
Protein Kinases C-Mediated Regulations of Drug Transporter Activity, Localization and Expression


Abstract
:1. Introduction
2. The Drug Transportome
3. The Protein Kinases C (PKCs) Family
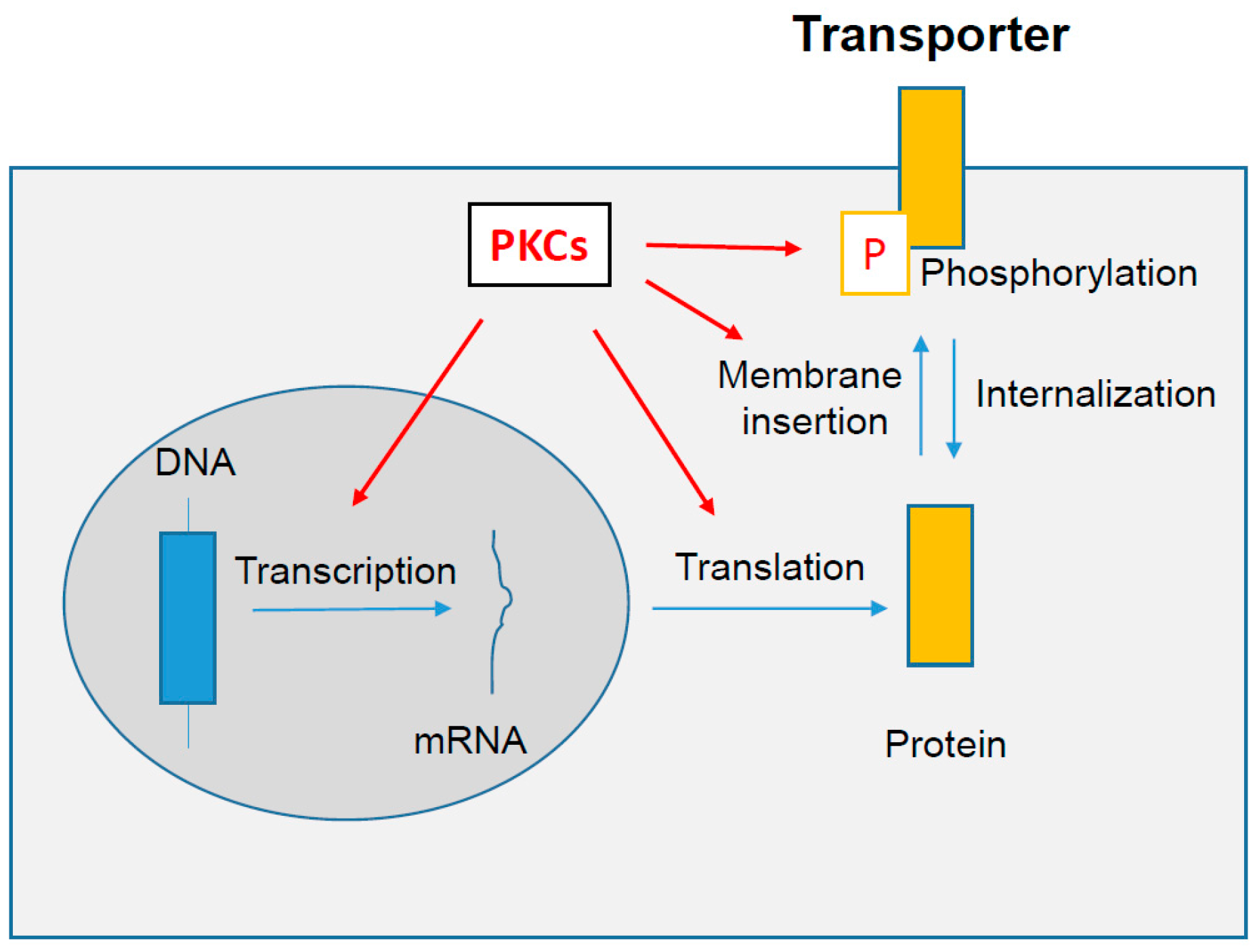
4. PKCs-Dependent Regulation of Drug Transporter Activity
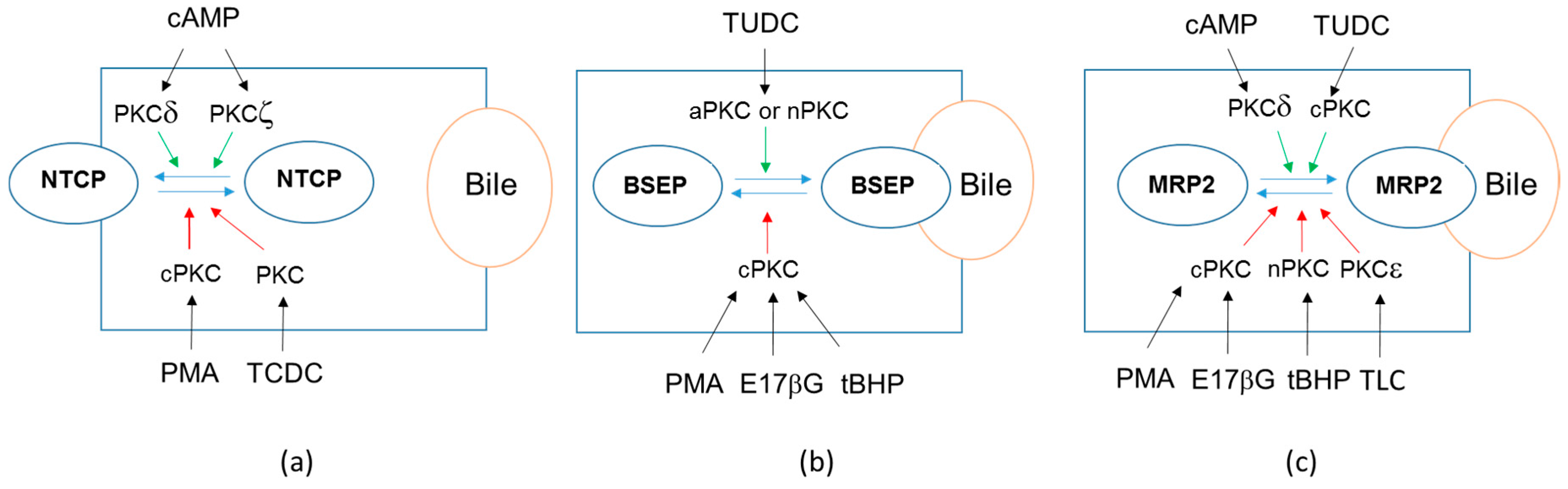
5. PKCs-Dependent Regulation of Drug Transporter Localization
6. PKCs-Dependent Regulation of Drug Transporter Expression
7. Putative Clinical Relevance of PKCs-Related Alteration of Transporter Activity, Localization and/or Expression
8. Conclusions
Author Contributions
Conflicts of Interest
Abbreviations
| PKC | Protein kinase |
| mRNA | Messenger ribonucleic acid |
| cPKC | Classical/conventional PKC |
| nPKC | Novel PKC |
| aPKC | Atypical PKC |
| DAG | Diacylglycerol |
| SLC | Solute carrier |
| ABC | ATP-binding cassette |
| OATP | Organic anion transporting polypeptide |
| OAT | Organic anion transporter |
| OCT | Organic cation transporter |
| NTCP | Sodium-taurocholate co-transporting polypeptide |
| MATE | Multidrug and toxin extrusion protein |
| OCTN | Organic cation/carnitine transporter |
| PEPT | Peptide transporter |
| CNT | Concentrative nucleoside transporter |
| ENT | Equilibrative nucleoside transporter |
| P-gp | P-glycoprotein |
| MRP | Multidrug resistance-associate protein |
| BCRP | Breast cancer resistance protein |
| BSEP | Bile salt export pump |
| PMA | Phorbol myristate acetate |
| BIM | Bisindolylmaleimide |
| PK15-NTD | Pig kidney epithelial nucleoside transporter deficient |
| cAMP | 3′,5′-cyclic adenosine monophosphate |
References
- Szakacs, G.; Varadi, A.; Ozvegy-Laczka, C.; Sarkadi, B. The role of ABC transporters in drug absorption, distribution, metabolism, excretion and toxicity (ADME-Tox). Drug Discov. Today 2008, 13, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef] [PubMed]
- DeGorter, M.K.; Xia, C.Q.; Yang, J.J.; Kim, R.B. Drug transporters in drug efficacy and toxicity. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 249–273. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; Yu, A.M. ABC transporters in multidrug resistance and pharmacokinetics, and strategies for drug development. Curr. Pharm. Des. 2014, 20, 793–807. [Google Scholar] [CrossRef] [PubMed]
- Konig, J.; Muller, F.; Fromm, M.F. Transporters and drug-drug interactions: Important determinants of drug disposition and effects. Pharmacol. Rev. 2013, 65, 944–966. [Google Scholar] [CrossRef] [PubMed]
- Prueksaritanont, T.; Chu, X.; Gibson, C.; Cui, D.; Yee, K.L.; Ballard, J.; Cabalu, T.; Hochman, J. Drug-drug interaction studies: Regulatory guidance and an industry perspective. AAPS J. 2013, 15, 629–645. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.I.; Williams, R.T.; Henderson, M.J.; Norris, M.D.; Haber, M. ABC transporters as mediators of drug resistance and contributors to cancer cell biology. Drug Resist. Updat. 2016, 26, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.X.; Hu, H.H.; Zhou, Q.; Yu, A.M.; Zeng, S. An overview of ABC and SLC drug transporter gene regulation. Curr. Drug Metab. 2013, 14, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Lecureur, V.; Courtois, A.; Payen, L.; Verhnet, L.; Guillouzo, A.; Fardel, O. Expression and regulation of hepatic drug and bile acid transporters. Toxicology 2000, 153, 203–219. [Google Scholar] [CrossRef]
- Fardel, O.; Payen, L.; Courtois, A.; Vernhet, L.; Lecureur, V. Regulation of biliary drug efflux pump expression by hormones and xenobiotics. Toxicology 2001, 167, 37–46. [Google Scholar] [CrossRef]
- Gandhi, A.; Moorthy, B.; Ghose, R. Drug disposition in pathophysiological conditions. Curr. Drug Metab. 2012, 13, 1327–1344. [Google Scholar] [CrossRef] [PubMed]
- Muller, J.; Keiser, M.; Drozdzik, M.; Oswald, S. Expression, regulation and function of intestinal drug transporters: An update. Biol. Chem. 2017, 398, 175–192. [Google Scholar] [CrossRef] [PubMed]
- Chambers, T.C.; Zheng, B.; Kuo, J.F. Regulation by phorbol ester and protein kinase C inhibitors, and by a protein phosphatase inhibitor (okadaic acid), of P-glycoprotein phosphorylation and relationship to drug accumulation in multidrug-resistant human KB cells. Mol. Pharmacol. 1992, 41, 1008–1015. [Google Scholar] [PubMed]
- Powell, J.; Farasyn, T.; Kock, K.; Meng, X.; Pahwa, S.; Brouwer, K.L.; Yue, W. Novel mechanism of impaired function of organic anion-transporting polypeptide 1B3 in human hepatocytes: Post-translational regulation of OATP1B3 by protein kinase C activation. Drug Metab. Dispos. 2014, 42, 1964–1970. [Google Scholar] [CrossRef] [PubMed]
- Schonhoff, C.M.; Gillin, H.; Webster, C.R.; Anwer, M.S. Protein kinase Cδ mediates cyclic adenosine monophosphate-stimulated translocation of sodium taurocholate cotransporting polypeptide and multidrug resistant associated protein 2 in rat hepatocytes. Hepatology 2008, 47, 1309–1316. [Google Scholar] [CrossRef] [PubMed]
- Anwer, M.S. Role of protein kinase C isoforms in bile formation and cholestasis. Hepatology 2014, 60, 1090–1097. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.M.; Roninson, I.B. Activation of MDR1 (P-glycoprotein) gene expression in human cells by protein kinase C agonists. Oncol. Res. 1992, 4, 281–290. [Google Scholar] [PubMed]
- Mayati, A.; Le Vee, M.; Moreau, A.; Jouan, E.; Bucher, S.; Stieger, B.; Denizot, C.; Parmentier, Y.; Fardel, O. Protein kinase C-dependent regulation of human hepatic drug transporter expression. Biochem. Pharmacol. 2015, 98, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Mackay, H.J.; Twelves, C.J. Targeting the protein kinase C family: Are we there yet? Nat. Rev. Cancer 2007, 7, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Benedetti, L.G.; Abera, M.B.; Wang, H.; Abba, M.; Kazanietz, M.G. Protein kinase C and cancer: What we know and what we do not. Oncogene 2014, 33, 5225–5237. [Google Scholar] [CrossRef] [PubMed]
- Mochly-Rosen, D.; Das, K.; Grimes, K.V. Protein kinase C, an elusive therapeutic target? Nat. Rev. Drug Discov. 2012, 11, 937–957. [Google Scholar] [CrossRef] [PubMed]
