
Revisiting the Metabolism and Bioactivation of Ketoconazole in Human and Mouse Using Liquid Chromatography–Mass Spectrometry-Based Metabolomics



Abstract
:
1. Introduction
2. Results
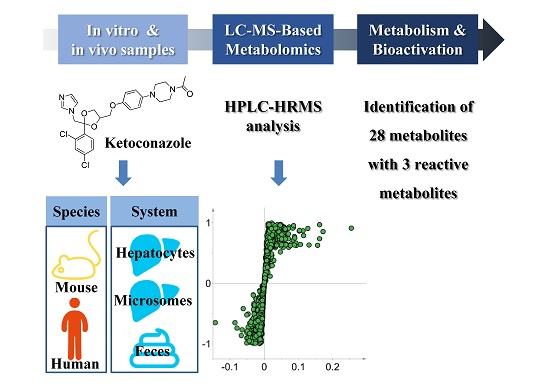
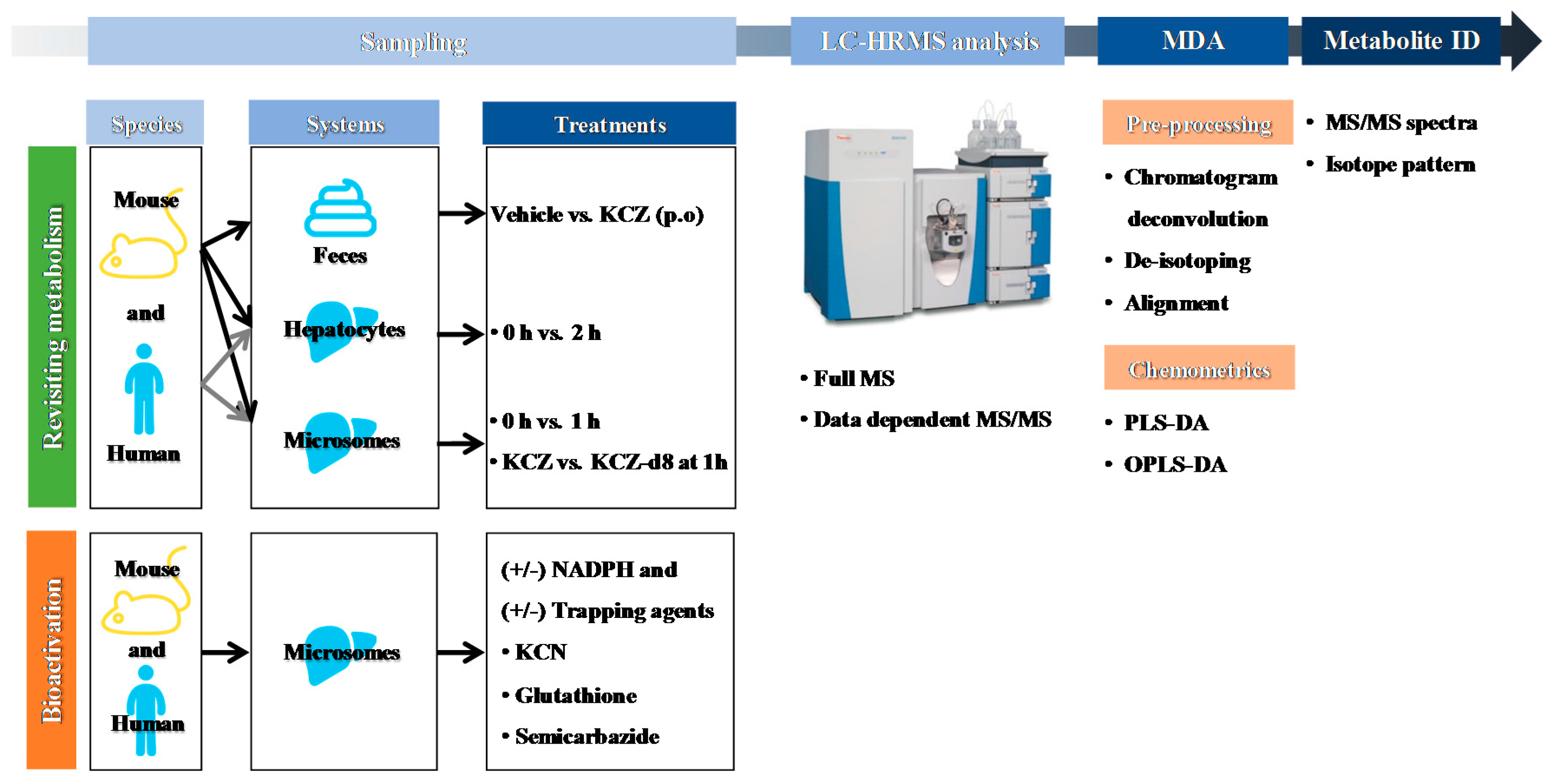
2.1. Overall Strategy for KCZ Metabolite Profiling
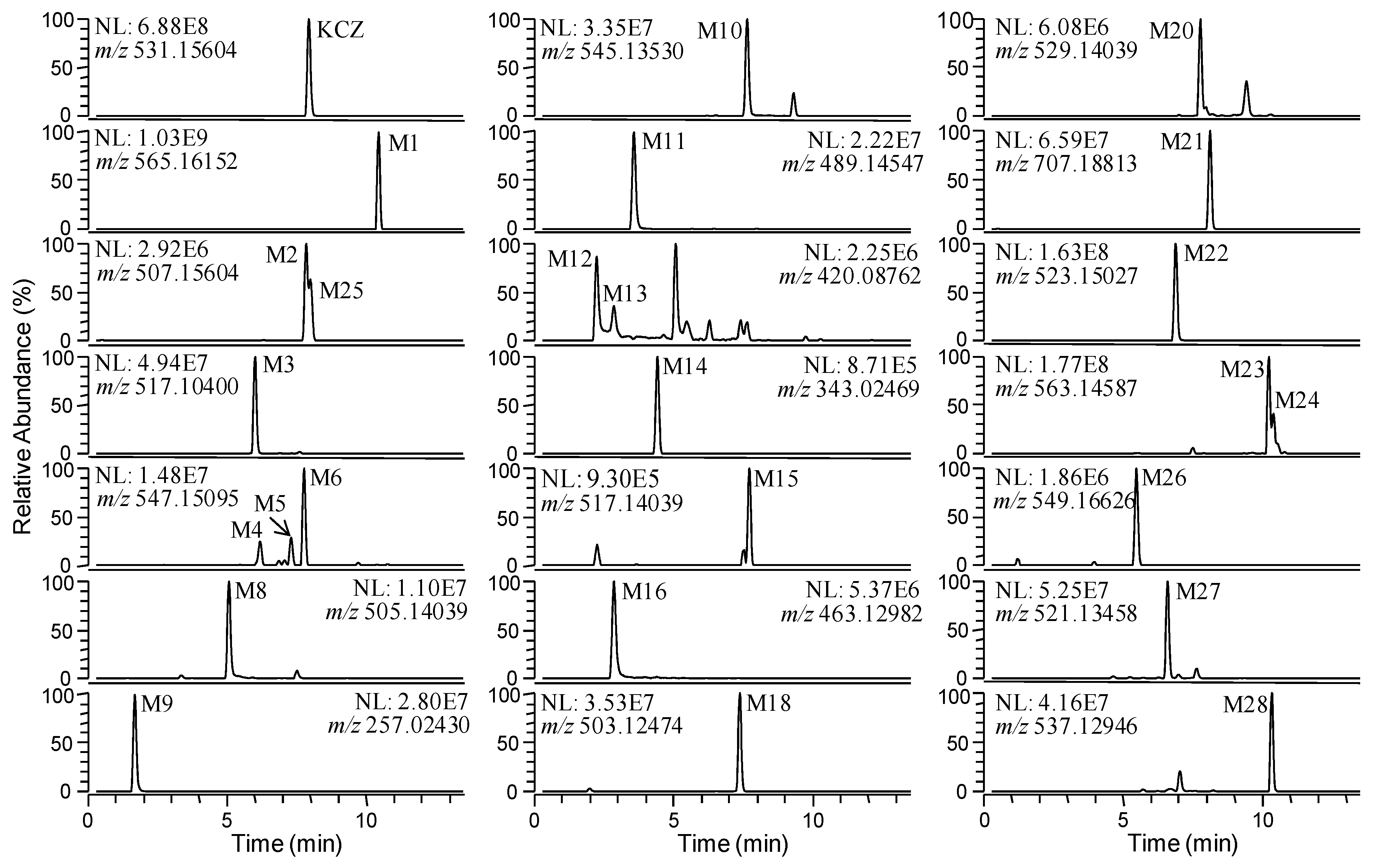
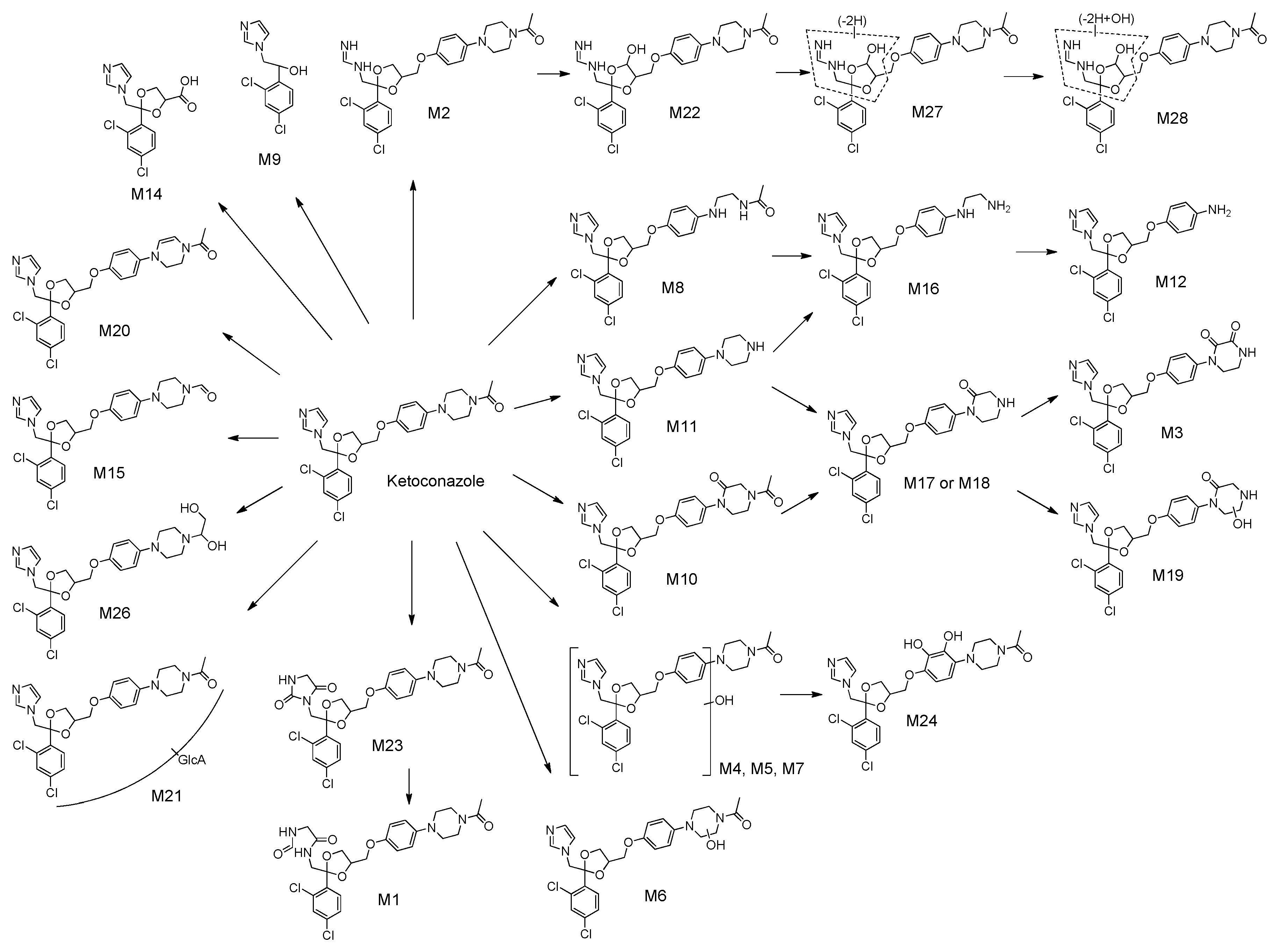
2.2. Identification of KCZ Metabolites
2.2.1. Mouse and Human Liver Microsomes
2.2.2. Mouse or Human Hepatocytes
2.2.3. Mouse Feces
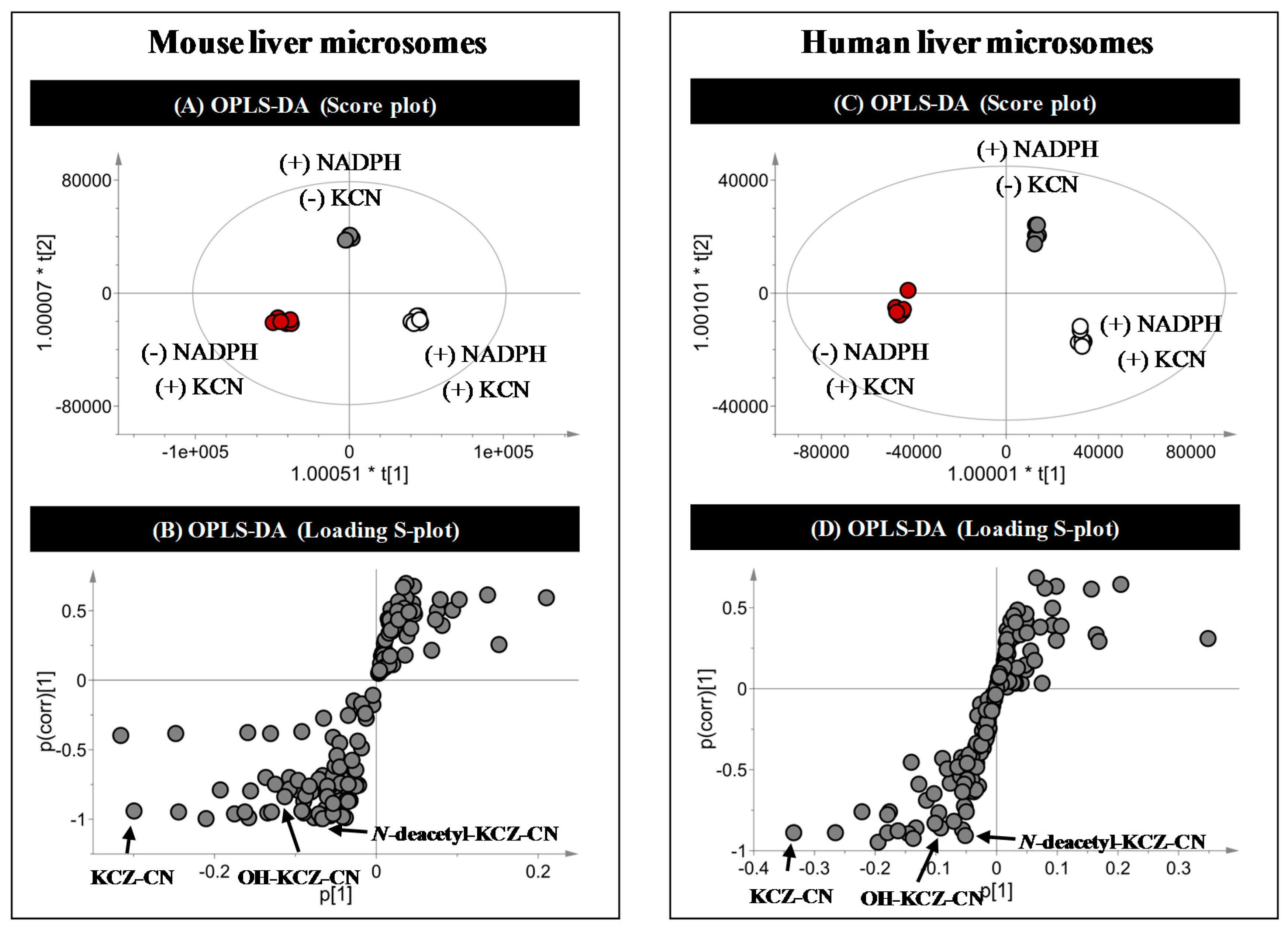
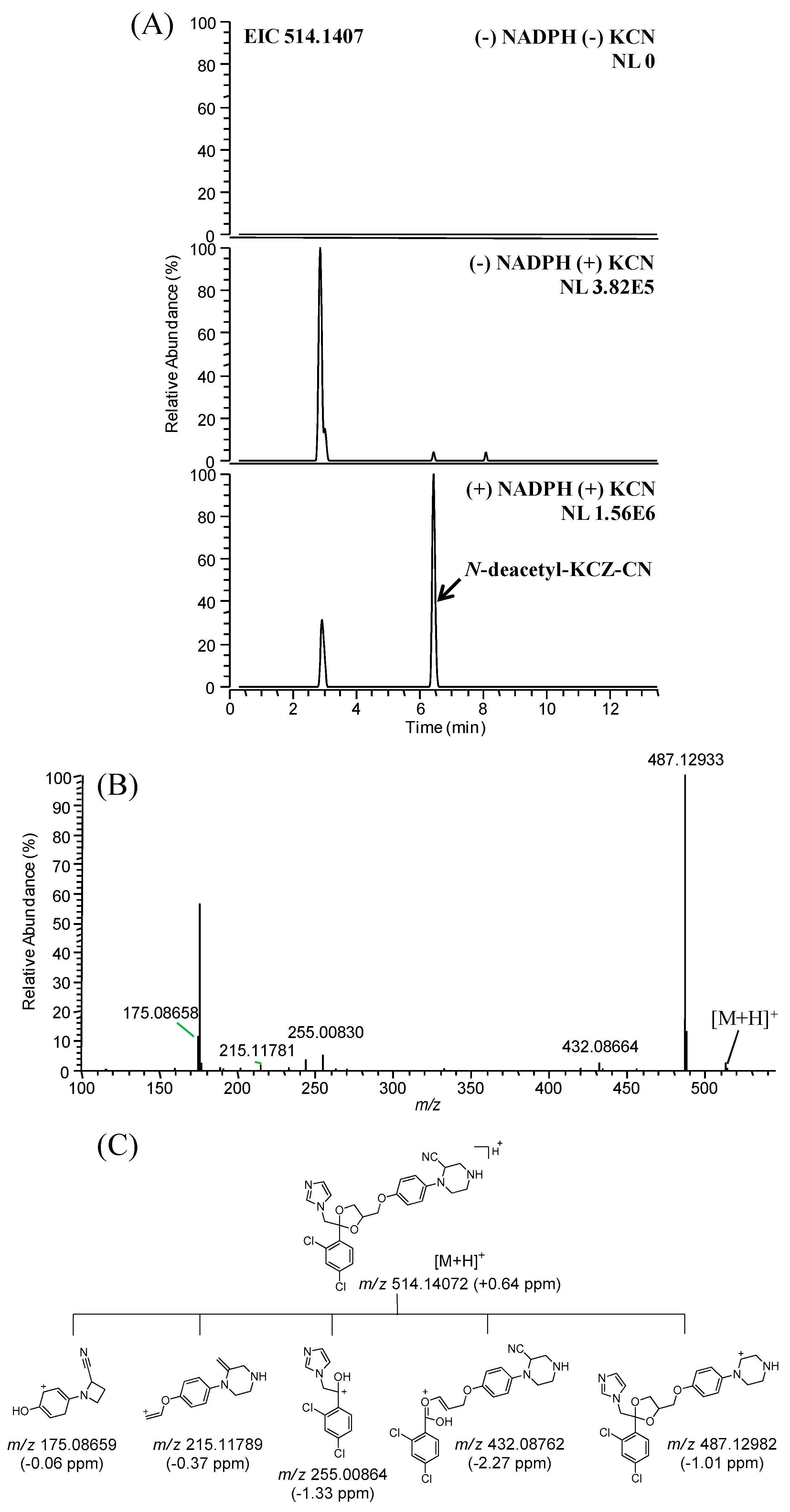
2.3. Identification of Reactive Metabolites
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. In Vitro Incubation of KCZ in Liver Microsomes
4.3. In Vitro Incubation of KCZ in Hepatocytes
4.4. In Vivo Metabolite Profiling of KCZ in Mouse Feces
4.5. Screening of KCZ Reactive Metabolites in Liver Microsomes
4.6. High-Performance Liquid Chromatography (HPLC)–High Resolution Mass Spectrometry (HRMS) Analysis
4.7. Data Pre-Processing and Multivariate Data Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gupta, A.K.; Daigle, D.; Foley, K.A. Drug safety assessment of oral formulations of ketoconazole. Expert Opin. Drug Saf. 2015, 14, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Ghannoum, M.A.; Rice, L.B. Antifungal agents: Mode of action, mechanisms of resistance, and correlation of these mechanisms with bacterial resistance. Clin. Microbiol. Rev. 1999, 12, 501–517. [Google Scholar] [PubMed]
- Lewis, R.E. Current concepts in antifungal pharmacology. Mayo Clin. Proc. 2011, 86, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Gascoigne, E.; Barton, G.; Michaels, M.; Meuldermans, W.; Heykants, J. The kinetics of ketoconazole in animals and man. Clin. Res. Rev. 1981, 1, 177–187. [Google Scholar]
- Daneshmend, T.K.; Warnock, D.W. Clinical pharmacokinetics of ketoconazole. Clin. Pharmacokinet. 1988, 14, 13–34. [Google Scholar] [CrossRef] [PubMed]
