
Endoplasmic Reticulum Stress and Homeostasis in Reproductive Physiology and Pathology



Abstract
:1. Introduction
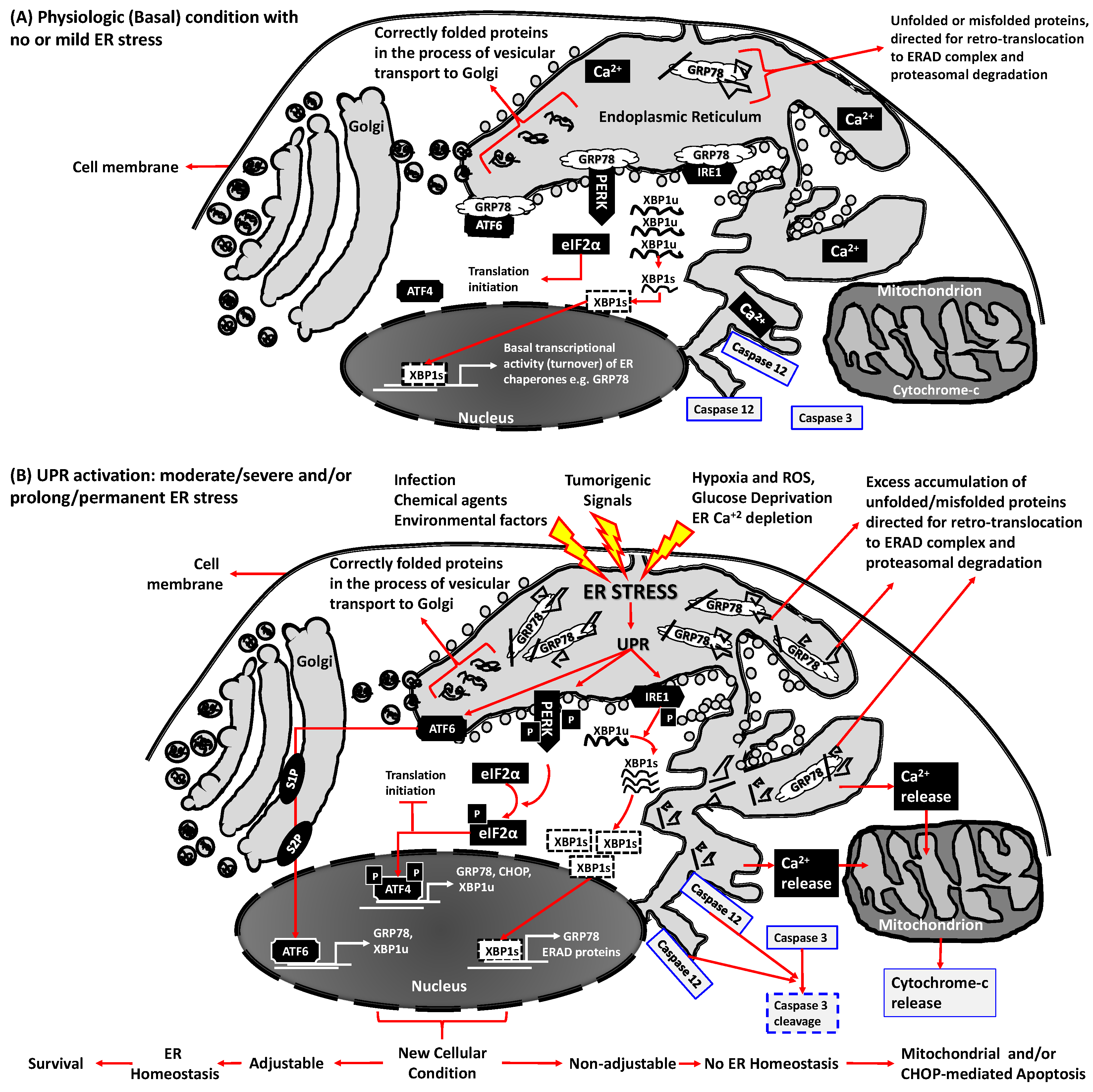
2. The Endoplasmic Reticulum (ER) Stress Induced Unfolded Protein Response (UPR) Signaling Cascades are Vital to Sustain ER Homeostasis
3. ER Stress, UPR Signaling and ER Homeostasis
4. UPR Signaling, ER Stress in Reproductive Physiopathology
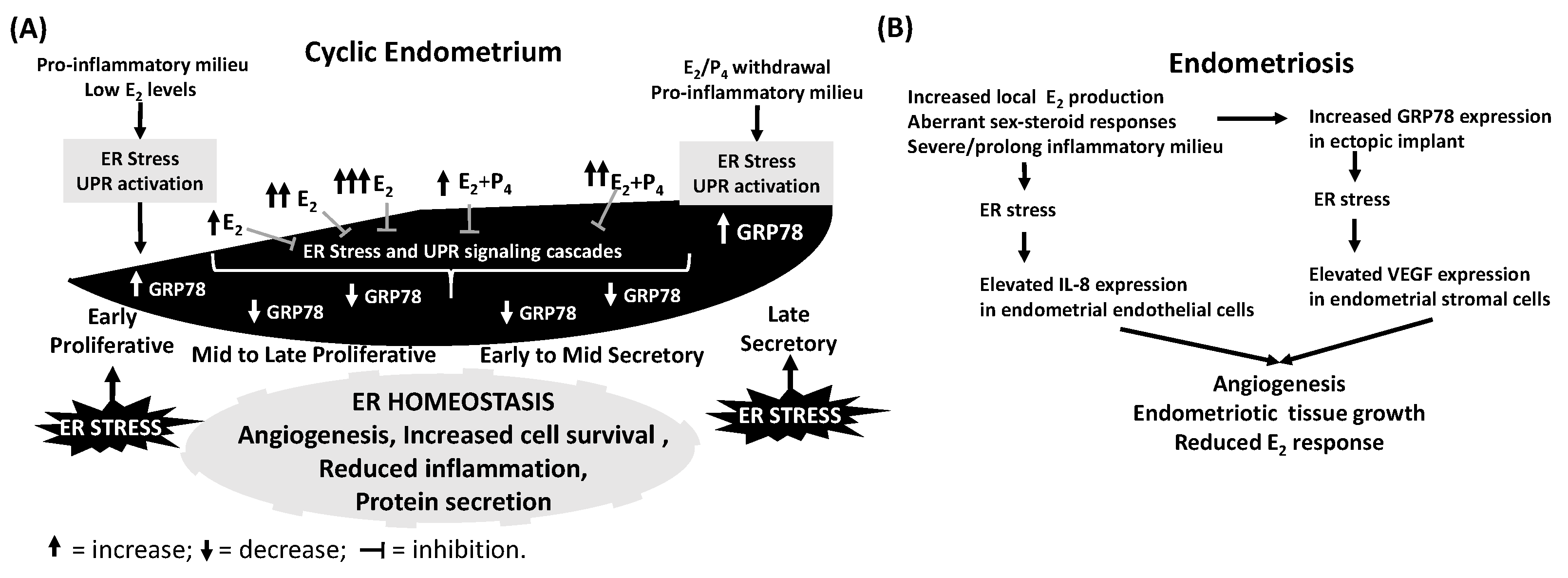
4.1. The Menstrual Cycle Endometrium
4.2. Endometriosis
4.3. Endometrial and Other Reproductive Tissue Cancers
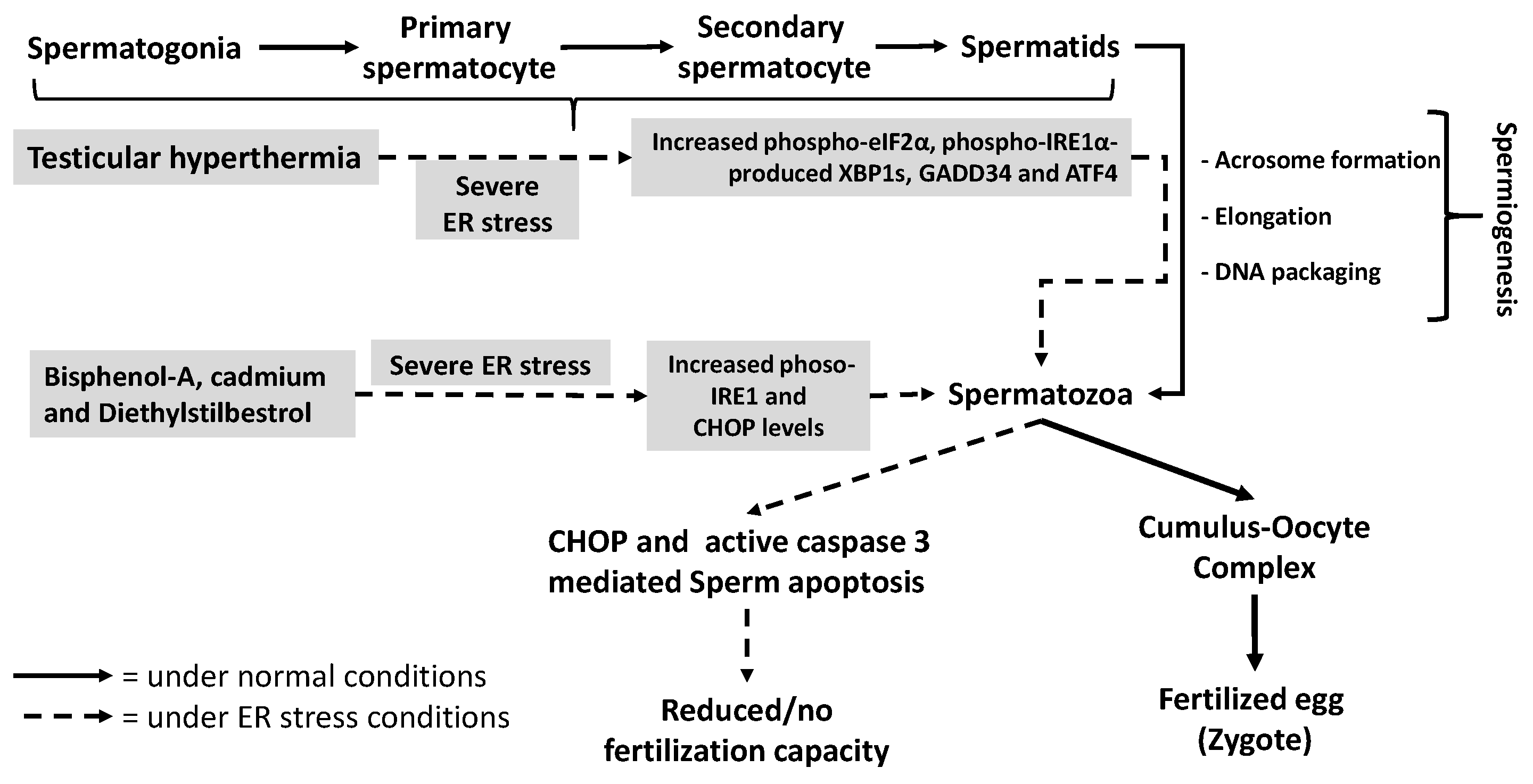
4.4. Sperm
4.5. Oocytes
4.6. Preimplantation Embryo Development
4.7. Implantation
5. Role of ER Stress in Pregnancy Complications and Preterm Birth
6. Conclusion
Conflicts of Interest
Abbreviations
| AN3CA | An endometrial adenocarcinoma cell line |
| ATF6 | Activating transcription factor 6 |
| ATF6N | Activated transcription factor |
| Ca2+ | Calcium |
| CHOP | C/EBP homologous protein |
| DNA | Deoxyribonucleic acid |
| DRP1 | Dynamin-related protein 1 |
| E2 | Estradiol |
| EIF2AK3 | Eukaryotic translation initiation factor 2-α kinase 3 |
| EMT | Epithelial-mesenchymal transition |
| ER | Endoplasmic reticulum |
| ERAD | ER-associated degradation |
| FGR | Fetal growth restriction |
| GADD34 | Growth arrest and DNA damage-inducible protein 34 |
| GRP78 | Glucose-regulated protein 78 |
| GRP94 | Glucose-regulated protein 94 |
| H2O2 | Hydrogen peroxide |
| HSPA5 | Heat shock 70 protein 5 |
| HeLa | A cervical adenocarcinoma cell line |