- Roffey, J.; Rosse, C.; Linch, M.; Hibbert, A.; McDonald, N.Q.; Parker, P.J. Protein kinase C intervention: The state of play. Curr. Opin. Cell Biol. 2009, 21, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Hampson, P.; Chahal, H.; Khanim, F.; Hayden, R.; Mulder, A.; Assi, L.K.; Bunce, C.M.; Lord, J.M. PEP005, a selective small-molecule activator of protein kinase C, has potent antileukemic activity mediated via the δ isoform of PKC. Blood 2005, 106, 1362–1368. [Google Scholar] [CrossRef] [PubMed]
- Hediger, M.A.; Clemencon, B.; Burrier, R.E.; Bruford, E.A. The ABCs of membrane transporters in health and disease (SLC series): Introduction. Mol. Asp. Med. 2013, 34, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Perland, E.; Fredriksson, R. Classification Systems of Secondary Active Transporters. Trends Pharmacol. Sci. 2017, 38, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Cesar-Razquin, A.; Snijder, B.; Frappier-Brinton, T.; Isserlin, R.; Gyimesi, G.; Bai, X.; Reithmeier, R.A.; Hepworth, D.; Hediger, M.A.; Edwards, A.M.; et al. A Call for Systematic Research on Solute Carriers. Cell 2015, 162, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Blood-brain barrier endogenous transporters as therapeutic targets: A new model for small molecule CNS drug discovery. Expert Opin. Ther. Targets 2015, 19, 1059–1072. [Google Scholar] [CrossRef] [PubMed]
- Obaidat, A.; Roth, M.; Hagenbuch, B. The expression and function of organic anion transporting polypeptides in normal tissues and in cancer. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 135–151. [Google Scholar] [CrossRef] [PubMed]
- Sai, Y.; Kaneko, Y.; Ito, S.; Mitsuoka, K.; Kato, Y.; Tamai, I.; Artursson, P.; Tsuji, A. Predominant contribution of organic anion transporting polypeptide OATP-B (OATP2B1) to apical uptake of estrone-3-sulfate by human intestinal Caco-2 cells. Drug Metab. Dispos. 2006, 34, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, H.; Lips, K.; Volk, C. Polyspecific organic cation transporters: Structure, function, physiological roles, and biopharmaceutical implications. Pharm. Res. 2007, 24, 1227–1251. [Google Scholar] [CrossRef] [PubMed]
- Burckhardt, G. Drug transport by Organic Anion Transporters (OATs). Pharmacol. Ther. 2012, 136, 106–130. [Google Scholar] [CrossRef] [PubMed]
- Pochini, L.; Scalise, M.; Galluccio, M.; Indiveri, C. OCTN cation transporters in health and disease: Role as drug targets and assay development. J. Biomol. Screen 2013, 18, 851–867. [Google Scholar] [CrossRef] [PubMed]
- Nies, A.T.; Damme, K.; Kruck, S.; Schaeffeler, E.; Schwab, M. Structure and function of multidrug and toxin extrusion proteins (MATEs) and their relevance to drug therapy and personalized medicine. Arch. Toxicol. 2016, 90, 1555–1584. [Google Scholar] [CrossRef] [PubMed]
- Ho, R.H.; Tirona, R.G.; Leake, B.F.; Glaeser, H.; Lee, W.; Lemke, C.J.; Wang, Y.; Kim, R.B. Drug and bile acid transporters in rosuvastatin hepatic uptake: Function, expression, and pharmacogenetics. Gastroenterology 2006, 130, 1793–1806. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.E.; Clemencon, B.; Hediger, M.A. Proton-coupled oligopeptide transporter family SLC15: Physiological, pharmacological and pathological implications. Mol. Asp. Med. 2013, 34, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Pastor-Anglada, M.; Perez-Torras, S. Nucleoside transporter proteins as biomarkers of drug responsiveness and drug targets. Front. Pharmacol. 2015, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Sheps, J.A.; Ling, V. Preface: The concept and consequences of multidrug resistance. Pflug. Arch. 2007, 453, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Ling, V. The molecular basis of multidrug resistance in cancer: The early years of P-glycoprotein research. FEBS Lett. 2006, 580, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Fardel, O.; Lecureur, V.; Guillouzo, A. The P-glycoprotein multidrug transporter. Gen. Pharmacol. 1996, 27, 1283–1291. [Google Scholar] [CrossRef]
- Cordon-Cardo, C.; O'Brien, J.P.; Casals, D.; Rittman-Grauer, L.; Biedler, J.L.; Melamed, M.R.; Bertino, J.R. Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc. Natl. Acad. Sci. USA 1989, 86, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Stieger, B.; Gao, B. Drug transporters in the central nervous system. Clin Pharmacokinet 2015, 54, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.P. Multidrug resistance protein 1 (MRP1, ABCC1), a “multitasking” ATP-binding cassette (ABC) transporter. J. Biol. Chem. 2014, 289, 30880–30888. [Google Scholar] [CrossRef] [PubMed]
- Keppler, D. Multidrug resistance proteins (MRPs, ABCCs): Importance for pathophysiology and drug therapy. Handb. Exp. Pharmacol. 2011, 299–323. [Google Scholar]
- Wen, J.; Luo, J.; Huang, W.; Tang, J.; Zhou, H.; Zhang, W. The Pharmacological and Physiological Role of Multidrug-Resistant Protein 4. J. Pharmacol. Exp. Ther. 2015, 354, 358–375. [Google Scholar] [CrossRef] [PubMed]
- Slot, A.J.; Molinski, S.V.; Cole, S.P. Mammalian multidrug-resistance proteins (MRPs). Essays Biochem. 2011, 50, 179–207. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q.; Unadkat, J.D. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport—An update. AAPS J. 2015, 17, 65–82. [Google Scholar] [CrossRef] [PubMed]
- Stieger, B. The role of the sodium-taurocholate cotransporting polypeptide (NTCP) and of the bile salt export pump (BSEP) in physiology and pathophysiology of bile formation. Handb. Exp. Pharmacol. 2011, 205–259. [Google Scholar]
- Hirano, M.; Maeda, K.; Hayashi, H.; Kusuhara, H.; Sugiyama, Y. Bile salt export pump (BSEP/ABCB11) can transport a nonbile acid substrate, pravastatin. J. Pharmacol. Exp. Ther. 2005, 314, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Newton, A.C. Protein kinase C: Poised to signal. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E395–E402. [Google Scholar] [CrossRef] [PubMed]