- Remmel, R.P.; Amoh, K.; Abdel-Monem, M.M. The disposition and pharmacokinetics of ketoconazole in the rat. Drug Metab. Dispos. 1987, 15, 735–739. [Google Scholar] [PubMed]
- Whitehouse, L.W.; Menzies, A.; Dawson, B.; Zamecnik, J.; Sy, W.W. Deacetylated ketoconazole: A major ketoconazole metabolite isolated from mouse liver. J. Pharm. Biomed. Anal. 1990, 8, 603–606. [Google Scholar] [CrossRef]
- Whitehouse, L.W.; Menzies, A.; Dawson, B.; Cyr, T.D.; By, A.W.; Black, D.B.; Zamecnik, J. Mouse hepatic metabolites of ketoconazole: Isolation and structure elucidation. J. Pharm. Biomed. Anal. 1994, 12, 1425–1441. [Google Scholar] [CrossRef]
- Rodriguez, R.J.; Acosta, D., Jr. Metabolism of ketoconazole and deacetylated ketoconazole by rat hepatic microsomes and flavin-containing monooxygenases. Drug Metab. Dispos. 1997, 25, 772–777. [Google Scholar] [PubMed]
- Rodriguez, R.J.; Proteau, P.J.; Marquez, B.L.; Hetherington, C.L.; Buckholz, C.J.; O’Connell, K.L. Flavin-containing monooxygenase-mediated metabolism of N-deacetyl ketoconazole by rat hepatic microsomes. Drug Metab. Dispos. 1999, 27, 880–886. [Google Scholar] [PubMed]
- Fitch, W.; Tran, T.; Young, M.; Liu, L.; Chen, Y. Revisiting the metabolism of ketoconazole using accurate mass. Drug Metab. Lett. 2009, 3, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Wewering, F.; Jouy, F.; Wissenbach, D.K.; Gebauer, S.; Bluher, M.; Gebhardt, R.; Pirow, R.; von Bergen, M.; Kalkhof, S.; Luch, A.; et al. Characterization of chemical-induced sterile inflammation in vitro: Application of the model compound ketoconazole in a human hepatic co-culture system. Arch. Toxicol. 2017, 91, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Bourcier, K.; Hyland, R.; Kempshall, S.; Jones, R.; Maximilien, J.; Irvine, N.; Jones, B. Investigation into UDP-glucuronosyltransferase (UGT) enzyme kinetics of imidazole- and triazole-containing antifungal drugs in human liver microsomes and recombinant ugt enzymes. Drug Metab. Dispos. 2010, 38, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Shehu, A.I.; Ma, X.; Venkataramanan, R. Mechanisms of drug-induced hepatotoxicity. Clin. Liver Dis. 2017, 21, 35–54. [Google Scholar] [CrossRef] [PubMed]