| ICSI | Intra-cytoplasmic sperm injection |
| IL-3 | Interleukin-3 |
| IL-8 | Interleukin-8 |
| IL-6 | Interleukin-6 |
| IRE1 | Inositol-requiring enzyme 1 |
| LPS | Lipopolysaccharide |
| MAP1LC3B | Microtubule-associated proteins 1A/1B light chain 3B |
| MDM2 | Mouse double minute 2 homolog |
| MET | Mesenchymal-epithelial transition |
| mRNA | Messenger RNA |
| mtDNA | Mitochondrial DNA |
| NOS | Nitric oxide synthase |
| CDKN1A | Cyclin-dependent kinase inhibitor 1 |
| P-ATF4 | Phosphorylated-activating transcription factor 4 |
| PDI | Protein disulfide isomerase |
| P-eIF2α | Phosphorylated-eukaryotic initiation factor 2α |
| PERK | Phosphorylated endoplasmic reticulum kinase |
| P-JNK | Phosphorylated-c-Jun NH2-terminal kinases |
| PKR | Protein kinase R |
| PPI | Peptidyl-prolyl isomerase |
| ROS | Reactive oxygen species |
| S1P | Site-1 protease |
| S2P | Site-2 protease |
| TFAM | Mitochondrial transcription factor A |
| TNFα | Tumor necrosis factor α |
| TUDCA | Tauroursodeoxycholic acid |
| UPR | Unfolded protein response |
| VCP | Valosin-containing protein |
| VEGF | Vascular endothelial growth factor |
| XBP1 | X-box binding protein 1 |
References
- Porter, K.R.; Claude, A.; Fullam, E.F. A study of tissue culture cells by electron microscopy: Methods and preliminary observations. J. Exp. Med. 1945, 81, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.R.; Kallman, F.L. Significance of cell particulates as seen by electron microscopy. Ann. N. Y. Acad. Sci. 1952, 54, 882–891. [Google Scholar] [CrossRef] [PubMed]
