
The Five Immune Forces Impacting DNA-Based Cancer Immunotherapeutic Strategy



Abstract
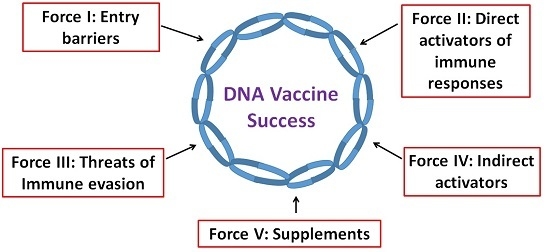
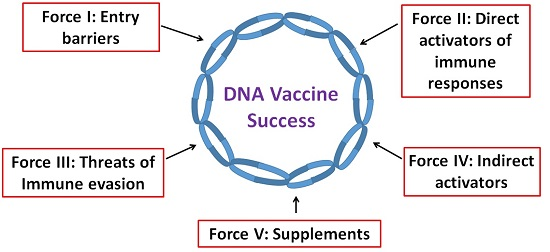
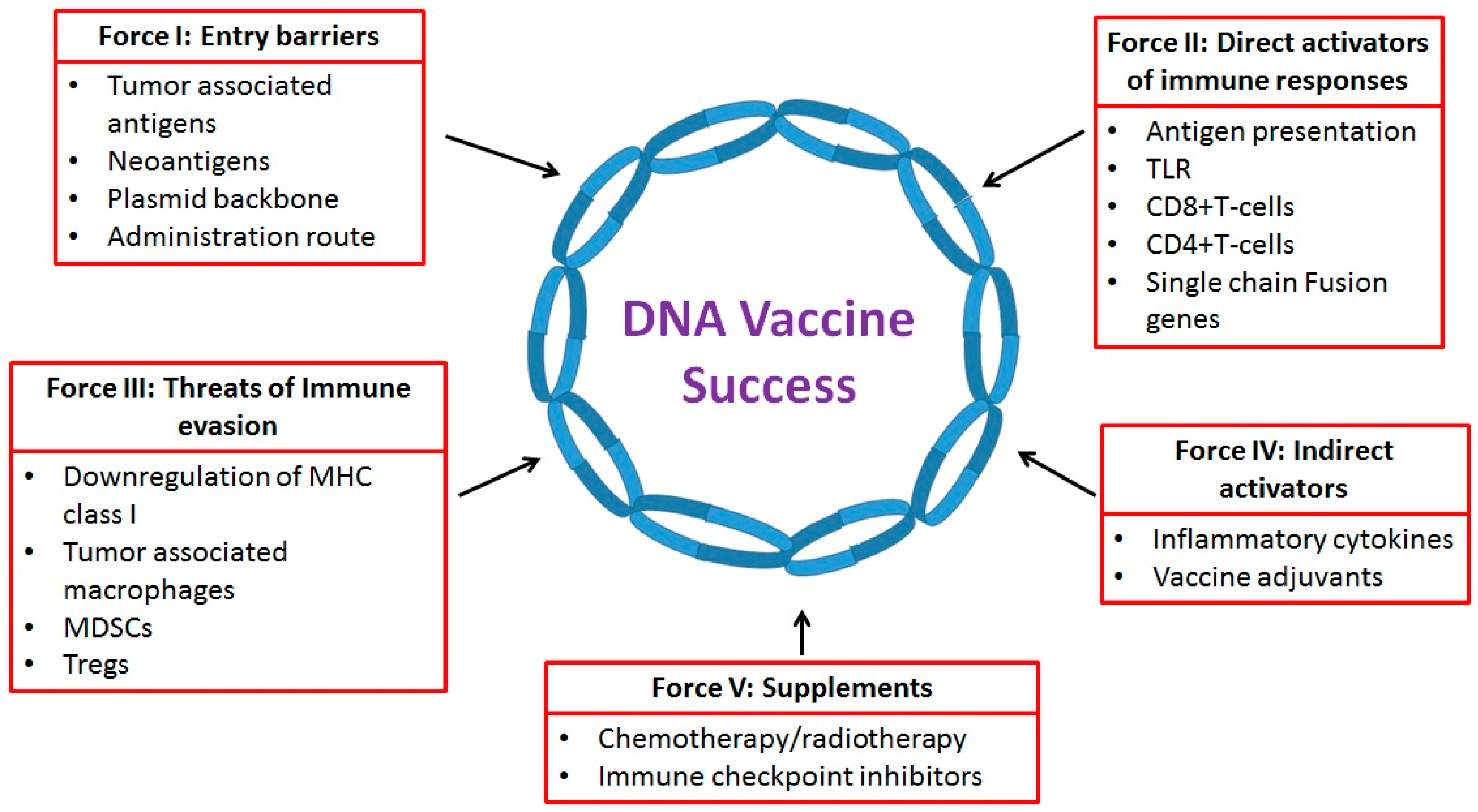
:
1. Introduction
2. Force I: Entry Barriers—Target Choice and Delivery Techniques
2.1. Tumor-Associated Antigens
2.2. Neoantigens
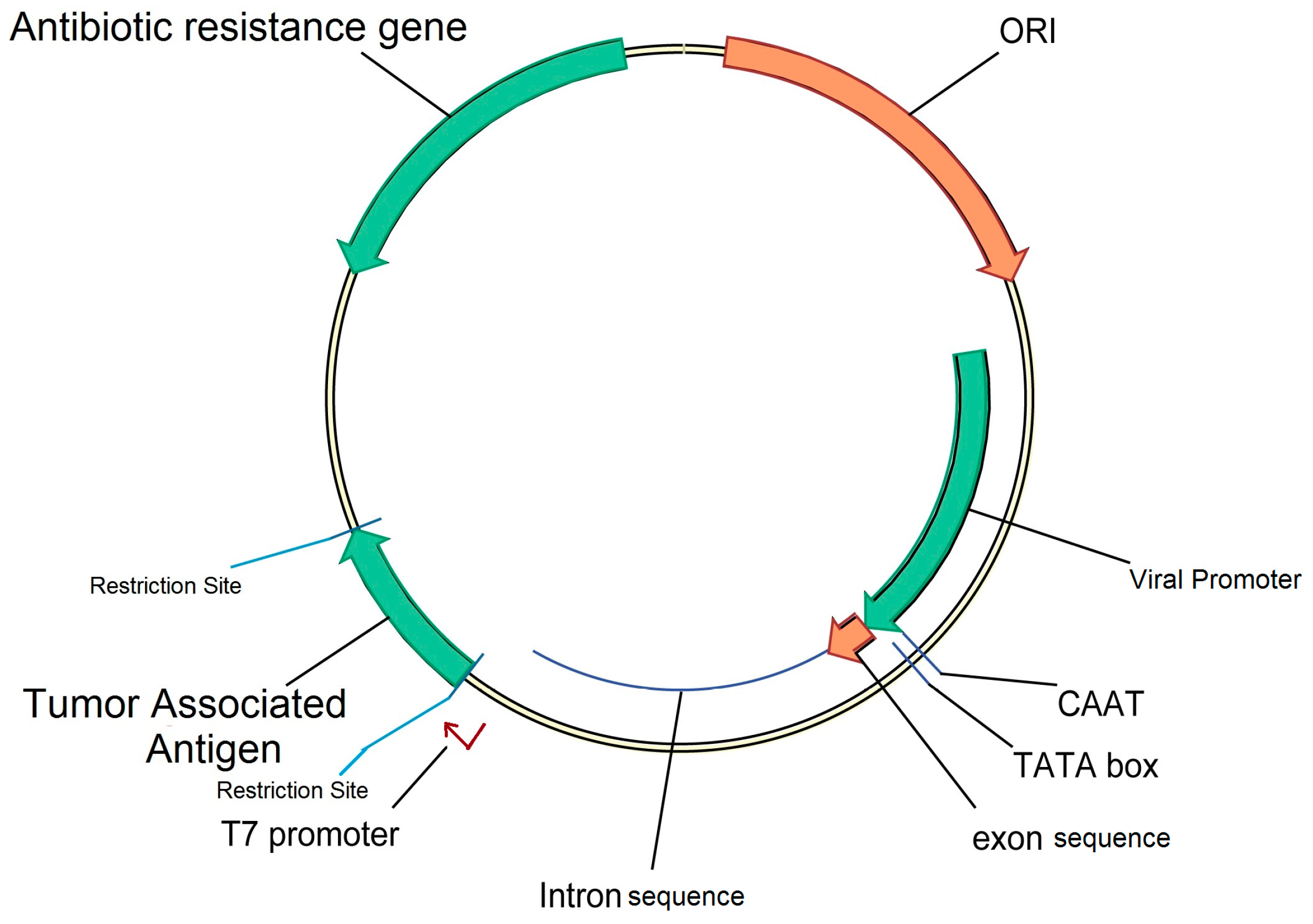
2.3. Plasmid Backbone
2.4. Administration Site
3. Force II: Direct Activators of T-Cell Responses
3.1. Role of Antigen Presenting Cells
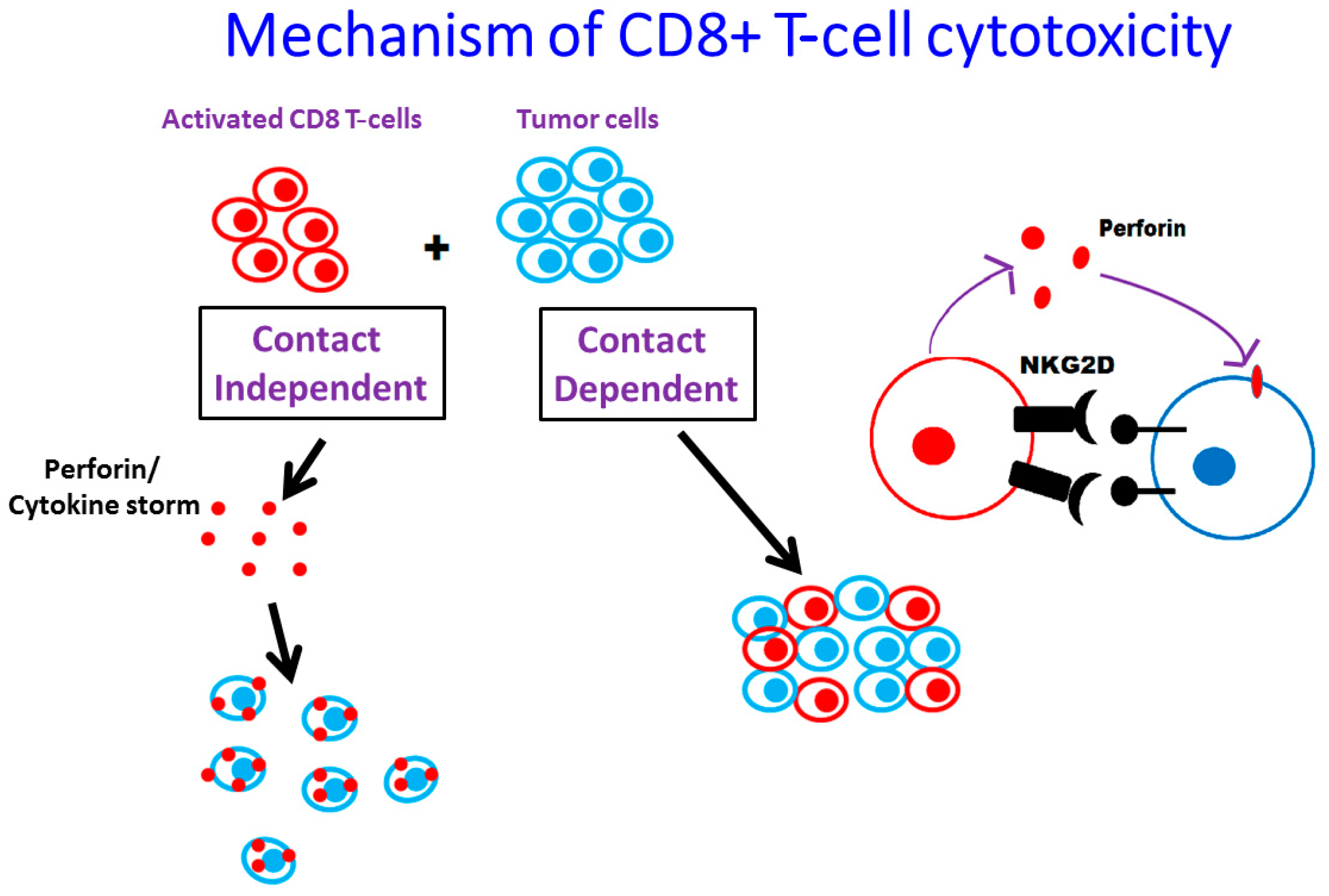
3.2. Role of CD8+ T-Cells
3.3. Role of CD4+ T-Cells
3.4. Role of Fusion Genes on T-Cell Activation
4. Force III: The Threat of Immune Evasion
4.1. Downregulation of MHC Class I on Tumor Cell
4.2. Tumor Associated Macrophages
4.3. Myeloid-Derived Suppressor Cells (MDSCs)
4.4. Regulatory T-Cells (Tregs)
5. Force IV: Indirect Activators
5.1. Inflammatory Cytokines
5.2. Adjuvants
6. Force V: Supplements
6.1. Combination with Chemotherapy and Radiotherapy
6.2. Combination with Immune-Check Point Inhibitors
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Guo, C.; Manjili, M.H.; Subjeck, J.R.; Sarkar, D.; Fisher, P.B.; Wang, X.Y. Therapeutic cancer vaccines: Past, present, and future. Adv. Cancer Res. 2013, 119, 421–475. [Google Scholar] [PubMed]
- Ferraro, B.; Morrow, M.P.; Hutnick, N.A.; Shin, T.H.; Lucke, C.E.; Weiner, D.B. Clinical applications of DNA vaccines: Current progress. Clin. Infect. Dis. 2011, 53, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Buonaguro, L.; Petrizzo, A.; Tornesello, M.L.; Buonaguro, F.M. Translating tumor antigens into cancer vaccines. Clin. Vaccine Immunol. 2011, 18, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Roosinovich, E.; Ma, B.; Hung, C.F.; Wu, T.C. Therapeutic HPV DNA vaccines. Immunol. Res. 2010, 47, 86–112. [Google Scholar] [CrossRef] [PubMed]
- Porter, M.E. The five competitive forces that shape strategy. Harv. Bus. Rev. 2008, 86, 78–93,137. [Google Scholar] [PubMed]
- Melero, I.; Gaudernack, G.; Gerritsen, W.; Huber, C.; Parmiani, G.; Scholl, S.; Thatcher, N.; Wagstaff, J.; Zielinski, C.; Faulkner, I.; et al. Therapeutic vaccines for cancer: An overview of clinical trials. Nat. Rev. Clin. Oncol. 2014, 11, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Finn, O.J. Cancer immunology. N. Engl. J. Med. 2008, 358, 2704–2715. [Google Scholar] [CrossRef] [PubMed]
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M.; et al. The prioritization of cancer antigens: A national cancer institute pilot project for the acceleration of translational research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.R.; Sorensen, M.R.; Buus, S.; Christensen, J.P.; Thomsen, A.R. Comparison of vaccine-induced effector CD8 T cell responses directed against self- and non-self-tumor antigens: Implications for cancer immunotherapy. J. Immunol. 2013, 191, 3955–3967. [Google Scholar] [CrossRef] [PubMed]