- Benichou, C. Criteria of drug-induced liver disorders. Report of an international consensus meeting. J. Hepatol. 1990, 11, 272–276. [Google Scholar] [PubMed]
- Danan, G.; Benichou, C. Causality assessment of adverse reactions to drugs—I. A novel method based on the conclusions of international consensus meetings: Application to drug-induced liver injuries. J. Clin. Epidemiol. 1993, 46, 1323–1330. [Google Scholar] [CrossRef]
- Danan, G.; Teschke, R. RUCAM in drug and herb induced liver injury: The update. Int. J. Mol. Sci. 2015, 17, 14. [Google Scholar] [CrossRef] [PubMed]
- Weiler, S.; Merz, M.; Kullak-Ublick, G.A. Drug-induced liver injury: The dawn of biomarkers? F1000Prime Rep. 2015, 7, 34. [Google Scholar] [CrossRef] [PubMed]
- Garcia Rodriguez, L.A.; Duque, A.; Castellsague, J.; Perez-Gutthann, S.; Stricker, B.H. A cohort study on the risk of acute liver injury among users of ketoconazole and other antifungal drugs. Br. J. Clin. Pharmacol. 1999, 48, 847–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, W.Y.; Su, C.W.; Huang, Y.S.; Chou, Y.C.; Chen, Y.C.; Chung, W.H.; Hou, M.C.; Lin, H.C.; Lee, F.Y.; Wu, J.C. Risk of oral antifungal agent-induced liver injury in taiwanese. Br. J. Clin. Pharmacol. 2014, 77, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.Y.; Nie, X.L.; Tao, Q.M.; Zhan, S.Y.; Zhang, Y.D. Ketoconazole associated hepatotoxicity: A systematic review and meta-analysis. Biomed. Environ. Sci. 2013, 26, 605–610. [Google Scholar] [PubMed]
- Rodriguez, R.J.; Acosta, D. N-deacetyl ketoconazole-induced hepatotoxicity in a primary culture system of rat hepatocytes. Toxicology 1997, 117, 123–131. [Google Scholar] [CrossRef]
