
Microvesicles Contribute to the Bystander Effect of DNA Damage


Abstract
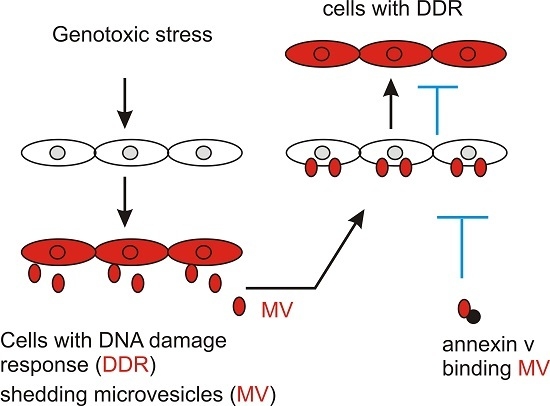
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
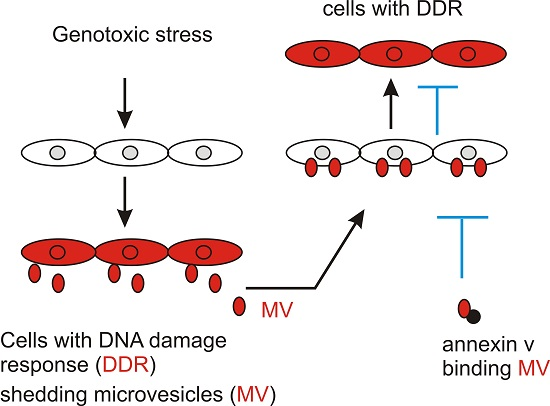
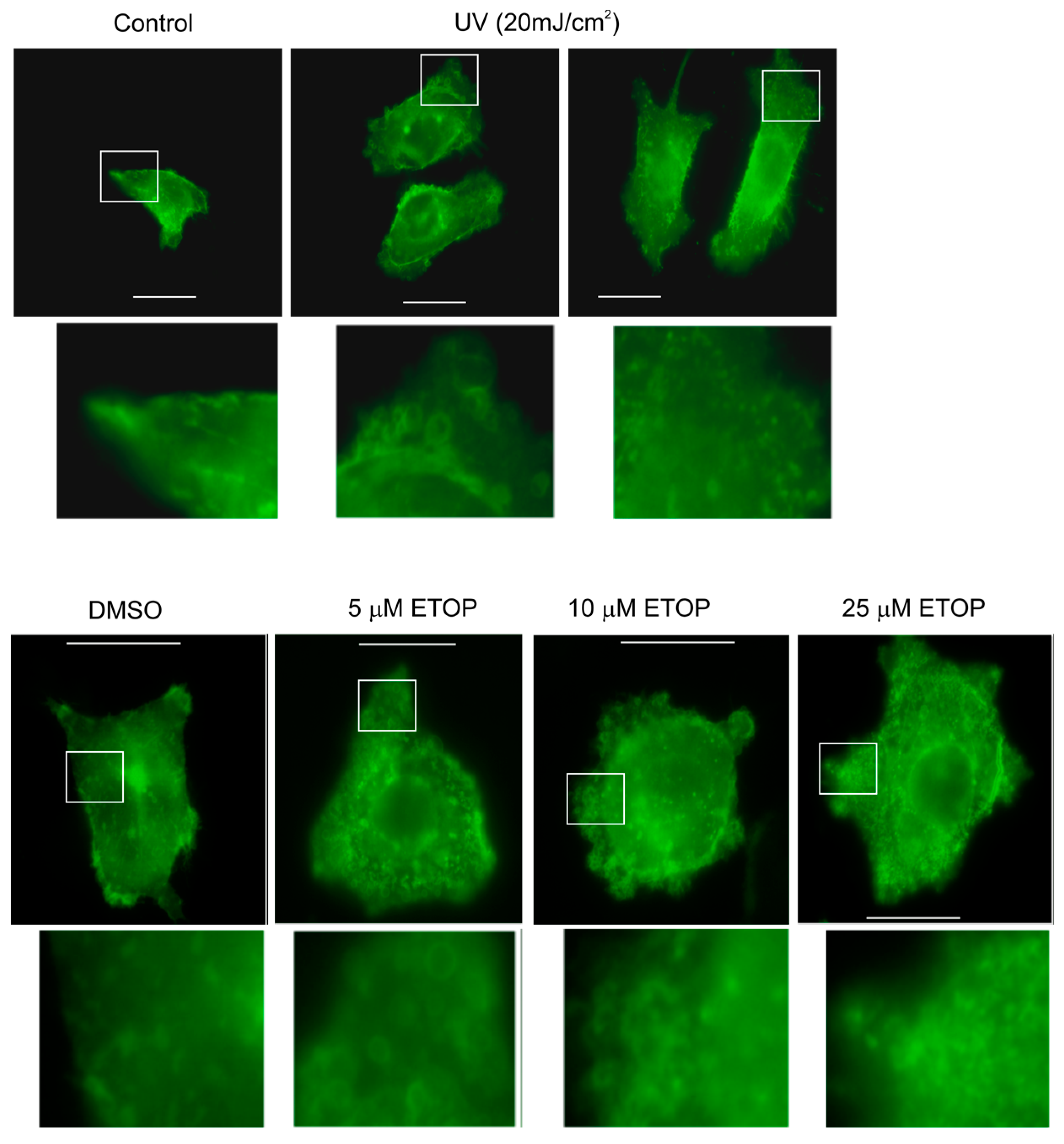
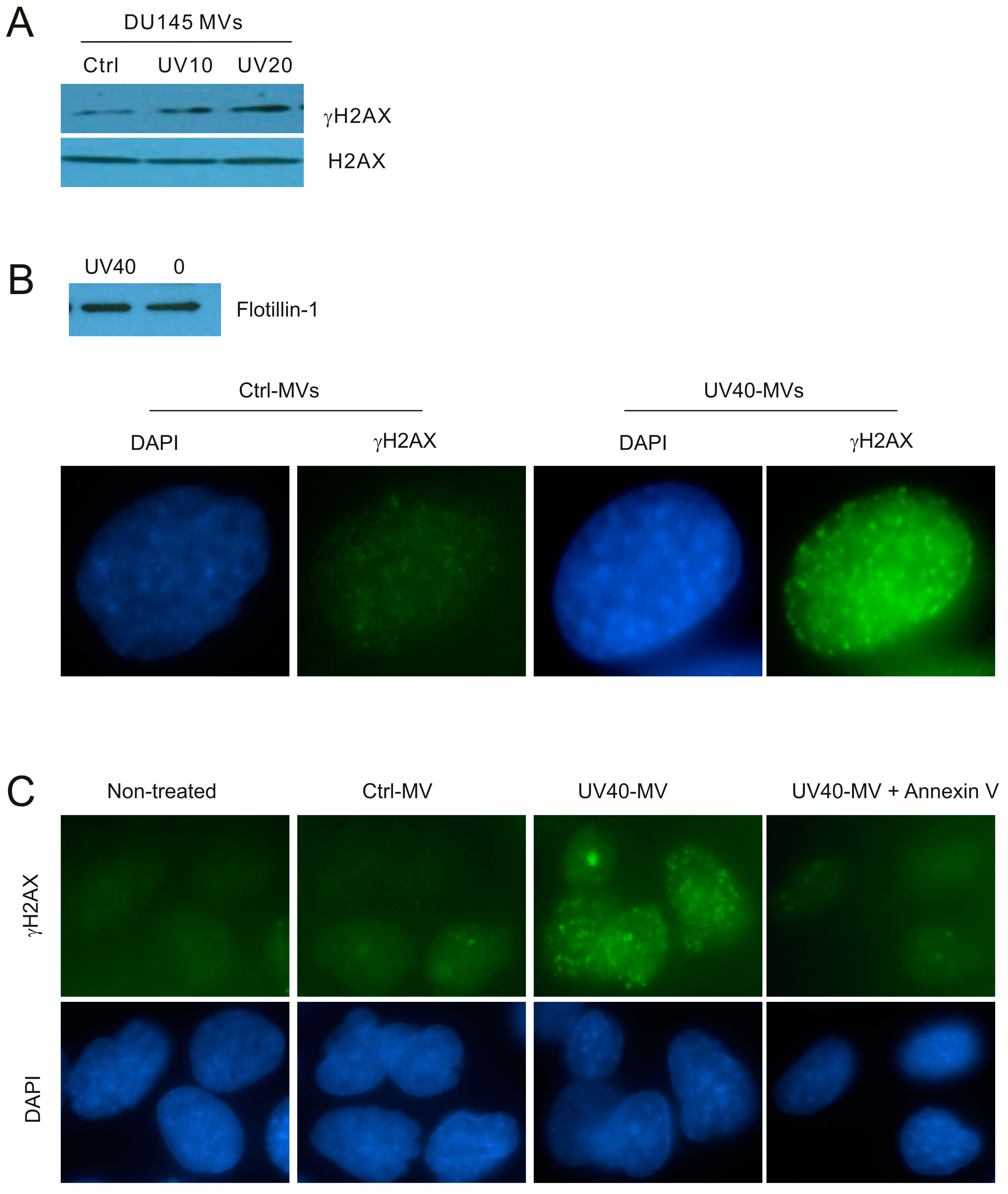
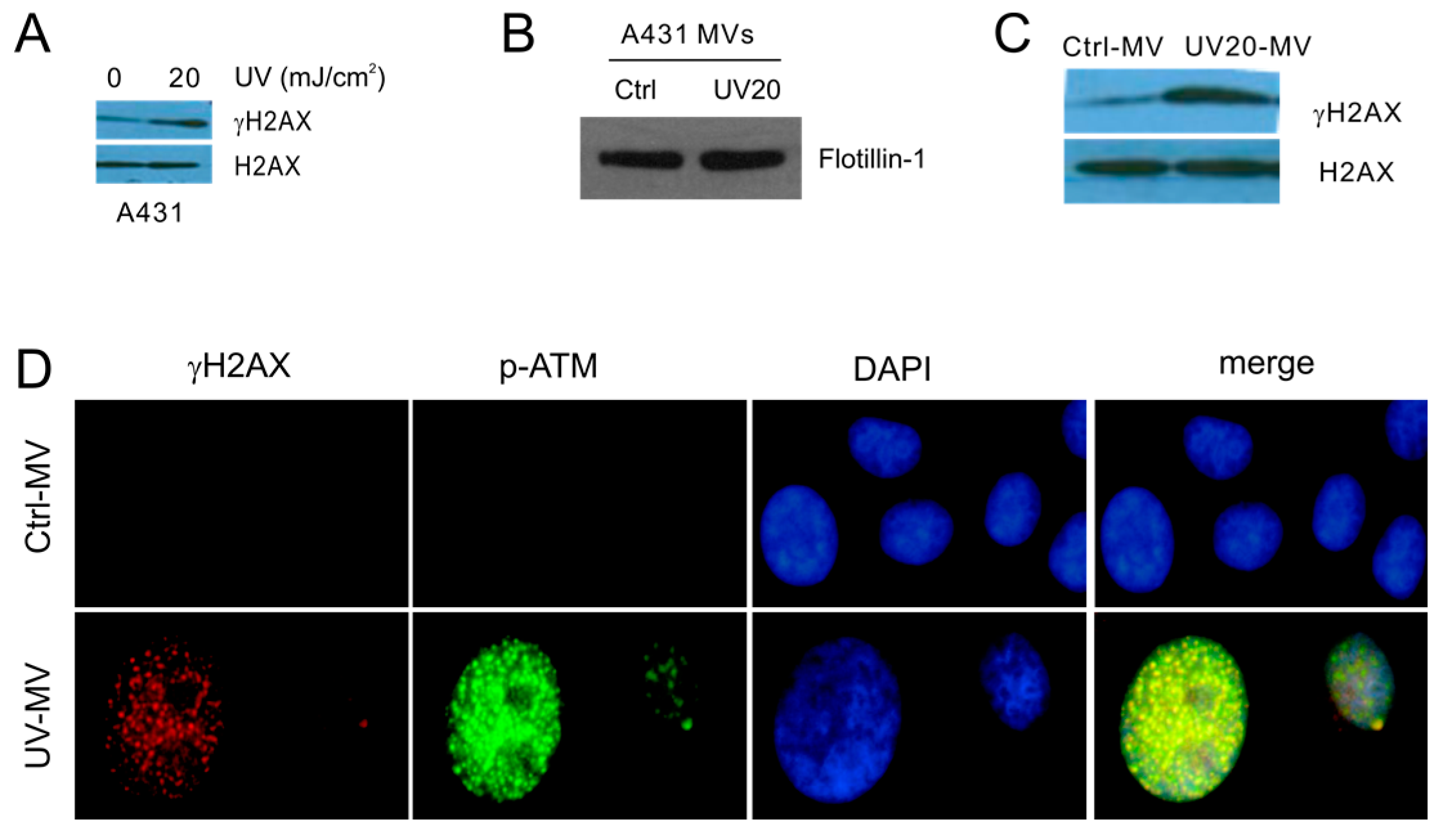
2.1. DNA Damage Enhances Microvesicles (MV) Formation
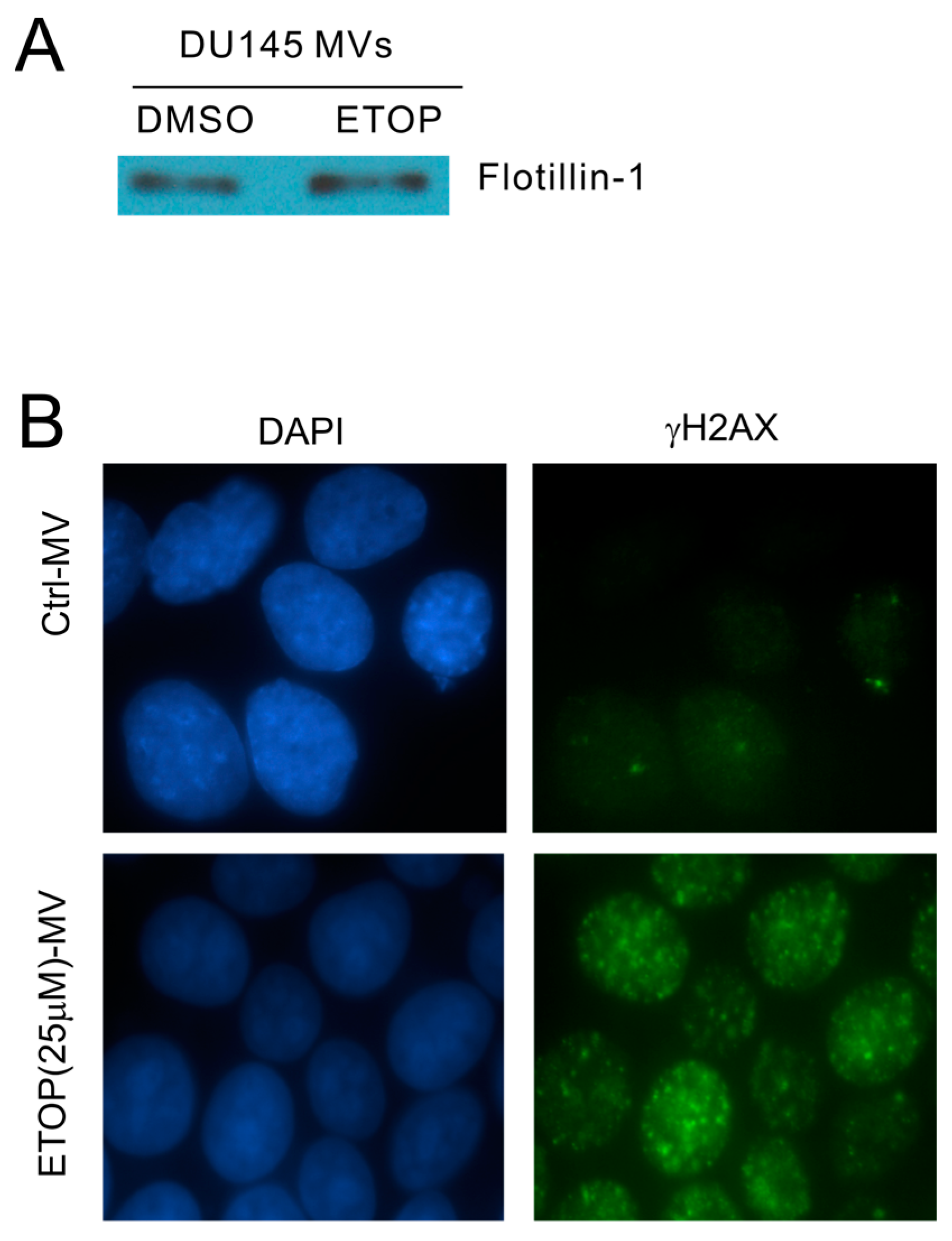
2.2. MVs Contributes to DNA Damage-Induced Bystander Effect (BE)
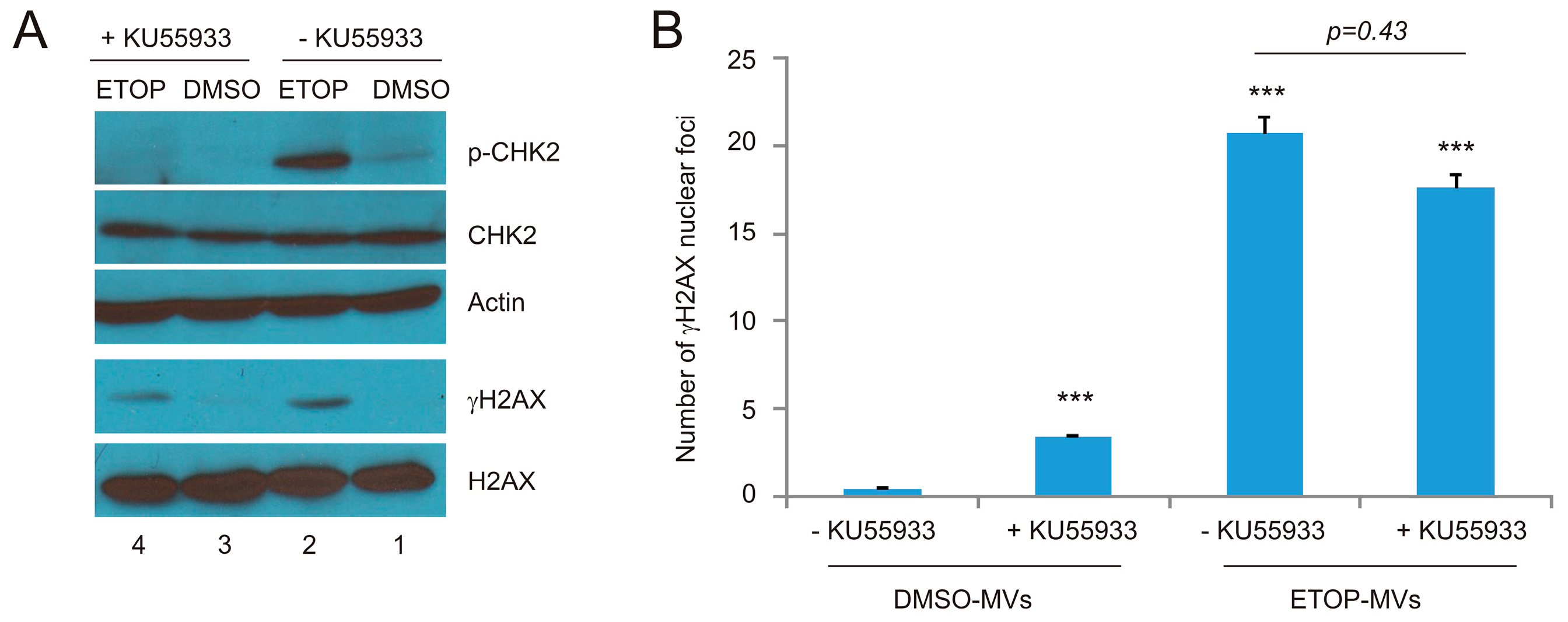
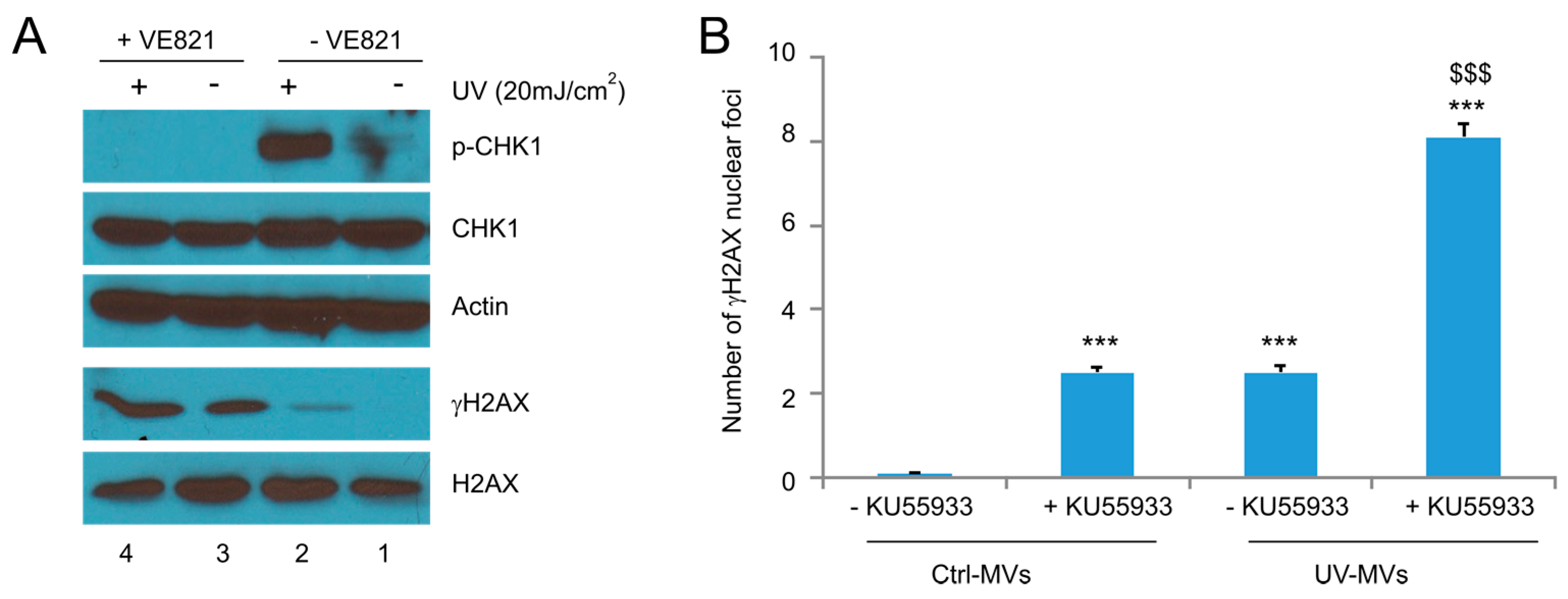
2.3. MV Induces BE in Recipient Cells Upstream of Ataxia-Telangiectasia Mutated (ATM)
3. Discussion
4. Materials and Methods
4.1. Chemicals, Cell Lines, and Plasmids
4.2. Retroviral Infection
4.3. Immunofluorescence Staining
4.4. Western Blot Analysis
4.5. Microvesicle Isolation and Treatment of Cells with MVs
4.6. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- El Ghissassi, F.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; Bouvard, V.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part D: Radiation. Lancet Oncol. 2009, 10, 751–752. [Google Scholar] [CrossRef]
- Pawel, D.; Preston, D.; Pierce, D.; Cologne, J. Improved estimates of cancer site-specific risks for A-bomb survivors. Radiat. Res. 2008, 169, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Yan, J.; Tang, D. Extracellular signal-regulated kinases modulate DNA damage response—A contributing factor to using MEK inhibitors in cancer therapy. Curr. Med. Chem. 2011, 18, 5476–5482. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Yan, J.; Tang, D. ERK kinases modulate the activation of PI3 kinase related kinases (PIKKs) in DNA damage response. Histol. Histopathol. 2013, 28, 1547–1554. [Google Scholar] [PubMed]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [PubMed]
- Jiang, H.; Wang, B.; Zhang, F.; Qian, Y.; Chuang, C.C.; Ying, M.; Wang, Y.; Zuo, L. The Expression and Clinical Outcome of pCHK2-Thr68 and pCDC25C-Ser216 in Breast Cancer. Int. J. Mol. Sci. 2016, 17, 1803. [Google Scholar] [CrossRef] [PubMed]