- Klein, L.; Hinterberger, M.; Wirnsberger, G.; Kyewski, B. Antigen presentation in the thymus for positive selection and central tolerance induction. Nat. Rev. Immunol. 2009, 9, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Madan, R.A.; Arlen, P.M.; Mohebtash, M.; Hodge, J.W.; Gulley, J.L. Prostvac-VF: A vector-based vaccine targeting PSA in prostate cancer. Expert Opin. Investig. Drugs 2009, 18, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Gnjatic, S.; Altorki, N.K.; Tang, D.N.; Tu, S.M.; Kundra, V.; Ritter, G.; Old, L.J.; Logothetis, C.J.; Sharma, P. NY-ESO-1 DNA vaccine induces T-cell responses that are suppressed by regulatory T cells. Clin. Cancer Res. 2009, 15, 2130–2139. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Hoai, T.; Kobelt, D.; Hohn, O.; Vu, M.D.; Schlag, P.M.; Dorken, B.; Norley, S.; Lipp, M.; Walther, W.; Pezzutto, A.; et al. HER2/neu DNA vaccination by intradermal gene delivery in a mouse tumor model: Gene gun is superior to jet injector in inducing CTL responses and protective immunity. Oncoimmunology 2012, 1, 1537–1545. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Xie, D.; Zhang, W.; Xiao, G.; Wen, J. Fusion of Hsp70 to Mage-a1 enhances the potency of vaccine-specific immune responses. J. Transl. Med. 2013, 11, 300. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Ku, G.Y.; Adamow, M.; Mu, Z.; Tandon, S.; Hannaman, D.; Chapman, P.; Schwartz, G.; Carvajal, R.; Panageas, K.S.; et al. Immunologic responses to xenogeneic tyrosinase DNA vaccine administered by electroporation in patients with malignant melanoma. J. Immunother. Cancer 2013, 1, 20. [Google Scholar] [CrossRef] [PubMed]
- Snyder, L.A.; Goletz, T.J.; Gunn, G.R.; Shi, F.F.; Harris, M.C.; Cochlin, K.; McCauley, C.; McCarthy, S.G.; Branigan, P.J.; Knight, D.M. A MUC1/IL-18 DNA vaccine induces anti-tumor immunity and increased survival in MUC1 transgenic mice. Vaccine 2006, 24, 3340–3352. [Google Scholar] [CrossRef] [PubMed]
- Xiang, R.; Silletti, S.; Lode, H.N.; Dolman, C.S.; Ruehlmann, J.M.; Niethammer, A.G.; Pertl, U.; Gillies, S.D.; Primus, F.J.; Reisfeld, R.A. Protective immunity against human carcinoembryonic antigen (CEA) induced by an oral DNA vaccine in CEA-transgenic mice. Clin. Cancer Res. 2001, 7, 856s–864s. [Google Scholar] [PubMed]
- Tiriveedhi, V.; Tucker, N.; Herndon, J.; Li, L.; Sturmoski, M.; Ellis, M.; Ma, C.; Naughton, M.; Lockhart, A.C.; Gao, F.; et al. Safety and preliminary evidence of biologic efficacy of a mammaglobin-a DNA vaccine in patients with stable metastatic breast cancer. Clin. Cancer Res. 2014, 20, 5964–5975. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Pankhong, P.; Shin, T.H.; Obeng-Adjei, N.; Morrow, M.P.; Walters, J.N.; Khan, A.S.; Sardesai, N.Y.; Weiner, D.B. Highly optimized DNA vaccine targeting human telomerase reverse transcriptase stimulates potent antitumor immunity. Cancer Immunol. Res. 2013, 1, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, L.R.; Carrascal, M.A.; Barbas, A.; Ramalho, J.S.; Novo, C.; Delannoy, P.; Videira, P.A. Challenges in Antibody Development against Tn and Sialyl-Tn Antigens. Biomolecules 2015, 5, 1783–1809. [Google Scholar] [CrossRef] [PubMed]