- Rodriguez, R.J.; Buckholz, C.J. Hepatotoxicity of ketoconazole in sprague-dawley rats: Glutathione depletion, flavin-containing monooxygenases-mediated bioactivation and hepatic covalent binding. Xenobiotica 2003, 33, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Park, B.K.; Boobis, A.; Clarke, S.; Goldring, C.E.; Jones, D.; Kenna, J.G.; Lambert, C.; Laverty, H.G.; Naisbitt, D.J.; Nelson, S.; et al. Managing the challenge of chemically reactive metabolites in drug development. Nat. Rev. Drug Discov. 2011, 10, 292–306. [Google Scholar] [CrossRef] [PubMed]
- Dalvie, D.; Kalgutkar, A.S.; Chen, W. Practical approaches to resolving reactive metabolite liabilities in early discovery. Drug Metab. Rev. 2015, 47, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Schadt, S.; Simon, S.; Kustermann, S.; Boess, F.; McGinnis, C.; Brink, A.; Lieven, R.; Fowler, S.; Youdim, K.; Ullah, M.; et al. Minimizing dili risk in drug discovery—A screening tool for drug candidates. Toxicol. In Vitro 2015, 30, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.A.; Isin, E.M.; Ogese, M.O.; Mettetal, J.T.; Williams, D.P. Reactive metabolites: Current and emerging risk and hazard assessments. Chem. Res. Toxicol. 2016, 29, 505–533. [Google Scholar] [CrossRef] [PubMed]
- Argoti, D.; Liang, L.; Conteh, A.; Chen, L.; Bershas, D.; Yu, C.P.; Vouros, P.; Yang, E. Cyanide trapping of iminium ion reactive intermediates followed by detection and structure identification using liquid chromatography—Tandem mass spectrometry (LC-MS/MS). Chem. Res. Toxicol. 2005, 18, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Rousu, T.; Pelkonen, O.; Tolonen, A. Rapid detection and characterization of reactive drug metabolites in vitro using several isotope-labeled trapping agents and ultra-performance liquid chromatography/time-of-flight mass spectrometry. Rapid Commun. Mass Spectr. 2009, 23, 843–855. [Google Scholar] [CrossRef] [PubMed]