- Rykx, A.; De Kimpe, L.; Mikhalap, S.; Vantus, T.; Seufferlein, T.; Vandenheede, J.R.; van Lint, J. Protein kinase D: A family affair. FEBS Lett. 2003, 546, 81–86. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, S.F. Structural basis of protein kinase C isoform function. Physiol. Rev. 2008, 88, 1341–1378. [Google Scholar] [CrossRef] [PubMed]
- Newton, A.C. Protein kinase C: Structure, function, and regulation. J. Biol. Chem. 1995, 270, 28495–28498. [Google Scholar] [CrossRef] [PubMed]
- Wu-Zhang, A.X.; Newton, A.C. Protein kinase C pharmacology: Refining the toolbox. Biochem. J. 2013, 452, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Liu, M. Atypical protein kinase C in cell motility. Cell. Mol. Life Sci. 2013, 70, 3057–3066. [Google Scholar] [CrossRef] [PubMed]
- Goel, G.; Makkar, H.P.; Francis, G.; Becker, K. Phorbol esters: Structure, biological activity, and toxicity in animals. Int. J. Toxicol. 2007, 26, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Vorhagen, S.; Niessen, C.M. Mammalian aPKC/Par polarity complex mediated regulation of epithelial division orientation and cell fate. Exp. Cell Res. 2014, 328, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Drummond, M.L.; Prehoda, K.E. Molecular Control of Atypical Protein Kinase C: Tipping the Balance between Self-Renewal and Differentiation. J. Mol. Biol. 2016, 428, 1455–1464. [Google Scholar] [CrossRef] [PubMed]
- Aftab, D.T.; Yang, J.M.; Hait, W.N. Functional role of phosphorylation of the multidrug transporter (P-glycoprotein) by protein kinase C in multidrug-resistant MCF-7 cells. Oncol. Res. 1994, 6, 59–70. [Google Scholar] [PubMed]
- Chambers, T.C.; Chalikonda, I.; Eilon, G. Correlation of protein kinase C translocation, P-glycoprotein phosphorylation and reduced drug accumulation in multidrug resistant human KB cells. Biochem. Biophys. Res. Commun. 1990, 169, 253–259. [Google Scholar] [CrossRef]
- Tsuruoka, S.; Sugimoto, K.; Fujimura, A.; Imai, M.; Asano, Y.; Muto, S. Protein kinase C and phosphatidylinositol 3-kinase independently contribute to P-glycoprotein-mediated drug secretion in the mouse proximal tubule. Pflug. Arch. 2001, 442, 321–328. [Google Scholar] [CrossRef]
- Chaudhary, P.M.; Roninson, I.B. Induction of multidrug resistance in human cells by transient exposure to different chemotherapeutic drugs. J. Natl. Cancer Inst. 1993, 85, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Bi, H.; Zhong, G.; Huang, L.; Li, G.; Xia, Y.; Chen, X.; Huang, M. Effect of phorbol 12-myristate 13-acetate on function and gene expression of P-glycoprotein in adriamycin-resistant K562/ADM cells. Pharmacology 2013, 92, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Kubitz, R.; Saha, N.; Kuhlkamp, T.; Dutta, S.; vom Dahl, S.; Wettstein, M.; Haussinger, D. Ca2+-dependent protein kinase C isoforms induce cholestasis in rat liver. J. Biol. Chem. 2004, 279, 10323–10330. [Google Scholar] [CrossRef] [PubMed]
- Kubitz, R.; Huth, C.; Schmitt, M.; Horbach, A.; Kullak-Ublick, G.; Haussinger, D. Protein kinase C-dependent distribution of the multidrug resistance protein 2 from the canalicular to the basolateral membrane in human HepG2 cells. Hepatology 2001, 34, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Lee, A.C.; Krafczyk, K.; Zhu, L.; Murray, M. Protein kinase C regulates the internalization and function of the human organic anion transporting polypeptide 1A2. Br. J. Pharmacol. 2011, 162, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Hong, W.; Ni, C.; Huang, J.; Zhou, C. Protein kinase C affects the internalization and recycling of organic anion transporting polypeptide 1B1. Biochim. Biophys. Acta 2015, 1848, 2022–2030. [Google Scholar] [CrossRef] [PubMed]
- Kock, K.; Koenen, A.; Giese, B.; Fraunholz, M.; May, K.; Siegmund, W.; Hammer, E.; Volker, U.; Jedlitschky, G.; Kroemer, H.K.; et al. Rapid modulation of the organic anion transporting polypeptide 2B1 (OATP2B1, SLCO2B1) function by protein kinase C-mediated internalization. J. Biol. Chem. 2010, 285, 11336–11347. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, S.; Patterson, C.; You, G. Lysine 48-linked polyubiquitination of organic anion transporter-1 is essential for its protein kinase C-regulated endocytosis. Mol. Pharmacol. 2013, 83, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Soodvilai, S.; Chatsudthipong, V.; Evans, K.K.; Wright, S.H.; Dantzler, W.H. Acute regulation of OAT3-mediated estrone sulfate transport in isolated rabbit renal proximal tubules. Am. J. Physiol. Ren. Physiol. 2004, 287, F1021–F1029. [Google Scholar] [CrossRef] [PubMed]
- Stross, C.; Helmer, A.; Weissenberger, K.; Gorg, B.; Keitel, V.; Haussinger, D.; Kubitz, R. Protein kinase C induces endocytosis of the sodium taurocholate cotransporting polypeptide. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G320–G328. [Google Scholar] [CrossRef] [PubMed]
- Czeredys, M.; Samluk, L.; Michalec, K.; Tulodziecka, K.; Skowronek, K.; Nalecz, K.A. Caveolin-1—A novel interacting partner of organic cation/carnitine transporter (Octn2): Effect of protein kinase C on this interaction in rat astrocytes. PLoS ONE 2013, 8, e82105. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.J.; Cravetchi, X.; Vilas, G.; Hammond, J.R. Adenosine A1 receptor activation modulates human equilibrative nucleoside transporter 1 (hENT1) activity via PKC-mediated phosphorylation of serine-281. Cell. Signal. 2015, 27, 1008–1018. [Google Scholar] [CrossRef] [PubMed]
- Brandsch, M.; Miyamoto, Y.; Ganapathy, V.; Leibach, F.H. Expression and protein kinase C-dependent regulation of peptide/H+ co-transport system in the Caco-2 human colon carcinoma cell line. Biochem. J. 1994, 299 Pt 1, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, U.; Diehl, D.; Herget, M.; Kuntz, S.; Daniel, H. Regulation of the high-affinity H+/peptide cotransporter in renal LLC-PK1 cells. J. Cell. Physiol. 1999, 178, 341–348. [Google Scholar] [CrossRef]