- Chaise, C.; Buchan, S.L.; Rice, J.; Marquet, J.; Rouard, H.; Kuentz, M.; Vittes, G.E.; Molinier-Frenkel, V.; Farcet, J.P.; Stauss, H.J.; et al. DNA vaccination induces WT1-specific T-cell responses with potential clinical relevance. Blood 2008, 112, 2956–2964. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hong, Y.; Mizejewski, G.J. Engineering alpha-fetoprotein-based gene vaccines to prevent and treat hepatocellular carcinoma: Review and future prospects. Immunotherapy 2014, 6, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Yin, B.W.; Lloyd, K.O. Molecular cloning of the CA125 ovarian cancer antigen: Identification as a new mucin, MUC16. J. Biol. Chem. 2001, 276, 27371–27375. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Ku, G.Y.; Gallardo, H.F.; Orlandi, F.; Manukian, G.; Rasalan, T.S.; Xu, Y.; Li, H.; Vyas, S.; Mu, Z.; et al. Safety and immunogenicity of a human and mouse gp100 DNA vaccine in a phase I trial of patients with melanoma. Cancer Immunol. 2009, 9, 5. [Google Scholar]
- Yang, B.; Jeang, J.; Yang, A.; Wu, T.C.; Hung, C.F. DNA vaccine for cancer immunotherapy. Hum. Vaccines Immunother. 2014, 10, 3153–3164. [Google Scholar] [CrossRef] [PubMed]
- Peoples, G.E.; Goedegebuure, P.S.; Smith, R.; Linehan, D.C.; Yoshino, I.; Eberlein, T.J. Breast and ovarian cancer-specific cytotoxic T lymphocytes recognize the same HER2/neu-derived peptide. Proc. Natl. Acad. Sci. USA 1995, 92, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.A.; Burchell, J.M.; Taylor-Papadimitriou, J. The polymorphic epithelial mucin: Potential as an immunogen for a cancer vaccine. Cancer Immunol. Immunother. 1996, 42, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Lohrisch, C.; Piccart, M. HER2/neu as a predictive factor in breast cancer. Clin. Breast Cancer 2001, 2, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.A.; Dintzis, S.; Darrow, C.M.; Voss, L.E.; DiPersio, J.; Jensen, R.; Fleming, T.P. Mammaglobin expression in primary, metastatic, and occult breast cancer. Cancer Res. 1999, 59, 3028–3031. [Google Scholar] [PubMed]
- Grunewald, K.; Haun, M.; Urbanek, M.; Fiegl, M.; Muller-Holzner, E.; Gunsilius, E.; Dunser, M.; Marth, C.; Gastl, G. Mammaglobin gene expression: A superior marker of breast cancer cells in peripheral blood in comparison to epidermal-growth-factor receptor and cytokeratin-19. Lab. Investig. 2000, 80, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, A.; Narayanan, K.; Campbell, L.G.; Benshoff, N.D.; Lybarger, L.; Hansen, T.H.; Fleming, T.P.; Dietz, J.R.; Mohanakumar, T. Recognition of HLA-A2-restricted mammaglobin-A-derived epitopes by CD8+ cytotoxic T lymphocytes from breast cancer patients. Breast Cancer Res. Treat. 2004, 88, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, K.; Jaramillo, A.; Benshoff, N.D.; Campbell, L.G.; Fleming, T.P.; Dietz, J.R.; Mohanakumar, T. Response of established human breast tumors to vaccination with mammaglobin-A cDNA. J. Natl. Cancer Inst. 2004, 96, 1388–1396. [Google Scholar] [CrossRef] [PubMed]