- Plumb, R.S.; Stumpf, C.L.; Granger, J.H.; Castro-Perez, J.; Haselden, J.N.; Dear, G.J. Use of liquid chromatography/time-of-flight mass spectrometry and multivariate statistical analysis shows promise for the detection of drug metabolites in biological fluids. Rapid Commun. Mass Spectr. 2003, 17, 2632–2638. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Gonzalez, F.J.; Idle, J.R. LC-MS-based metabolomics in drug metabolism. Drug Metab. Rev. 2007, 39, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.Z.; Gonzalez, F.J. LC-MS-based metabolomics: An update. Arch. Toxicol. 2014, 88, 1491–1502. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Lechon, M.J.; Tolosa, L.; Donato, M.T. Metabolic activation and drug-induced liver injury: In vitro approaches for the safety risk assessment of new drugs. J. Appl. Toxicol. 2016, 36, 752–768. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.W.; Lee, G.; Coukos, J.S.; Moellering, R.E. Profiling reactive metabolites via chemical trapping and targeted mass spectrometry. Anal. Chem. 2016, 88, 6658–6661. [Google Scholar] [CrossRef] [PubMed]
- Kalgutkar, A.S.; Dalvie, D. Predicting toxicities of reactive metabolite-positive drug candidates. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 35–54. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Lu, J.; Ma, X. Metabolomic screening and identification of the bioactivation pathways of ritonavir. Chem. Res. Toxicol. 2011, 24, 2109–2114. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.Z.; Krausz, K.W.; Li, F.; Cheng, J.; Tanaka, N.; Gonzalez, F.J. Metabolic map and bioactivation of the anti-tumour drug noscapine. Br. J. Pharmacol. 2012, 167, 1271–1286. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.J.; Fang, Z.Z.; Ma, X. Transgenic mice and metabolomics for study of hepatic xenobiotic metabolism and toxicity. Expert Opin. Drug Metab. Toxicol. 2015, 11, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Gonzalez, F.J.; Ma, X. LC-MS-based metabolomics in profiling of drug metabolism and bioactivation. Acta Pharm. Sin. B 2012, 2, 118–125. [Google Scholar] [CrossRef]