- Chambers, T.C.; Pohl, J.; Glass, D.B.; Kuo, J.F. Phosphorylation by protein kinase C and cyclic AMP-dependent protein kinase of synthetic peptides derived from the linker region of human P-glycoprotein. Biochem. J. 1994, 299 Pt 1, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Fine, R.L.; Chambers, T.C.; Sachs, C.W. P-Glycoprotein, Multidrug Resistance and Protein Kinase C. Oncologist 1996, 1, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Chambers, T.C.; McAvoy, E.M.; Jacobs, J.W.; Eilon, G. Protein kinase C phosphorylates P-glycoprotein in multidrug resistant human KB carcinoma cells. J. Biol. Chem. 1990, 265, 7679–7686. [Google Scholar] [PubMed]
- Zhang, Z.; Wu, J.Y.; Hait, W.N.; Yang, J.M. Regulation of the stability of P-glycoprotein by ubiquitination. Mol. Pharmacol. 2004, 66, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Posada, J.A.; McKeegan, E.M.; Worthington, K.F.; Morin, M.J.; Jaken, S.; Tritton, T.R. Human multidrug resistant KB cells overexpress protein kinase C: Involvement in drug resistance. Cancer Commun. 1989, 1, 285–292. [Google Scholar] [PubMed]
- Gupta, K.P.; Ward, N.E.; Gravitt, K.R.; Bergman, P.J.; O’Brian, C.A. Partial reversal of multidrug resistance in human breast cancer cells by an N-myristoylated protein kinase C-α pseudosubstrate peptide. J. Biol. Chem. 1996, 271, 2102–2111. [Google Scholar] [PubMed]
- Hofmann, J. Modulation of protein kinase C in antitumor treatment. Rev. Physiol. Biochem. Pharmacol. 2001, 142, 1–96. [Google Scholar] [PubMed]
- Chambers, T.C.; Pohl, J.; Raynor, R.L.; Kuo, J.F. Identification of specific sites in human P-glycoprotein phosphorylated by protein kinase C. J. Biol. Chem. 1993, 268, 4592–4595. [Google Scholar] [PubMed]
- Sachs, C.W.; Chambers, T.C.; Fine, R.L. Differential phosphorylation of sites in the linker region of P-glycoprotein by protein kinase C isozymes α, βI, βII, γ, δ, ε, η, and ζ. Biochem. Pharmacol. 1999, 58, 1587–1592. [Google Scholar] [CrossRef]
- Yang, J.M.; Chin, K.V.; Hait, W.N. Interaction of P-glycoprotein with protein kinase C in human multidrug resistant carcinoma cells. Cancer Res. 1996, 56, 3490–3494. [Google Scholar] [PubMed]
- Ahmad, S.; Glazer, R.I. Expression of the antisense cDNA for protein kinase C α attenuates resistance in doxorubicin-resistant MCF-7 breast carcinoma cells. Mol. Pharmacol. 1993, 43, 858–862. [Google Scholar] [PubMed]
- Zhan, M.; Yu, D.; Liu, J.; Glazer, R.I.; Hannay, J.; Pollock, R.E. Transcriptional repression of protein kinase Cα via Sp1 by wild type p53 is involved in inhibition of multidrug resistance 1 P-glycoprotein phosphorylation. J. Biol. Chem. 2005, 280, 4825–4833. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Shehzad, A.; Jung, J.C.; Sonn, J.K.; Lee, J.T.; Park, J.W.; Lee, Y.S. Protein kinase Cα protects against multidrug resistance in human colon cancer cells. Mol. Cells 2012, 34, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.W.; Asai, D.; Kang, J.H.; Kishimura, A.; Mori, T.; Katayama, Y. Reversal of efflux of an anticancer drug in human drug-resistant breast cancer cells by inhibition of protein kinase Cα (PKCα) activity. Tumour Biol. 2016, 37, 1901–1908. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Ahmad, S.; Aquino, A.; Fairchild, C.R.; Trepel, J.B.; Ohno, S.; Suzuki, K.; Tsuruo, T.; Cowan, K.H.; Glazer, R.I. Transfection with protein kinase Cα confers increased multidrug resistance to MCF-7 cells expressing P-glycoprotein. Cancer Commun. 1991, 3, 181–189. [Google Scholar] [PubMed]
- Blobe, G.C.; Sachs, C.W.; Khan, W.A.; Fabbro, D.; Stabel, S.; Wetsel, W.C.; Obeid, L.M.; Fine, R.L.; Hannun, Y.A. Selective regulation of expression of protein kinase C (PKC) isoenzymes in multidrug-resistant MCF-7 cells. Functional significance of enhanced expression of PKCα. J. Biol. Chem. 1993, 268, 658–664. [Google Scholar] [PubMed]
- Goodfellow, H.R.; Sardini, A.; Ruetz, S.; Callaghan, R.; Gros, P.; McNaughton, P.A.; Higgins, C.F. Protein kinase C-mediated phosphorylation does not regulate drug transport by the human multidrug resistance P-glycoprotein. J. Biol. Chem. 1996, 271, 13668–13674. [Google Scholar] [PubMed]
- Germann, U.A.; Chambers, T.C.; Ambudkar, S.V.; Licht, T.; Cardarelli, C.O.; Pastan, I.; Gottesman, M.M. Characterization of phosphorylation-defective mutants of human P-glycoprotein expressed in mammalian cells. J. Biol. Chem. 1996, 271, 1708–1716. [Google Scholar] [CrossRef] [PubMed]
- Castro, A.F.; Horton, J.K.; Vanoye, C.G.; Altenberg, G.A. Mechanism of inhibition of P-glycoprotein-mediated drug transport by protein kinase C blockers. Biochem. Pharmacol. 1999, 58, 1723–1733. [Google Scholar] [CrossRef]
- Michaelis, M.; Rothweiler, F.; Loschmann, N.; Sharifi, M.; Ghafourian, T.; Cinatl, J., Jr. Enzastaurin inhibits ABCB1-mediated drug efflux independently of effects on protein kinase C signalling and the cellular p53 status. Oncotarget 2015, 6, 17605–17620. [Google Scholar] [CrossRef] [PubMed]
- Merritt, J.E.; Sullivan, J.A.; Drew, L.; Khan, A.; Wilson, K.; Mulqueen, M.; Harris, W.; Bradshaw, D.; Hill, C.H.; Rumsby, M.; et al. The bisindolylmaleimide protein kinase C inhibitor, Ro 32–2241, reverses multidrug resistance in KB tumour cells. Cancer Chemother. Pharmacol. 1999, 43, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Gekeler, V.; Boer, R.; Uberall, F.; Ise, W.; Schubert, C.; Utz, I.; Hofmann, J.; Sanders, K.H.; Schachtele, C.; Klemm, K.; et al. Effects of the selective bisindolylmaleimide protein kinase C inhibitor GF 109203X on P-glycoprotein-mediated multidrug resistance. Br. J. Cancer 1996, 74, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Scala, S.; Dickstein, B.; Regis, J.; Szallasi, Z.; Blumberg, P.M.; Bates, S.E. Bryostatin 1 affects P-glycoprotein phosphorylation but not function in multidrug-resistant human breast cancer cells. Clin. Cancer Res. 1995, 1, 1581–1587. [Google Scholar] [PubMed]
- Miller, D.S.; Sussman, C.R.; Renfro, J.L. Protein kinase C regulation of p-glycoprotein-mediated xenobiotic secretion in renal proximal tubule. Am. J. Physiol. 1998, 275, F785–F795. [Google Scholar] [PubMed]