- Bharat, A.; Benshoff, N.; Fleming, T.P.; Dietz, J.R.; Gillanders, W.E.; Mohanakumar, T. Characterization of the role of CD8+ T cells in breast cancer immunity following mammaglobin-A DNA vaccination using HLA-class-I tetramers. Breast Cancer Res. Treat. 2008, 110, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Tiriveedhi, V.; Fleming, T.P.; Goedegebuure, P.S.; Naughton, M.; Ma, C.; Lockhart, C.; Gao, F.; Gillanders, W.E.; Mohanakumar, T. Mammaglobin-A cDNA vaccination of breast cancer patients induces antigen-specific cytotoxic CD4+ICOShi T cells. Breast Cancer Res. Treat. 2013, 138, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Parmiani, G.; De Filippo, A.; Novellino, L.; Castelli, C. Unique human tumor antigens: Immunobiology and use in clinical trials. J. Immunol. 2007, 178, 1975–1979. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sharma, P.K.; Peter Goedegebuure, S.; Gillanders, W.E. Personalized cancer vaccines: Targeting the cancer mutanome. Vaccine 2017, 35, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, S.; Castle, J.C.; Tureci, O.; Sahin, U. Targeting the tumor mutanome for personalized vaccination therapy. Oncoimmunology 2012, 1, 768–769. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.C.; Yao, X.; Crystal, J.S.; Li, Y.F.; El-Gamil, M.; Gross, C.; Davis, L.; Dudley, M.E.; Yang, J.C.; Samuels, Y.; et al. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin. Cancer Res. 2014, 20, 3401–3410. [Google Scholar] [CrossRef] [PubMed]
- Manthorpe, M.; Cornefert-Jensen, F.; Hartikka, J.; Felgner, J.; Rundell, A.; Margalith, M.; Dwarki, V. Gene therapy by intramuscular injection of plasmid DNA: Studies on firefly luciferase gene expression in mice. Hum. Gene Ther. 1993, 4, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Carnes, A.E.; Hodgson, C.P. Plasmid DNA vaccine vector design: Impact on efficacy, safety and upstream production. Biotechnol. Adv. 2009, 27, 353–370. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. Recognition of AUG and alternative initiator codons is augmented by G in position +4 but is not generally affected by the nucleotides in positions +5 and +6. EMBO J. 1997, 16, 2482–2492. [Google Scholar] [CrossRef] [PubMed]
- Spies, B.; Hochrein, H.; Vabulas, M.; Huster, K.; Busch, D.H.; Schmitz, F.; Heit, A.; Wagner, H. Vaccination with plasmid DNA activates dendritic cells via Toll-like receptor 9 (TLR9) but functions in TLR9-deficient mice. J. Immunol. 2003, 171, 5908–5912. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T.; Akira, S. Recognition of nucleic acids by pattern-recognition receptors and its relevance in autoimmunity. Immunol. Rev. 2011, 243, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Lladser, A.; Mougiakakos, D.; Tufvesson, H.; Ligtenberg, M.A.; Quest, A.F.; Kiessling, R.; Ljungberg, K. DAI (DLM-1/ZBP1) as a genetic adjuvant for DNA vaccines that promotes effective antitumor CTL immunity. Mol. Ther. 2011, 19, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Azzoni, A.R.; Ribeiro, S.C.; Monteiro, G.A.; Prazeres, D.M. The impact of polyadenylation signals on plasmid nuclease-resistance and transgene expression. J. Gene Med. 2007, 9, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, S.C.; Oliveira, P.H.; Prazeres, D.M.; Monteiro, G.A. High frequency plasmid recombination mediated by 28 bp direct repeats. Mol. Biotechnol. 