- Chen, C.; Kim, S. LC-MS-based metabolomics of xenobiotic-induced toxicities. Comput. Struct. Biotechnol. J. 2013, 4, e201301008. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Fang, Z.Z.; Zheng, Y.; Zhou, K.; Hu, C.; Krausz, K.W.; Sun, D.; Idle, J.R.; Gonzalez, F.J. Metabolic profiling of praziquantel enantiomers. Biochem. Pharmacol. 2014, 90, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Lu, J.; Ma, X. CYP3A4-mediated α-hydroxyaldehyde formation in saquinavir metabolism. Drug Metab. Dispos. 2014, 42, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Riley, R.J.; Roberts, P.; Kitteringham, N.R.; Park, B.K. Formation of cytotoxic metabolites from phenytoin, imipramine, desipramine, amitriptyline and mianserin by mouse and human hepatic microsomes. Biochem. Pharmacol. 1990, 39, 1951–1958. [Google Scholar] [CrossRef]
- Doss, G.A.; Miller, R.R.; Zhang, Z.; Teffera, Y.; Nargund, R.P.; Palucki, B.; Park, M.K.; Tang, Y.S.; Evans, D.C.; Baillie, T.A.; et al. Metabolic activation of a 1,3-disubstituted piperazine derivative: Evidence for a novel ring contraction to an imidazoline. Chem. Res. Toxicol. 2005, 18, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Bauman, J.N.; Frederick, K.S.; Sawant, A.; Walsky, R.L.; Cox, L.M.; Obach, R.S.; Kalgutkar, A.S. Comparison of the bioactivation potential of the antidepressant and hepatotoxin nefazodone with aripiprazole, a structural analog and marketed drug. Drug Metab. Dispos. 2008, 36, 1016–1029. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Lu, J.; Ma, X. Profiling the reactive metabolites of xenobiotics using metabolomic technologies. Chem. Res. Toxicol. 2011, 24, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Oresic, M. Mzmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Formula | [M + H]+ (m/z) | Error (ppm) | tR (min) | Identified in | References and Comments | |
|---|---|---|---|---|---|---|---|
| Theoretical | Observed | ||||||
| KCZ | C26H28Cl2N4O4 | 531.1560 | 531.1552 | −1.56 | 7.9 | - | Parent |