- Mothersill, C.; Seymour, C.B. Radiation-induced bystander effects—Implications for cancer. Nat. Rev. Cancer 2004, 4, 158–164. [Google Scholar] [PubMed]
- Martin, O.A.; Redon, C.E.; Nakamura, A.J.; Dickey, J.S.; Georgakilas, A.G.; Bonner, W.M. Systemic DNA damage related to cancer. Cancer Res. 2011, 71, 3437–3441. [Google Scholar] [CrossRef] [PubMed]
- Prise, K.M.; O’Sullivan, J.M. Radiation-induced bystander signalling in cancer therapy. Nat. Rev. Cancer 2009, 9, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Choy, A.; Barr, L.C.; Serpell, J.W.; Baum, M. Radiation-induced sarcoma of the retained breast after conservative surgery and radiotherapy for early breast cancer. Eur. J. Surg. Oncol. 1993, 19, 376–377. [Google Scholar] [PubMed]
- Kleinerman, R.A.; Boice, J.D., Jr.; Storm, H.H.; Sparen, P.; Andersen, A.; Pukkala, E.; Lynch, C.F.; Hankey, B.F.; Flannery, J.T. Second primary cancer after treatment for cervical cancer. An international cancer registries study. Cancer 1995, 76, 442–452. [Google Scholar] [CrossRef]
- Mills, T.D.; Vinnicombe, S.J.; Wells, C.A.; Carpenter, R. Angiosarcoma of the breast after wide local excision and radiotherapy for breast carcinoma. Clin. Radiol. 2002, 57, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Ohno, T.; Sakurai, H.; Saito, Y.; Takahashi, M.; Fukusato, T.; Mitsuhashi, N.; Niibe, H. Secondary malignant fibrous histiocytoma following radiation therapy for carcinoma of the uterine cervix: Report of two cases. Radiat. Med. 1997, 15, 229–233. [Google Scholar] [PubMed]
- Senkus, E.; Konefka, T.; Nowaczyk, M.; Jassem, J. Second lower genital tract squamous cell carcinoma following cervical cancer. A clinical study of 46 patients. Acta Obstet. Gynecol. Scand. 2000, 79, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Wijnmaalen, A.; van Ooijen, B.; van Geel, B.N.; Henzen-Logmans, S.C.; Treurniet-Donker, A.D. Angiosarcoma of the breast following lumpectomy, axillary lymph node dissection, and radiotherapy for primary breast cancer: Three case reports and a review of the literature. Int. J. Radiat. Oncol. Biol. Phys. 1993, 26, 135–139. [Google Scholar] [CrossRef]