- Simonyi, I.; Pataki, S.; Kalman, K.; Buda, L. Determination of the active ingredient content in Tavegyl tablets. Acta Pharm. Hung. 1975, 45, 237–244. [Google Scholar] [PubMed]
- Wischnitzer, S. The nuclear envelope: Its ultrastructure and functional significance. Endeavour 1974, 33, 137–142. [Google Scholar] [CrossRef]
- Krebs, J.; Agellon, L.B.; Michalak, M. Ca2+ homeostasis and endoplasmic reticulum (ER) stress: An integrated view of calcium signaling. Biochem. Biophys. Res. Commun. 2015, 460, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, R.J.; Scheuner, D.; Schroder, M.; Shen, X.; Lee, K.; Liu, C.Y.; Arnold, S.M. The unfolded protein response in nutrient sensing and differentiation. Nat. Rev. Mol. Cell Biol. 2002, 3, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Goodman, J.M. The lipid droplet-a well-connected organelle. Front. Cell Dev. Biol. 2015, 3, 49. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. Phospholipid synthesis and transport in mammalian cells. Traffic 2015, 16, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Fagone, P.; Jackowski, S. Membrane phospholipid synthesis and endoplasmic reticulum function. J. Lipid Res. 2009, 50, S311–S316. [Google Scholar] [CrossRef] [PubMed]
- Dill, K.A.; Ozkan, S.B.; Shell, M.S.; Weikl, T.R. The protein folding problem. Annu. Rev. Biophys. 2008, 37, 289–316. [Google Scholar] [CrossRef] [PubMed]