2008, 40, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.J.; Coban, C.; Kato, H.; Takahashi, K.; Torii, Y.; Takeshita, F.; Ludwig, H.; Sutter, G.; Suzuki, K.; Hemmi, H.; et al. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat. Immunol. 2006, 7, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Cooke, J.R.; McKie, E.A.; Ward, J.M.; Keshavarz-Moore, E. Impact of intrinsic DNA structure on processing of plasmids for gene therapy and DNA vaccines. J. Biotechnol. 2004, 114, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Bolhassani, A.; Safaiyan, S.; Rafati, S. Improvement of different vaccine delivery systems for cancer therapy. Mol. Cancer 2011, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Haynes, J.R.; McCabe, D.E.; Swain, W.F.; Widera, G.; Fuller, J.T. Particle-mediated nucleic acid immunization. J. Biotechnol. 1996, 44, 37–42. [Google Scholar] [CrossRef]
- Rao, S.S.; Gomez, P.; Mascola, J.R.; Dang, V.; Krivulka, G.R.; Yu, F.; Lord, C.I.; Shen, L.; Bailer, R.; Nabel, G.J.; et al. Comparative evaluation of three different intramuscular delivery methods for DNA immunization in a nonhuman primate animal model. Vaccine 2006, 24, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Schwendener, R.A.; Ludewig, B.; Cerny, A.; Engler, O. Liposome-based vaccines. Methods Mol. Biol. 2010, 605, 163–175. [Google Scholar] [PubMed]
- Torrieri-Dramard, L.; Lambrecht, B.; Ferreira, H.L.; Van den Berg, T.; Klatzmann, D.; Bellier, B. Intranasal DNA vaccination induces potent mucosal and systemic immune responses and cross-protective immunity against influenza viruses. Mol. Ther. 2011, 19, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ewald, B.A.; Lynch, D.M.; Nanda, A.; Sumida, S.M.; Barouch, D.H. Modulation of DNA vaccine-elicited CD8+ T-lymphocyte epitope immunodominance hierarchies. J. Virol. 2006, 80, 11991–11997. [Google Scholar] [CrossRef] [PubMed]
- Vittes, G.E.; Harden, E.L.; Ottensmeier, C.H.; Rice, J.; Stevenson, F.K. DNA fusion gene vaccines induce cytotoxic T-cell attack on naturally processed peptides of human prostate-specific membrane antigen. Eur. J. Immunol. 2011, 41, 2447–2456. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Hendriks, W.; Althage, A.; Hemmi, S.; Bluethmann, H.; Kamijo, R.; Vilcek, J.; Zinkernagel, R.M.; Aguet, M. Immune response in mice that lack the interferon-gamma receptor. Science 1993, 259, 1742–1745. [Google Scholar] [CrossRef] [PubMed]
- Podack, E.R.; Lowrey, D.M.; Lichtenheld, M.; Hameed, A. Function of granule perforin and esterases in T cell-mediated reactions. Components required for delivery of molecules to target cells. Ann. N. Y. Acad. Sci. 1988, 532, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Groh, V.; Wu, J.; Yee, C.; Spies, T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 2002, 419, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Billadeau, D.D.; Upshaw, J.L.; Schoon, R.A.; Dick, C.J.; Leibson, P.J. NKG2D-DAP10 triggers human NK cell-mediated killing via a Syk-independent regulatory pathway. Nat. Immunol. 2003, 4, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Janssen, E.M.; Lemmens, E.E.; Wolfe, T.; Christen, U.; von Herrath, M.G.; Schoenberger, S.P. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 2003, 421, 852–856. [Google Scholar] [CrossRef] [PubMed]