- Sountoulides, P.; Koletsas, N.; Kikidakis, D.; Paschalidis, K.; Sofikitis, N. Secondary malignancies following radiotherapy for prostate cancer. Ther. Adv. Urol. 2010, 2, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Moon, K.; Stukenborg, G.J.; Keim, J.; Theodorescu, D. Cancer incidence after localized therapy for prostate cancer. Cancer 2006, 107, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Parsons, W.B., Jr.; Watkins, C.H.; Pease, G.L.; Childs, D.S., Jr. Changes in sternal marrow following roentgen-ray therapy to the spleen in chronic granulocytic leukemia. Cancer 1954, 7, 179–189. [Google Scholar] [CrossRef]
- Goh, K.; Sumner, H. Breaks in normal human chromosomes: Are they induced by a transferable substance in the plasma of persons exposed to total-body irradiation? Radiat. Res. 1968, 35, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Hollowell, J.G., Jr.; Littlefield, L.G. Chromosome damage induced by plasma of X-rayed patients: An indirect effect of X-ray. Proc. Soc. Exp. Biol. Med. 1968, 129, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Emerit, I.; Levy, A.; Cernjavski, L.; Arutyunyan, R.; Oganesyan, N.; Pogosian, A.; Mejlumian, H.; Sarkisian, T.; Gulkandanian, M.; Quastel, M.; et al. Transferable clastogenic activity in plasma from persons exposed as salvage personnel of the Chernobyl reactor. J. Cancer Res. Clin. Oncol. 1994, 120, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, H.; Little, J.B. Induction of sister chromatid exchanges by extremely low doses of α-particles. Cancer Res. 1992, 52, 6394–6396. [Google Scholar] [PubMed]
- Deshpande, A.; Goodwin, E.H.; Bailey, S.M.; Marrone, B.L.; Lehnert, B.E. Alpha-particle-induced sister chromatid exchange in normal human lung fibroblasts: Evidence for an extranuclear target. Radiat. Res. 1996, 145, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Mothersill, C.; Seymour, C. Medium from irradiated human epithelial cells but not human fibroblasts reduces the clonogenic survival of unirradiated cells. Int. J. Radiat. Biol. 1997, 71, 421–427. [Google Scholar] [PubMed]
