). J. Endocrinol. 2008, 199, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, F.J.; Ansari, M.; Braunholz, D.; Gil-Rodriguez, M.C.; Decroos, C.; Wilde, J.J.; Fincher, C.T.; Kaur, M.; Bando, M.; Amor, D.J.; et al. Loss-of-function HDAC mutations cause a phenotypic spectrum of Cornelia de Lange syndrome-like features, ocular hypertelorism, large fontanelle and X-linked inheritance. Hum. Mol. Genet. 2014, 23, 2888–2900. [Google Scholar] [CrossRef]
- LOVD—Leiden Open Variation Database. Available online: http://www.lovd.nl/3.0/home (accessed on 21 December 2016).
- Vrouwe, M.G.; Elghalbzouri-Maghrani, E.; Meijers, M.; Schouten, P.; Godthelp, B.C.; Bhuiyan, Z.A.; Redeker, E.J.; Mannens, M.M.; Mullenders, L.H.; Pastink, A.; et al. Increased DNA damage sensitivity of Cornelia de Lange syndrome cells: Evidence for impaired recombinational repair. Hum. Mol. Genet. 2007, 16, 1478–1487. [Google Scholar] [CrossRef] [PubMed]
- Human Genome Variation Society. Available online: http://www.hgvs.org/ (accessed on 21 December 2016).



{kind=link}
{kind=link}
| Clinical Data | Patient 1 | Patient 2 | |
|---|---|---|---|
| Anthropometric data (newborn) | Gestational age | 36 weeks | 35 weeks |
| Birth weight (g) | 2600 (P10–25) | 2420 (P50) | |
| Birth length (cm) | 46 (P10–25) | 46.7 (P50) | |
| Birth OFC (cm) | 31 (P10–25) | 30.5 (P10–25) | |
| Intrauterine growth retardation | No | No | |
| Anthropometric data (last evaluation) | Age at evaluation | 14 years 3 month | 10 years 8 month |
| Weight (kg) | 54 (P25–50) | 36.3 (P25–50) | |
| Length (cm) | 159.5 (P25–50) | 136.5 (P3–25) | |
| OFC (cm) | 52.5 (P3–25) | 52.5 (P25–50) | |
| Postnatal growth retardation | No | No | |
| Limb abnomalities | Bilateral brachydactyly—clinodactyly 5th finger Bilateral 2–3 syndactyly (feet) | 2–3 partial syndactyly (feet) | |
| Developmental delay | + | + | |
| Intellectual disability | + | + | |
| Microcephaly | + | - | |
| Behavior problems | + | + | |
| Atypical wide nose with broad nasal bridge | + | + | |
| Hypoacusia | − | - | |
| Gastroesophageal reflux disease | − | + | |
| Feeding (swallowing) problems | − | + | |
| Hirsutism | + | - | |
| Cutis marmorata | − | - | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puisac, B.; Teresa-Rodrigo, M.-E.; Hernández-Marcos, M.; Baquero-Montoya, C.; Gil-Rodríguez, M.-C.; Visnes, T.; Bot, C.; Gómez-Puertas, P.; Kaiser, F.J.; Ramos, F.J.; et al. mRNA Quantification of NIPBL Isoforms A and B in Adult and Fetal Human Tissues, and a Potentially Pathological Variant Affecting Only Isoform A in Two Patients with Cornelia de Lange Syndrome. Int. J. Mol. Sci. 2017, 18, 481. https://doi.org/10.3390/ijms18030481
Puisac B, Teresa-Rodrigo M-E, Hernández-Marcos M, Baquero-Montoya C, Gil-Rodríguez M-C, Visnes T, Bot C, Gómez-Puertas P, Kaiser FJ, Ramos FJ, et al. mRNA Quantification of NIPBL Isoforms A and B in Adult and Fetal Human Tissues, and a Potentially Pathological Variant Affecting Only Isoform A in Two Patients with Cornelia de Lange Syndrome. International Journal of Molecular Sciences. 2017; 18(3):481. https://doi.org/10.3390/ijms18030481
Chicago/Turabian StylePuisac, Beatriz, María-Esperanza Teresa-Rodrigo, María Hernández-Marcos, Carolina Baquero-Montoya, María-Concepción Gil-Rodríguez, Torkild Visnes, Christopher Bot, Paulino Gómez-Puertas, Frank J. Kaiser, Feliciano J. Ramos, and et al. 2017. "mRNA Quantification of NIPBL Isoforms A and B in Adult and Fetal Human Tissues, and a Potentially Pathological Variant Affecting Only Isoform A in Two Patients with Cornelia de Lange Syndrome" International Journal of Molecular Sciences 18, no. 3: 481. https://doi.org/10.3390/ijms18030481
APA StylePuisac, B., Teresa-Rodrigo, M. -E., Hernández-Marcos, M., Baquero-Montoya, C., Gil-Rodríguez, M. -C., Visnes, T., Bot, C., Gómez-Puertas, P., Kaiser, F. J., Ramos, F. J., Ström, L., & Pié, J. (2017). mRNA Quantification of NIPBL Isoforms A and B in Adult and Fetal Human Tissues, and a Potentially Pathological Variant Affecting Only Isoform A in Two Patients with Cornelia de Lange Syndrome. International Journal of Molecular Sciences, 18(3), 481. https://doi.org/10.3390/ijms18030481