- Susskind, B.; Shornick, M.D.; Iannotti, M.R.; Duffy, B.; Mehrotra, P.T.; Siegel, J.P.; Mohanakumar, T. Cytolytic effector mechanisms of human CD4+ cytotoxic T lymphocytes. Hum. Immunol. 1996, 45, 64–75. [Google Scholar] [CrossRef]
- Zhang, F.; Tang, Z.; Hou, X.; Lennartsson, J.; Li, Y.; Koch, A.W.; Scotney, P.; Lee, C.; Arjunan, P.; Dong, L.; et al. VEGF-B is dispensable for blood vessel growth but critical for their survival, and VEGF-B targeting inhibits pathological angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 6152–6157. [Google Scholar] [CrossRef] [PubMed]
- Aslan, N.; Yurdaydin, C.; Wiegand, J.; Greten, T.; Ciner, A.; Meyer, M.F.; Heiken, H.; Kuhlmann, B.; Kaiser, T.; Bozkaya, H.; et al. Cytotoxic CD4 T cells in viral hepatitis. J. Viral Hepat. 2006, 13, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, S.; Cervera, A.; MacLeod, M.; Fillatreau, S.; Perona-Wright, G.; Meek, S.; Smith, A.; MacDonald, A.; Gray, D. The role of ICOS in the development of CD4 T cell help and the reactivation of memory T cells. Eur. J. Immunol. 2007, 37, 1796–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strauss, L.; Bergmann, C.; Szczepanski, M.J.; Lang, S.; Kirkwood, J.M.; Whiteside, T.L. Expression of ICOS on human melanoma-infiltrating CD4+ CD25highFoxp3+ T regulatory cells: Implications and impact on tumor-mediated immune suppression. J. Immunol. 2008, 180, 2967–2980. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.M.; Lundberg, K.; Ozenci, V.; Banham, A.H.; Hellstrom, M.; Egevad, L.; Pisa, P. CD4+ CD25high T cells are enriched in the tumor and peripheral blood of prostate cancer patients. J. Immunol. 2006, 177, 7398–7405. [Google Scholar] [CrossRef] [PubMed]
- Liakou, C.I.; Kamat, A.; Tang, D.N.; Chen, H.; Sun, J.; Troncoso, P.; Logothetis, C.; Sharma, P. CTLA-4 blockade increases IFNgamma-producing CD4+ ICOShi cells to shift the ratio of effector to regulatory T cells in cancer patients. Proc. Natl. Acad. Sci. USA 2008, 105, 14987–14992. [Google Scholar] [CrossRef] [PubMed]
- King, C.A.; Spellerberg, M.B.; Zhu, D.; Rice, J.; Sahota, S.S.; Thompsett, A.R.; Hamblin, T.J.; Radl, J.; Stevenson, F.K. DNA vaccines with single-chain Fv fused to fragment C of tetanus toxin induce protective immunity against lymphoma and myeloma. Nat. Med. 1998, 4, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.; Buchan, S.; Stevenson, F.K. Critical components of a DNA fusion vaccine able to induce protective cytotoxic T cells against a single epitope of a tumor antigen. J. Immunol. 2002, 169, 3908–3913. [Google Scholar] [CrossRef] [PubMed]