- Lorimore, S.A.; Kadhim, M.A.; Pocock, D.A.; Papworth, D.; Stevens, D.L.; Goodhead, D.T.; Wright, E.G. Chromosomal instability in the descendants of unirradiated surviving cells after α-particle irradiation. Proc. Natl. Acad. Sci. USA 1998, 95, 5730–5733. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, H.; Little, J.B. Bystander effect for chromosomal aberrations induced in wild-type and repair deficient CHO cells by low fluences of alpha particles. Mutat. Res. 2002, 508, 121–129. [Google Scholar] [CrossRef]
- Sawant, S.G.; Randers-Pehrson, G.; Geard, C.R.; Brenner, D.J.; Hall, E.J. The bystander effect in radiation oncogenesis: I. Transformation in C3H 10T1/2 cells in vitro can be initiated in the unirradiated neighbors of irradiated cells. Radiat. Res. 2001, 155, 397–401. [Google Scholar] [CrossRef]
- Zhou, H.; Randers-Pehrson, G.; Waldren, C.A.; Vannais, D.; Hall, E.J.; Hei, T.K. Induction of a bystander mutagenic effect of α particles in mammalian cells. Proc. Natl. Acad. Sci. USA 2000, 97, 2099–2104. [Google Scholar] [CrossRef] [PubMed]
- Koturbash, I.; Loree, J.; Kutanzi, K.; Koganow, C.; Pogribny, I.; Kovalchuk, O. In vivo bystander effect: Cranial X-irradiation leads to elevated DNA damage, altered cellular proliferation and apoptosis, and increased p53 levels in shielded spleen. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Tamminga, J.; Koturbash, I.; Baker, M.; Kutanzi, K.; Kathiria, P.; Pogribny, I.P.; Sutherland, R.J.; Kovalchuk, O. Paternal cranial irradiation induces distant bystander DNA damage in the germline and leads to epigenetic alterations in the offspring. Cell Cycle 2008, 7, 1238–1245. [Google Scholar] [CrossRef] [PubMed]
- Koturbash, I.; Rugo, R.E.; Hendricks, C.A.; Loree, J.; Thibault, B.; Kutanzi, K.; Pogribny, I.; Yanch, J.C.; Engelward, B.P.; Kovalchuk, O. Irradiation induces DNA damage and modulates epigenetic effectors in distant bystander tissue in vivo. Oncogene 2006, 25, 4267–4275. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, M.; Pasquali, E.; Leonardi, S.; Tanori, M.; Rebessi, S.; Di Majo, V.; Pazzaglia, S.; Toni, M.P.; Pimpinella, M.; Covelli, V.; et al. Oncogenic bystander radiation effects in Patched heterozygous mouse cerebellum. Proc. Natl. Acad. Sci. USA 2008, 105, 12445–12450. [Google Scholar] [CrossRef] [PubMed]