- Stromskaya, T.P.; Grigorian, I.A.; Ossovskaya, V.S.; Rybalkina, E.Y.; Chumakov, P.M.; Kopnin, B.P. Cell-specific effects of RAS oncogene and protein kinase C agonist TPA on P-glycoprotein function. FEBS Lett. 1995, 368, 373–376. [Google Scholar] [CrossRef]
- Rigor, R.R.; Hawkins, B.T.; Miller, D.S. Activation of PKC isoform βI at the blood-brain barrier rapidly decreases p-glycoprotein activity and enhances drug delivery to the brain. J. Cereb. Blood Flow Metab. 2010, 30, 1373–1383. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Huls, M.; Cornelius, M.G.; Fricker, G. St. John’s Wort constituents modulate P-glycoprotein transport activity at the blood-brain barrier. Pharm. Res. 2010, 27, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Wielinga, P.R.; Heijn, M.; Broxterman, H.J.; Lankelma, J. P-glycoprotein-independent decrease in drug accumulation by phorbol ester treatment of tumor cells. Biochem. Pharmacol. 1997, 54, 791–799. [Google Scholar] [CrossRef]
- Wimmer, R.; Hohenester, S.; Pusl, T.; Denk, G.U.; Rust, C.; Beuers, U. Tauroursodeoxycholic acid exerts anticholestatic effects by a cooperative cPKC α-/PKA-dependent mechanism in rat liver. Gut 2008, 57, 1448–1454. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.S.; Masereeuw, R.; Karnaky, K.J., Jr. Regulation of MRP2-mediated transport in shark rectal salt gland tubules. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 282, R774–R781. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.W.; Shukla, S.; Steadman, K.; Obrzut, T.; Finley, E.M.; Ambudkar, S.V.; Bates, S.E. Inhibition of ABCG2-mediated transport by protein kinase inhibitors with a bisindolylmaleimide or indolocarbazole structure. Mol. Cancer Ther. 2007, 6, 1877–1885. [Google Scholar] [CrossRef] [PubMed]
- Gekeler, V.; Boer, R.; Ise, W.; Sanders, K.H.; Schachtele, C.; Beck, J. The specific bisindolylmaleimide PKC-inhibitor GF 109203X efficiently modulates MRP-associated multiple drug resistance. Biochem. Biophys. Res. Commun. 1995, 206, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Ciarimboli, G.; Struwe, K.; Arndt, P.; Gorboulev, V.; Koepsell, H.; Schlatter, E.; Hirsch, J.R. Regulation of the human organic cation transporter hOCT1. J. Cell. Physiol. 2004, 201, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Ciarimboli, G.; Koepsell, H.; Iordanova, M.; Gorboulev, V.; Durner, B.; Lang, D.; Edemir, B.; Schroter, R.; van Le, T.; Schlatter, E. Individual PKC-phosphorylation sites in organic cation transporter 1 determine substrate selectivity and transport regulation. J. Am. Soc. Nephrol. 2005, 16, 1562–1570. [Google Scholar] [CrossRef] [PubMed]
- Cetinkaya, I.; Ciarimboli, G.; Yalcinkaya, G.; Mehrens, T.; Velic, A.; Hirsch, J.R.; Gorboulev, V.; Koepsell, H.; Schlatter, E. Regulation of human organic cation transporter hOCT2 by PKA, PI3K, and calmodulin-dependent kinases. Am. J. Physiol. Ren. Physiol. 2003, 284, F293–F302. [Google Scholar] [CrossRef] [PubMed]
- Mayati, A.; Bruyere, A.; Moreau, A.; Jouan, E.; Denizot, C.; Parmentier, Y.; Fardel, O. Protein Kinase C-Independent Inhibition of Organic Cation Transporter 1 Activity by the Bisindolylmaleimide Ro 31–8220. PLoS ONE 2015, 10, e0144667. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.L.; Klaassen, C.D. Protein kinase C suppresses rat organic anion transporting polypeptide 1- and 2-mediated uptake. J. Pharmacol. Exp. Ther. 2001, 299, 551–557. [Google Scholar] [PubMed]
- Reyes, G.; Nivillac, N.M.; Karim, M.Z.; Desouza, L.; Siu, K.W.; Coe, I.R. The Equilibrative Nucleoside Transporter (ENT1) can be phosphorylated at multiple sites by PKC and PKA. Mol. Membr. Biol. 2011, 28, 412–426. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Wang, Y.; Cogut, S.B.; Mitchell, B.S.; Graves, L.M. Inhibition of nucleoside transport by protein kinase inhibitors. J. Pharmacol. Exp. Ther. 2003, 304, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Wang, Y.; Mitchell, B.S.; Graves, L.M. Regulation of equilibrative nucleoside uptake by protein kinase inhibitors. Nucleosides Nucleotides Nucleic Acids 2004, 23, 1445–1450. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S. Intercellular junctions and cellular polarity: The PAR-aPKC complex, a conserved core cassette playing fundamental roles in cell polarity. Curr. Opin. Cell Biol. 2001, 13, 641–648. [Google Scholar] [CrossRef]
- Goehring, N.W. PAR polarity: From complexity to design principles. Exp. Cell Res. 2014, 328, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Roman-Fernandez, A.; Bryant, D.M. Complex Polarity: Building Multicellular Tissues Through Apical Membrane Traffic. Traffic 2016, 17, 1244–1261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Hong, M.; Duan, P.; Pan, Z.; Ma, J.; You, G. Organic anion transporter OAT1 undergoes constitutive and protein kinase C-regulated trafficking through a dynamin- and clathrin-dependent pathway. J. Biol. Chem. 2008, 283, 32570–32579. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.; Cheung, F.S.; Zheng, J.; Lu, X.; Zhu, L.; Grewal, T.; Murray, M.; Zhou, F. Casein Kinase 2 Is a Novel Regulator of the Human Organic Anion Transporting Polypeptide 1A2 (OATP1A2) Trafficking. Mol. Pharm. 2016, 13, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Kipp, H.; Arias, I.M. Intracellular trafficking and regulation of canalicular ATP-binding cassette transporters. Semin. Liver Dis. 2000, 20, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Porcelli, L.; Lemos, C.; Peters, G.J.; Paradiso, A.; Azzariti, A. Intracellular trafficking of MDR transporters and relevance of SNPs. Curr. Top. Med. Chem. 2009, 9, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.P.; Sanchez-Covarubias, L.; Tome, M.E. P-glycoprotein trafficking as a therapeutic target to optimize CNS drug delivery. Adv. Pharmacol. 2014, 71, 25–44. [Google Scholar] [PubMed]