| M1 | C26H30Cl2N4O6 | 565.1615 | 565.1599 | −2.85 | 10.5 | MLM, HLM, MHP, HHP, MF | [11] |
| M2 | C24H28Cl2N4O4 | 507.1560 | 507.1554 | −1.2 | 7.9 | MLM, HLM, HHP, MF | [11] |
| M3 | C24H22Cl2N4O5 | 517.1040 | 517.1032 | −1.64 | 6.0 | MLM, MHP, MF | [11] |
| M4 | C26H28Cl2N4O5 | 547.1510 | 547.1508 | −0.24 | 6.2 | MLM, HLM, MHP, HHP, MF | [11] |
| M5 | C26H28Cl2N4O5 | 547.1510 | 547.1502 | −1.46 | 7.3 | MLM, HLM, MHP, HHP, MF | [11] |
| M6 | C26H28Cl2N4O5 | 547.1510 | 547.1504 | −1.02 | 7.8 | MLM, HLM, MHP, HHP, MF | [11] |
| M7 | C26H28Cl2N4O5 | 547.1510 | 547.1508 | −0.24 | 9.2 | MLM, HLM, HHP | [11] |
| M8 | C24H26Cl2N4O4 | 505.1404 | 505.1401 | −0.5 | 5.1 | MLM, HLM, MHP, HHP, MF | [11] |
| M9 | C11H10Cl2N2O | 257.0243 | 257.0243 | +0.08 | 1.7 | MLM, HLM, MHP, HHP, MF | [11] |
| M10 | C26H26Cl2N4O5 | 545.1353 | 545.1349 | −0.75 | 7.0 | MLM, HLM, HHP, MF | [11] |
| M11 | C24H26Cl2N4O3 | 489.1455 | 489.1447 | −1.68 | 3.6 | MLM, HLM, MHP, HHP, MF | [11] |
| M12 | C20H19Cl2N3O3 | 420.0876 | 420.0878 | +0.71 | 1.8 | MLM, MHP, HHP | [11] |
| M13 | C20H19Cl2N3O3 | 420.0876 | 420.0872 | −1.1 | 2.9 | MLM, MHP, HHP, MF | [11] |
| M14 | C14H12Cl2N2O4 | 343.0247 | 343.0243 | −1.08 | 4.4 | MLM, MF | [11] |
| M15 | C25H26Cl2N4O4 | 517.1404 | 517.1394 | −1.91 | 7.7 | MLM | [8] |
| M16 | C22H24Cl2N4O3 | 463.1298 | 463.1293 | −1.06 | 2.9 | MLM, HLM, MHP, HHP, MF | [8] |
| M17 | C24H24Cl2N4O4 | 503.1247 | 503.1252 | +0.93 | 5.9 | MLM | Novel |
| M18 | C24H24Cl2N4O4 | 503.1247 | 503.1241 | −1.31 | 7.4 | MLM, HLM, MHP, HHP, MF | Novel |
| M19 | C24H24Cl2N4O5 | 519.1197 | 519.1204 | +1.48 | 5.8 | MLM | Novel |
| M20 | C26H26Cl2N4O4 | 529.1404 | 529.1397 | −1.4 | 7.8 | MLM, HLM, MHP, HHP, MF | Novel |
| M21 | C32H36Cl2N4O10 | 707.1881 | 707.1865 | −2.28 | 8.1 | MHP, HHP, MF | [13] |
| M22 | C24H28Cl2N4O5 | 523.1510 | 523.1503 | −1.3 | 6.9 | MHP, MF | Novel |
| M23 | C26H28Cl2N4O6 | 563.1459 | 563.1450 | −1.62 | 10.2 | HHP, MF | [11] |
| M24 | C26H28Cl2N4O6 | 563.1459 | 563.1453 | −0.98 | 10.4 | HHP, MF | [11] |