- Rak, J. Microparticles in cancer. Semin. Thromb. Hemost. 2010, 36, 888–906. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, M.Z.; Ratajczak, J. Horizontal transfer of RNA and proteins between cells by extracellular microvesicles: 14 years later. Clin. Transl. Med. 2016, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan-Chari, V.; Clancy, J.W.; Sedgwick, A.; D’Souza-Schorey, C. Microvesicles: Mediators of extracellular communication during cancer progression. J. Cell Sci. 2010, 123, 1603–1611. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Wu, D.; Hirao, A.; Lahti, J.M.; Liu, L.; Mazza, B.; Kidd, V.J.; Mak, T.W.; Ingram, A.J. ERK activation mediates cell cycle arrest and apoptosis after DNA damage independently of p53. J. Biol. Chem. 2002, 277, 12710–12717. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Xie, Y.; Tao, L.; Tang, D. Both ERK1 and ERK2 kinases promote G2/M arrest in etoposide-treated MCF7 cells by facilitating ATM activation. Cell Signal. 2010, 22, 1783–1789. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Kang, T.H. Transcriptional and posttranslational regulation of nucleotide excision repair: The guardian of the genome against ultraviolet radiation. Int. J. Mol. Sci. 2016, 17, 1840. [Google Scholar] [CrossRef] [PubMed]
- Lagerwerf, S.; Vrouwe, M.G.; Overmeer, R.M.; Fousteri, M.I.; Mullenders, L.H. DNA damage response and transcription. DNA Repair 2011, 10, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Batista, L.F.; Kaina, B.; Meneghini, R.; Menck, C.F. How DNA lesions are turned into powerful killing structures: Insights from UV-induced apoptosis. Mutat. Res. 2009, 681, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, P.; Foiani, M.; Kumar, A. ATM and ATR signaling at a glance. J. Cell Sci. 2015, 128, 4255–4262. [Google Scholar] [CrossRef] [PubMed]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Guntuku, S.; Cui, X.S.; Matsuoka, S.; Cortez, D.; Tamai, K.; Luo, G.; Carattini-Rivera, S.; DeMayo, F.; Bradley, A.; et al. Chk1 is an essential kinase that is regulated by Atr and required for the G2/M DNA damage checkpoint. Genes Dev. 2000, 14, 1448–1459. [Google Scholar] [PubMed]