- Jigorel, E.; Le Vee, M.; Boursier-Neyret, C.; Bertrand, M.; Fardel, O. Functional expression of sinusoidal drug transporters in primary human and rat hepatocytes. Drug Metab. Dispos. 2005, 33, 1418–1422. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, Q.; You, G. Three ubiquitination sites of organic anion transporter-1 synergistically mediate protein kinase C-dependent endocytosis of the transporter. Mol. Pharmacol. 2013, 84, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Duan, P.; You, G. Regulation of human organic anion transporter 1 by ANG II: Involvement of protein kinase Cα. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E378–E383. [Google Scholar] [CrossRef] [PubMed]
- Duan, P.; Li, S.; You, G. Angiotensin II inhibits activity of human organic anion transporter 3 through activation of protein kinase Cα: Accelerating endocytosis of the transporter. Eur. J. Pharmacol. 2010, 627, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Barros, S.A.; Srimaroeng, C.; Perry, J.L.; Walden, R.; Dembla-Rajpal, N.; Sweet, D.H.; Pritchard, J.B. Activation of protein kinase Cζ increases OAT1 (SLC22A6)- and OAT3 (SLC22A8)-mediated transport. J. Biol. Chem. 2009, 284, 2672–2679. [Google Scholar] [CrossRef] [PubMed]
- Le Vee, M.; Noel, G.; Jouan, E.; Stieger, B.; Fardel, O. Polarized expression of drug transporters in differentiated human hepatoma HepaRG cells. Toxicol. In Vitro 2013, 27, 1979–1986. [Google Scholar] [CrossRef] [PubMed]
- Anwer, M.S.; Stieger, B. Sodium-dependent bile salt transporters of the SLC10A transporter family: More than solute transporters. Pflug. Arch. 2014, 466, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Sarwar, Z.; Annaba, F.; Dwivedi, A.; Saksena, S.; Gill, R.K.; Alrefai, W.A. Modulation of ileal apical Na+-dependent bile acid transporter ASBT by protein kinase C. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G532–G538. [Google Scholar] [CrossRef] [PubMed]
- Muhlfeld, S.; Domanova, O.; Berlage, T.; Stross, C.; Helmer, A.; Keitel, V.; Haussinger, D.; Kubitz, R. Short-term feedback regulation of bile salt uptake by bile salts in rodent liver. Hepatology 2012, 56, 2387–2397. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.R.; Srinivasulu, U.; Ananthanarayanan, M.; Suchy, F.J.; Anwer, M.S. Protein kinase B/Akt mediates cAMP- and cell swelling-stimulated Na+/taurocholate cotransport and Ntcp translocation. J. Biol. Chem. 2002, 277, 28578–28583. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Schonhoff, C.M.; Webster, C.R.; Anwer, M.S. Protein kinase Cδ differentially regulates cAMP-dependent translocation of NTCP and MRP2 to the plasma membrane. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G657–G665. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Bananis, E.; Nath, S.; Anwer, M.S.; Wolkoff, A.W.; Murray, J.W. PKCζ is required for microtubule-based motility of vesicles containing the ntcp transporter. Traffic 2006, 7, 1078–1091. [Google Scholar] [CrossRef] [PubMed]
- Kubitz, R.; Sutfels, G.; Kuhlkamp, T.; Kolling, R.; Haussinger, D. Trafficking of the bile salt export pump from the Golgi to the canalicular membrane is regulated by the p38 MAP kinase. Gastroenterology 2004, 126, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Dombrowski, F.; Stieger, B.; Beuers, U. Tauroursodeoxycholic acid inserts the bile salt export pump into canalicular membranes of cholestatic rat liver. Lab. Investig. 2006, 86, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Beuers, U. Drug insight: Mechanisms and sites of action of ursodeoxycholic acid in cholestasis. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Stieger, B.; Beuers, U. The canalicular bile salt export pump BSEP (ABCB11) as a potential therapeutic target. Curr. Drug Targets 2011, 12, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Perez, L.M.; Milkiewicz, P.; Elias, E.; Coleman, R.; Sanchez Pozzi, E.J.; Roma, M.G. Oxidative stress induces internalization of the bile salt export pump, Bsep, and bile salt secretory failure in isolated rat hepatocyte couplets: A role for protein kinase C and prevention by protein kinase A. Toxicol. Sci. 2006, 91, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Crocenzi, F.A.; Sanchez Pozzi, E.J.; Ruiz, M.L.; Zucchetti, A.E.; Roma, M.G.; Mottino, A.D.; Vore, M. Ca2+-dependent protein kinase C isoforms are critical to estradiol 17β-d-glucuronide-induced cholestasis in the rat. Hepatology 2008, 48, 1885–1895. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Sekine, S.; Ito, K.; Horie, T. Correlation between apical localization of Abcc2/Mrp2 and phosphorylation status of ezrin in rat intestine. Drug Metab. Dispos. 2009, 37, 1521–1527. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Sekine, S.; Ito, K.; Horie, T. Ezrin regulates the expression of Mrp2/Abcc2 and Mdr1/Abcb1 along the rat small intestinal tract. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G807–G817. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.; Cai, S.Y.; Liu, X.; Lian, W.; Chen, S.; Zhang, L.; Feng, X.; Cheng, Y.; He, X.; He, Y.; et al. Canalicular membrane MRP2/ABCC2 internalization is determined by Ezrin Thr567 phosphorylation in human obstructive cholestasis. J. Hepatol. 2015, 63, 1440–1448. [Google Scholar] [CrossRef] [PubMed]
- Sekine, S.; Ito, K.; Saeki, J.; Horie, T. Interaction of Mrp2 with radixin causes reversible canalicular Mrp2 localization induced by intracellular redox status. Biochim. Biophys. Acta 2011, 1812, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Schonhoff, C.M.; Webster, C.R.; Anwer, M.S. Taurolithocholate-induced MRP2 retrieval involves MARCKS phosphorylation by protein kinase C in HUH-NTCP Cells. Hepatology 2013, 58, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Beuers, U.; Bilzer, M.; Chittattu, A.; Kullak-Ublick, G.A.; Keppler, D.; Paumgartner, G.; Dombrowski, F. Tauroursodeoxycholic acid inserts the apical conjugate export pump, Mrp2, into canalicular membranes and stimulates organic anion secretion by protein kinase C-dependent mechanisms in cholestatic rat liver. Hepatology 2001, 33, 1206–1216. [Google Scholar] [CrossRef] [PubMed]
- Osborn, M.T.; Berry, A.; Ruberu, M.S.; Ning, B.; Bell, L.M.; Chambers, T.C. Phorbol ester induced MDR1 expression in K562 cells occurs independently of mitogen-activated protein kinase signaling pathways. Oncogene 1999, 18, 5756–5764. [Google Scholar] [CrossRef] [PubMed]