- Gerloni, M.; Xiong, S.; Mukerjee, S.; Schoenberger, S.P.; Croft, M.; Zanetti, M. Functional cooperation between T helper cell determinants. Proc. Natl. Acad. Sci. USA 2000, 97, 13269–13274. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, Z.A.; Yeap, S.K.; Ali, A.M.; Ho, W.Y.; Alitheen, N.B.; Hamid, M. ScFv antibody: Principles and clinical application. Clin. Dev. Immunol. 2012, 2012, 980250. [Google Scholar] [CrossRef] [PubMed]
- Seliger, B.; Harders, C.; Lohmann, S.; Momburg, F.; Urlinger, S.; Tampe, R.; Huber, C. Down-regulation of the MHC class I antigen-processing machinery after oncogenic transformation of murine fibroblasts. Eur. J. Immunol. 1998, 28, 122–133. [Google Scholar] [CrossRef]
- Vertuani, S.; Triulzi, C.; Roos, A.K.; Charo, J.; Norell, H.; Lemonnier, F.; Pisa, P.; Seliger, B.; Kiessling, R. HER-2/neu mediated down-regulation of MHC class I antigen processing prevents CTL-mediated tumor recognition upon DNA vaccination in HLA-A2 transgenic mice. Cancer Immunol. Immunother. 2009, 58, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Van der Burg, S.H.; Arens, R.; Ossendorp, F.; van Hall, T.; Melief, C.J. Vaccines for established cancer: Overcoming the challenges posed by immune evasion. Nat. Rev. Cancer 2016, 16, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.E.; Pollard, J.W. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Van der Sluis, T.C.; Sluijter, M.; van Duikeren, S.; West, B.L.; Melief, C.J.; Arens, R.; van der Burg, S.H.; van Hall, T. Therapeutic Peptide Vaccine-Induced CD8 T Cells Strongly Modulate Intratumoral Macrophages Required for Tumor Regression. Cancer Immunol. Res. 2015, 3, 1042–1051. [Google Scholar] [CrossRef] [PubMed]
- Arina, A.; Bronte, V. Myeloid-derived suppressor cell impact on endogenous and adoptively transferred T cells. Curr. Opin. Immunol. 2015, 33, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Cagnon, L.; Costa-Nunes, C.M.; Baumgaertner, P.; Montandon, N.; Leyvraz, L.; Michielin, O.; Romano, E.; Speiser, D.E. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol. Immunother. 2014, 63, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.J.; Mirza, N.; Fricke, I.; Chiappori, A.; Thompson, P.; Williams, N.; Bepler, G.; Simon, G.; Janssen, W.; Lee, J.H.; et al. Combination of p53 cancer vaccine with chemotherapy in patients with extensive stage small cell lung cancer. Clin. Cancer Res. 2006, 12, 878–887. [Google Scholar] [CrossRef] [PubMed]
- Piersma, S.J.; Welters, M.J.; van der Burg, S.H. Tumor-specific regulatory T cells in cancer patients. Hum. Immunol. 2008, 69, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Drake, C.G.; Levitsky, H.I. Amplification of tumor-specific regulatory T cells following therapeutic cancer vaccines. Blood 2006, 107, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, W.R., Jr.; Wu, T.G. Development of an interferon-based cancer vaccine protocol: Application to several types of murine cancers. Methods Mol. Med. 2005, 116, 151–166. [Google Scholar] [PubMed]