- Zhao, H.; Piwnica-Worms, H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol. 2001, 21, 4129–4139. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T. Mechanisms of ATM Activation. Annu. Rev. Biochem. 2015, 84, 711–738. [Google Scholar] [CrossRef] [PubMed]
- Mahaney, B.L.; Meek, K.; Lees-Miller, S.P. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem. J. 2009, 417, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Antonyak, M.A.; Li, B.; Boroughs, L.K.; Johnson, J.L.; Druso, J.E.; Bryant, K.L.; Holowka, D.A.; Cerione, R.A. Cancer cell-derived microvesicles induce transformation by transferring tissue transglutaminase and fibronectin to recipient cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4852–4857. [Google Scholar] [CrossRef] [PubMed]
- Carroll, B.L.; Pulkoski-Gross, M.J.; Hannun, Y.A.; Obeid, L.M. CHK1 regulates NF-kappaB signaling upon DNA damage in p53- deficient cells and associated tumor-derived microvesicles. Oncotarget 2016, 7, 18159–18170. [Google Scholar] [PubMed]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Al-Nedawi, K.; Meehan, B.; Kerbel, R.S.; Allison, A.C.; Rak, J. Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR. Proc. Natl. Acad. Sci. USA 2009, 106, 3794–3799. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Jeggo, P.A. The heterochromatic barrier to DNA double strand break repair: How to get the entry visa. Int. J. Mol. Sci. 2012, 13, 11844–11860. [Google Scholar] [CrossRef] [PubMed]
- Berkey, F.J. Managing the adverse effects of radiation therapy. Am. Fam. Phys. 2010, 82, 381–388, 394. [Google Scholar]
- Smith, M.A.; Seibel, N.L.; Altekruse, S.F.; Ries, L.A.; Melbert, D.L.; O’Leary, M.; Smith, F.O.; Reaman, G.H. Outcomes for children and adolescents with cancer: Challenges for the twenty-first century. J. Clin. Oncol. 2010, 28, 2625–2634. [Google Scholar] [CrossRef] [PubMed]