- McCoy, C.; Smith, D.E.; Cornwell, M.M. 12-O-tetradecanoylphorbol-13-acetate activation of the MDR1 promoter is mediated by EGR1. Mol. Cell. Biol. 1995, 15, 6100–6108. [Google Scholar] [CrossRef] [PubMed]
- Flescher, E.; Rotem, R. Protein kinase C epsilon mediates the induction of P-glycoprotein in LNCaP prostate carcinoma cells. Cell. Signal. 2002, 14, 37–43. [Google Scholar] [CrossRef]
- Gill, P.K.; Gescher, A.; Gant, T.W. Regulation of MDR1 promoter activity in human breast carcinoma cells by protein kinase C isozymes α and θ. Eur. J. Biochem. 2001, 268, 4151–4157. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.J.; Xu, H.; Qu, J.W.; Zhao, W.Z.; Zhao, Y.B.; Wang, J.H. Modulation of drug resistance in ovarian cancer cells by inhibition of protein kinase C-α (PKC-α) with small interference RNA (siRNA) agents. Asian Pac. J. Cancer Prev. 2012, 13, 3631–3636. [Google Scholar] [CrossRef] [PubMed]
- Kameyama, N.; Arisawa, S.; Ueyama, J.; Kagota, S.; Shinozuka, K.; Hattori, A.; Tatsumi, Y.; Hayashi, H.; Takagi, K.; Wakusawa, S. Increase in P-glycoprotein accompanied by activation of protein kinase Cα and NF-κB p65 in the livers of rats with streptozotocin-induced diabetes. Biochim. Biophys. Acta 2008, 1782, 355–360. [Google Scholar] [CrossRef] [PubMed]
- La Porta, C.A.; Dolfini, E.; Comolli, R. Inhibition of protein kinase C-α isoform enhances the P-glycoprotein expression and the survival of LoVo human colon adenocarcinoma cells to doxorubicin exposure. Br. J. Cancer 1998, 78, 1283–1287. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Nachtigal, M.W.; Kardami, E.; Cattini, P.A. FGF-2 protects cardiomyocytes from doxorubicin damage via protein kinase C-dependent effects on efflux transporters. Cardiovasc. Res. 2013, 98, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Grden, M.; Podgorska, M.; Kocbuch, K.; Rzepko, R.; Szutowicz, A.; Pawelczyk, T. High glucose suppresses expression of equilibrative nucleoside transporter 1 (ENT1) in rat cardiac fibroblasts through a mechanism dependent on PKC-ζ and MAP kinases. J. Cell. Physiol. 2008, 215, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Griner, E.M.; Kazanietz, M.G. Protein kinase C and other diacylglycerol effectors in cancer. Nat. Rev. Cancer 2007, 7, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Tarafdar, A.; Michie, A.M. Protein kinase C in cellular transformation: A valid target for therapy? Biochem. Soc. Trans. 2014, 42, 1556–1562. [Google Scholar] [CrossRef] [PubMed]
- Reyland, M.E.; Jones, D.N. Multifunctional roles of PKCδ: Opportunities for targeted therapy in human disease. Pharmacol. Ther. 2016, 165, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sprowl, J.A.; Ong, S.S.; Gibson, A.A.; Hu, S.; Du, G.; Lin, W.; Li, L.; Bharill, S.; Ness, R.A.; Stecula, A.; et al. A phosphotyrosine switch regulates organic cation transporters. Nat. Commun. 2016, 7, 10880. [Google Scholar] [CrossRef] [PubMed]
- Gschwendt, M.; Muller, H.J.; Kielbassa, K.; Zang, R.; Kittstein, W.; Rincke, G.; Marks, F. Rottlerin, a novel protein kinase inhibitor. Biochem. Biophys. Res. Commun. 1994, 199, 93–98. [Google Scholar] [CrossRef] [PubMed]
- McGovern, S.L.; Shoichet, B.K. Kinase inhibitors: Not just for kinases anymore. J. Med. Chem. 2003, 46, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- Galvez, M.I. Protein kinase C inhibitors in the treatment of diabetic retinopathy. Review. Curr. Pharm. Biotechnol. 2011, 12, 386–391. [Google Scholar] [CrossRef] [PubMed]
- Vinik, A. The protein kinase C-β inhibitor, ruboxistaurin, for the treatment of diabetic microvascular complications. Expert Opin. Investig. Drugs 2005, 14, 1547–1559. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.B.; LaCasce, A.S. Enzastaurin. Expert Opin. Investig. Drugs 2008, 17, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Zarate, C.A.; Manji, H.K. Protein kinase C inhibitors: Rationale for use and potential in the treatment of bipolar disorder. CNS Drugs 2009, 23, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Friman, S.; Arns, W.; Nashan, B.; Vincenti, F.; Banas, B.; Budde, K.; Cibrik, D.; Chan, L.; Klempnauer, J.; Mulgaonkar, S.; et al. Sotrastaurin, a novel small molecule inhibiting protein-kinase C: Randomized phase II study in renal transplant recipients. Am. J. Transplant. 2011, 11, 1444–1455. [Google Scholar] [CrossRef] [PubMed]
- Skvara, H.; Dawid, M.; Kleyn, E.; Wolff, B.; Meingassner, J.G.; Knight, H.; Dumortier, T.; Kopp, T.; Fallahi, N.; Stary, G.; et al. The PKC inhibitor AEB071 may be a therapeutic option for psoriasis. J. Clin. Investig. 2008, 118, 3151–3159. [Google Scholar] [CrossRef] [PubMed]
- Bates, E.; Bode, C.; Costa, M.; Gibson, C.M.; Granger, C.; Green, C.; Grimes, K.; Harrington, R.; Huber, K.; Kleiman, N.; et al. Intracoronary KAI-9803 as an adjunct to primary percutaneous coronary intervention for acute ST-segment elevation myocardial infarction. Circulation 2008, 117, 886–896. [Google Scholar] [PubMed]
- Tvedt, T.H.; Nepstad, I.; Bruserud, O. Antileukemic effects of midostaurin in acute myeloid leukemia—The possible importance of multikinase inhibition in leukemic as well as nonleukemic stromal cells. Expert Opin. Investig. Drugs 2017, 26, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Paquet, M. Ingenol mebutate: A promising treatment for actinic keratoses and nonmelanoma skin cancers. J. Cutan. Med. Surg. 2013, 17, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Geenes, V.; Williamson, C. Intrahepatic cholestasis of pregnancy. World J. Gastroenterol. 2009, 15, 2049–2066. [Google Scholar] [CrossRef] [PubMed]
- Pauli-Magnus, C.; Meier, P.J.; Stieger, B. Genetic determinants of drug-induced cholestasis and intrahepatic cholestasis of pregnancy. Semin. Liver Dis. 2010, 30, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Bacq, Y.; Sentilhes, L.; Reyes, H.B.; Glantz, A.; Kondrackiene, J.; Binder, T.; Nicastri, P.L.; Locatelli, A.; Floreani, A.; Hernandez, I.; et al. Efficacy of ursodeoxycholic acid in treating intrahepatic cholestasis of pregnancy: A meta-analysis. Gastroenterology 2012, 143, 1492–1501. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012, 3, e00049. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Transporter Family | Transporter | Main Expression | Main Type of Substrates |