| M25 | C24H28Cl2N4O4 | 507.1560 | 507.1558 | −0.49 | 8.0 | HHP, MF | Novel |
| M26 | C26H30Cl2N4O5 | 549.1666 | 549.1663 | −0.62 | 5.5 | MF | Novel |
| M27 | C24H26Cl2N4O5 | 521.1353 | 521.1346 | −1.38 | 6.6 | MF | Novel |
| M28 | C24H26Cl2N4O6 | 537.1302 | 537.1295 | −1.41 | 7.0 | MF | Novel |
| Adducts | Formula | [M+H]+ (m/z) | Error (ppm) | tR (min) | Identified in | References and Comments | |
|---|---|---|---|---|---|---|---|
| Theoretical | Observed | ||||||
| KCZ-CN | C27H27Cl2N5O4 | 556.1513 | 556.1513 | −0.07 | 7.2 | MLM, HLM | [11,28,29] |
| OH-KCZ-CN | C27H27Cl2N5O5 | 572.1462 | 572.1457 | −0.89 | 7.0 | MLM, HLM | [11,28,29] |
| N-deacetyl-KCZ-CN | C25H25Cl2N5O3 | 514.1407 | 514.1416 | +1.71 | 6.5 | MLM, HLM | Novel |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-H.; Choi, W.-G.; Lee, S.; Lee, H.S. Revisiting the Metabolism and Bioactivation of Ketoconazole in Human and Mouse Using Liquid Chromatography–Mass Spectrometry-Based Metabolomics. Int. J. Mol. Sci. 2017, 18, 621. https://doi.org/10.3390/ijms18030621
Kim J-H, Choi W-G, Lee S, Lee HS. Revisiting the Metabolism and Bioactivation of Ketoconazole in Human and Mouse Using Liquid Chromatography–Mass Spectrometry-Based Metabolomics. International Journal of Molecular Sciences. 2017; 18(3):621. https://doi.org/10.3390/ijms18030621
Chicago/Turabian StyleKim, Ju-Hyun, Won-Gu Choi, Sangkyu Lee, and Hye Suk Lee. 2017. "Revisiting the Metabolism and Bioactivation of Ketoconazole in Human and Mouse Using Liquid Chromatography–Mass Spectrometry-Based Metabolomics" International Journal of Molecular Sciences 18, no. 3: 621. https://doi.org/10.3390/ijms18030621
APA StyleKim, J. -H., Choi, W. -G., Lee, S., & Lee, H. S. (2017). Revisiting the Metabolism and Bioactivation of Ketoconazole in Human and Mouse Using Liquid Chromatography–Mass Spectrometry-Based Metabolomics. International Journal of Molecular Sciences, 18(3), 621. https://doi.org/10.3390/ijms18030621