- Robison, L.L.; Armstrong, G.T.; Boice, J.D.; Chow, E.J.; Davies, S.M.; Donaldson, S.S.; Green, D.M.; Hammond, S.; Meadows, A.T.; Mertens, A.C.; et al. The Childhood Cancer Survivor Study: A National Cancer Institute-supported resource for outcome and intervention research. J. Clin. Oncol. 2009, 27, 2308–2318. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.L.; Whitton, J.; Leisenring, W.; Mertens, A.C.; Hammond, S.; Stovall, M.; Donaldson, S.S.; Meadows, A.T.; Robison, L.L.; Neglia, J.P. Subsequent neoplasms in 5-year survivors of childhood cancer: The Childhood Cancer Survivor Study. J. Natl. Cancer Inst. 2010, 102, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Oeffinger, K.C.; Mertens, A.C.; Sklar, C.A.; Kawashima, T.; Hudson, M.M.; Meadows, A.T.; Friedman, D.L.; Marina, N.; Hobbie, W.; Kadan-Lottick, N.S.; et al. Chronic health conditions in adult survivors of childhood cancer. N. Engl. J. Med. 2006, 355, 1572–1582. [Google Scholar] [CrossRef] [PubMed]
- Lawson, C.; Vicencio, J.M.; Yellon, D.M.; Davidson, S.M. Microvesicles and exosomes: New players in metabolic and cardiovascular disease. J. Endocrinol. 2016, 228, R57–R71. [Google Scholar] [CrossRef] [PubMed]
- Budnik, V.; Ruiz-Canada, C.; Wendler, F. Extracellular vesicles round off communication in the nervous system. Nat. Rev. Neurosci. 2016, 17, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Wang, J.; Ding, N.; Hu, W.; Zhang, X.; Wang, B.; Hua, J.; Wei, W.; Zhu, Q. Exosome-mediated microRNA transfer plays a role in radiation-induced bystander effect. RNA Biol. 2015, 12, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Ghosh, A.; Krishna, M. Role of ATM in bystander signaling between human monocytes and lung adenocarcinoma cells. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2015, 794, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Burdak-Rothkamm, S.; Rothkamm, K.; McClelland, K.; Al Rashid, S.T.; Prise, K.M. BRCA1, FANCD2 and Chk1 are potential molecular targets for the modulation of a radiation-induced DNA damage response in bystander cells. Cancer Lett. 2015, 356, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Scott, A.; Wang, Y.W.; Weiss, R.H.; Murray, D. Spontaneous γH2AX foci in human solid tumor-derived cell lines in relation to p21WAF1 and WIP1 expression. Int. J. Mol. Sci. 2015, 16, 11609–11628. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Fan, C.; Kapoor, A.; Ingram, A.J.; Rybak, A.P.; Austin, R.C.; Dickhout, J.; Cutz, J.C.; Scholey, J.; Tang, D. α-Mannosidase 2C1 attenuates PTEN function in prostate cancer cells. Nat. Commun. 2011, 2, 307. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Ingram, A.; Rybak, A.P.; Tang, D. Shank-interacting protein-like 1 promotes tumorigenesis via PTEN inhibition in human tumor cells. J. Clin. Investig. 2010, 120, 2094–2108. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Ojo, D.; Kapoor, A.; Lin, X.; Pinthus, J.H.; Aziz, T.; Bismar, T.A.; Wei, F.; Wong, N.; de Melo, J.; et al. Neural cell adhesion protein CNTN1 promotes the metastatic progression of prostate cancer. Cancer Res. 2016, 76, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, K.; Ingram, A.; Austin, R.; Kapoor, A.; Tang, D.; Majeed, F.; Qureshi, T.; Al-Nedawi, K. Regulation of the tumor suppressor PTEN through exosomes: A diagnostic potential for prostate cancer. PLoS ONE 2013, 8, e70047. [Google Scholar] [CrossRef] [PubMed]







© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, X.; Wei, F.; Major, P.; Al-Nedawi, K.; Al Saleh, H.A.; Tang, D. Microvesicles Contribute to the Bystander Effect of DNA Damage. Int. J. Mol. Sci. 2017, 18, 788. https://doi.org/10.3390/ijms18040788
Lin X, Wei F, Major P, Al-Nedawi K, Al Saleh HA, Tang D. Microvesicles Contribute to the Bystander Effect of DNA Damage. International Journal of Molecular Sciences. 2017; 18(4):788. https://doi.org/10.3390/ijms18040788
Chicago/Turabian StyleLin, Xiaozeng, Fengxiang Wei, Pierre Major, Khalid Al-Nedawi, Hassan A. Al Saleh, and Damu Tang. 2017. "Microvesicles Contribute to the Bystander Effect of DNA Damage" International Journal of Molecular Sciences 18, no. 4: 788. https://doi.org/10.3390/ijms18040788
APA StyleLin, X., Wei, F., Major, P., Al-Nedawi, K., Al Saleh, H. A., & Tang, D. (2017). Microvesicles Contribute to the Bystander Effect of DNA Damage. International Journal of Molecular Sciences, 18(4), 788. https://doi.org/10.3390/ijms18040788