|---|---|---|---|
| SLCOs | OATP1A2 (SLCO1A2) | Ubiquitous | Organic anions |
| OATP1B1 (SLCO1B1) | Liver | Organic anions | |
| OATP1B3 (SLCO1B3) | Liver | Organic anions | |
| OATP2B1 (SLCO2B1) | Liver, intestine | Organic anions | |
| SLC10A | NTCP (SLC10A1) | Liver | Bile acids |
| SLC15A | PEPT1 (SLC15A1) | Intestine | Peptides |
| PEPT2 (SLC15A2) | Kidney | Peptides | |
| SLC22A | OCT1 (SLC22A1) | Liver | Organic cations |
| OCT2 (SLC22A2) | Kidney | Organic cations | |
| OCTN1 (SLC22A4) | Kidney | Organic cations/carnitine | |
| OCTN2 (SLC22A5) | Kidney | Organic cations/carnitine | |
| OAT1 (SLC22A6) | Kidney | Organic anions | |
| OAT2 (SLC22A7) | Liver | Organic anions | |
| OAT3 (SLC22A8) | Kidney | Organic anions | |
| OAT4 (SLC22A11) | Kidney, placenta | Organic anions | |
| SLC28A | CNT1 (SLC28A1) | Kidney, liver, intestine | Nucleosides |
| CNT2 (SLC28A2) | Ubiquitous | Nucleosides | |
| CNT3 (SLC28A3) | Ubiquitous | Nucleosides | |
| SLC29A | ENT1 (SLC29A1) | Ubiquitous | Nucleosides |
| ENT2 (SLC29A2) | Ubiquitous | Nucleosides | |
| SLC47A | MATE1 (SLC47A1) | Liver, kidney | Organic cations |
| MATE2-K (SLC47A2) | Kidney | Organic cations | |
| ABCB | P-gp (ABCB1) | Intestine, liver, kidney, blood-brain barrier | Hydrophobic compounds |
| BSEP (ABCB11) | Liver | Bile acids | |
| ABCC | MRP1 (ABCC1) | Ubiquitous | Hydrophobic compounds, hydrophilic anions, conjugates |
| MRP2 (ABCC2) | Intestine, liver, kidney | Hydrophilic anions, conjugates | |
| MRP3 (ABCC3) | Liver, kidney | Hydrophilic anions, conjugates | |
| MRP4 (ABCC4) | Liver, kidney, blood-brain barrier | Nucleotides | |
| MRP5 (ABCC5) | Ubiquitous | Nucleotides | |
| ABCG | BCRP (ABCG2) | Intestine, liver, kidney, blood-brain barrier, stem cells | Hydrophobic compounds, hydrophilic anions, conjugates |
| Class | Dependence | Isoform | |
|---|---|---|---|
| Calcium | Diacylglycerol | ||
| Classical/Conventional cPKC (cPKC) | Yes | Yes | PKCα |
| PKCβ1 | |||
| PKCβ2 | |||
| PKCγ | |||
| Novel PKC (nPKC) | No | Yes | PKCδ |
| PKCε | |||
| PKCη | |||
| PKCθ | |||
| Atypical PKC (aPKC) | No | No | PKCζ |
| PKCλ/ι | |||
| Transporter | Activity | Localization | Expression |
|---|---|---|---|
| P-gp | Increase (human cancer cell lines, mouse renal proximal tubules) [13,59,60,61] | Increase (human cancer cells and primary human hepatocytes) [17,62,63] | |
| BSEP | Internalization (rat liver) [64] | Decrease (primary human hepatocytes) [18] | |
| MRP2 | Internalization (human hepatic HepG2 cell line) [65] | No change (primary human hepatocytes) [18] | |
| MRP3 | Increase (primary human hepatocytes) [18] | ||
| BCRP | No change (primary human hepatocytes) [18] | ||
| OATP1A2 | Internalization (OATP1A2- COS-7 cells) [66] | ||
| OATP1B1 | Internalization (OATP1B1-HEK293 cells) [67] | Decrease (primary human hepatocytes) [18] | |
| OATP1B3 | Decrease (primary human hepatocytes) [14] | Decrease (primary human hepatocytes) [18] | |
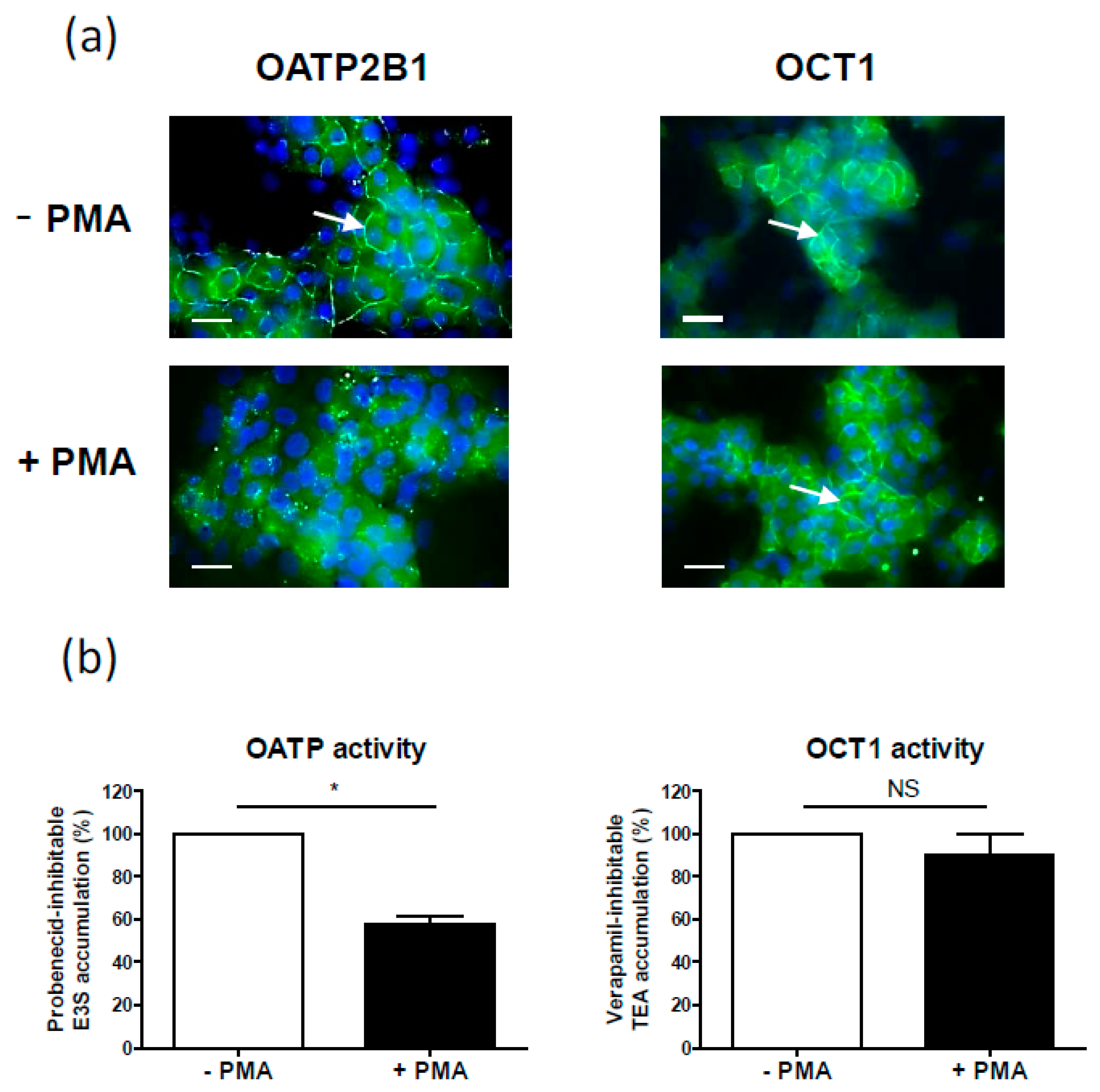
| OATP2B1 | Internalization (OATP2B1-MDCKII cells, Caco-2 cell line, human placenta, human hepatic HepaRG cell line) [68], (Figure 2a) | Decrease (primary human hepatocytes) [18] | |
| OAT1 | Internalization (OAT1-COS-7 cells) [69] | ||
| OAT3 | Decrease (rabbit renal proximal tubules) [70] | ||
| NTCP | Internalization (primary rat hepatocytes, NTCP-HepG2 cells) [64,71] | Decrease (primary human hepatocytes) [18] | |
| OCT1 | No change (Figure 2b) | Decrease (primary human hepatocytes) [18] | |
| OCTN2 | Increase in membrane expression (rat astrocytes) [72] | ||
| ENT1 | Increase (ENT1-PK15-NTD cells) [73] | Increase in membrane expression (ENT1- PK15-NTD cells) [73] | |
| PEPT1 | Decrease (human intestinal Caco-2 cell line) [74] | ||
| PEPT2 | Decrease (porcine kidney LLC-PK1 cell line) [75] |
| Drug | Nature of Effect | Targeted PKC(s) | Putative Therapeutic Indication |
|---|---|---|---|
| Rubixostaurin | PKC inhibition | PKCβ | Microvascular complications of diabetes [163,164] |
| Enzastaurin | PKC inhibition | PKCβ | Cancers [165] |
| Tamoxifen | PKC inhibition | Pan-PKC | Bipolar disorders [166] |
| Sotrastaurin (AEB071) | PKC inhibition | Pan-PKC | Organ transplantation [167], psoriasis [168] |
| KAI-9803 | PKC inhibition | PKCδ | Coronary intervention for myocardial infarction [169] |
| Midostaurin | PKC/FLT3/multikinase inhibition | Pan-PKC | Leukemias [170] |
| Ingenol mebutate | PKC activation | cPKCs/nPKCs | Actinic keratoses [171] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mayati, A.; Moreau, A.; Le Vée, M.; Stieger, B.; Denizot, C.; Parmentier, Y.; Fardel, O. Protein Kinases C-Mediated Regulations of Drug Transporter Activity, Localization and Expression. Int. J. Mol. Sci. 2017, 18, 764. https://doi.org/10.3390/ijms18040764
Mayati A, Moreau A, Le Vée M, Stieger B, Denizot C, Parmentier Y, Fardel O. Protein Kinases C-Mediated Regulations of Drug Transporter Activity, Localization and Expression. International Journal of Molecular Sciences. 2017; 18(4):764. https://doi.org/10.3390/ijms18040764
Chicago/Turabian StyleMayati, Abdullah, Amélie Moreau, Marc Le Vée, Bruno Stieger, Claire Denizot, Yannick Parmentier, and Olivier Fardel. 2017. "Protein Kinases C-Mediated Regulations of Drug Transporter Activity, Localization and Expression" International Journal of Molecular Sciences 18, no. 4: 764. https://doi.org/10.3390/ijms18040764
APA StyleMayati, A., Moreau, A., Le Vée, M., Stieger, B., Denizot, C., Parmentier, Y., & Fardel, O. (2017). Protein Kinases C-Mediated Regulations of Drug Transporter Activity, Localization and Expression. International Journal of Molecular Sciences, 18(4), 764. https://doi.org/10.3390/ijms18040764