- Jalah, R.; Patel, V.; Kulkarni, V.; Rosati, M.; Alicea, C.; Ganneru, B.; von Gegerfelt, A.; Huang, W.; Guan, Y.; Broderick, K.E.; et al. IL-12 DNA as molecular vaccine adjuvant increases the cytotoxic T cell responses and breadth of humoral immune responses in SIV DNA vaccinated macaques. Hum. Vaccines Immunother. 2012, 8, 1620–1629. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A. IL-2: The first effective immunotherapy for human cancer. J. Immunol. 2014, 192, 5451–5458. [Google Scholar] [CrossRef] [PubMed]
- Toussi, D.N.; Massari, P. Immune Adjuvant Effect of Molecularly-defined Toll-Like Receptor Ligands. Vaccines (Basel) 2014, 2, 323–353. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.H.; Mao, C.P.; Lee, S.Y.; Chen, A.; Lee, J.H.; Kim, T.W.; Alvarez, R.D.; Roden, R.B.; Pardoll, D.; Hung, C.F.; et al. Chemotherapy acts as an adjuvant to convert the tumor microenvironment into a highly permissive state for vaccination-induced antitumor immunity. Cancer Res. 2013, 73, 2493–2504. [Google Scholar] [CrossRef] [PubMed]
- Walter, S.; Weinschenk, T.; Stenzl, A.; Zdrojowy, R.; Pluzanska, A.; Szczylik, C.; Staehler, M.; Brugger, W.; Dietrich, P.Y.; Mendrzyk, R.; et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat. Med. 2012, 18, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Rudd, C.E.; Taylor, A.; Schneider, H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol. Rev. 2009, 229, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Vilgelm, A.E.; Johnson, D.B.; Richmond, A. Combinatorial approach to cancer immunotherapy: Strength in numbers. J. Leukoc. Biol. 2016, 100, 275–290. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TAAs | Organs | Reference |
|---|---|---|
| NYESO-1 | Prostate cancer, bladder cancer, esophagus cancer, non-small cell lung cancer, sarcoma | [12] |
| HER-2/Neu | Breast | [13] |
| MAGE-1 | Melanoma | [14] |
| Tyrosinase | Melanoma Leukemia | [15] |
| MUC1 | Breast cancer | [16] |
| CEA | Colon cancer, lung cancer | [17] |
| Mam-A | Breast cancer | [18] |
| hTERT | Melanoma, leukemia, reported several solid organs | [19] |
| Sialyl-Tn | gastric, colon, breast, lung, oesophageal, prostate and endometrial cancer | [20] |
| WT1 | Renal cancer | [21] |
| α-FetoProtein | Hepatic cancer | [22] |
| CA-125 | Ovarian cancer | [23] |
| gp-100 | Melanoma | [24] |
| p53, Ras, Src | reported in multiple cancers | [25] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amara, S.; Tiriveedhi, V. The Five Immune Forces Impacting DNA-Based Cancer Immunotherapeutic Strategy. Int. J. Mol. Sci. 2017, 18, 650. https://doi.org/10.3390/ijms18030650
Amara S, Tiriveedhi V. The Five Immune Forces Impacting DNA-Based Cancer Immunotherapeutic Strategy. International Journal of Molecular Sciences. 2017; 18(3):650. https://doi.org/10.3390/ijms18030650
Chicago/Turabian StyleAmara, Suneetha, and Venkataswarup Tiriveedhi. 2017. "The Five Immune Forces Impacting DNA-Based Cancer Immunotherapeutic Strategy" International Journal of Molecular Sciences 18, no. 3: 650. https://doi.org/10.3390/ijms18030650
APA StyleAmara, S., & Tiriveedhi, V. (2017). The Five Immune Forces Impacting DNA-Based Cancer Immunotherapeutic Strategy. International Journal of Molecular Sciences, 18(3), 650. https://doi.org/10.3390/ijms18030650