- Scriven, P.; Coulson, S.; Haines, R.; Balasubramanian, S.; Cross, S.; Wyld, L. Activation and clinical significance of the unfolded protein response in breast cancer. Br. J. Cancer 2009, 101, 1692–1698. [Google Scholar] [CrossRef] [PubMed]
- Bukau, B.; Weissman, J.; Horwich, A. Molecular chaperones and protein quality control. Cell 2006, 125, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Luo, K.L.; Shi, L. Endoplasmic reticulum stress interacts with inflammation in human diseases. J. Cell. Physiol. 2016, 231, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.M.; Yang, W.L.; Wang, P. Endoplasmic reticulum stress in sepsis. Shock 2015, 44, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Pei, X.; Jin, Y.; Wang, Y.; Zhang, C. The roles of endoplasmic reticulum stress response in female mammalian reproduction. Cell Tissue Res. 2016, 363, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, A.; Hotamisligil, G.S. Impact of ER protein homeostasis on metabolism. Diabetes 2013, 62, 691–693. [Google Scholar] [CrossRef] [PubMed]
- Buhimschi, I.A.; Nayeri, U.A.; Zhao, G.; Shook, L.L.; Pensalfini, A.; Funai, E.F.; Bernstein, I.M.; Glabe, C.G.; Buhimschi, C.S. Protein misfolding, congophilia, oligomerization, and defective amyloid processing in preeclampsia. Sci. Transl. Med. 2014, 6, 245ra292. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 2005, 35, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Lievremont, J.P.; Rizzuto, R.; Hendershot, L.; Meldolesi, J. BiP, a major chaperone protein of the endoplasmic reticulum lumen, plays a direct and important role in the storage of the rapidly exchanging pool of Ca2+. J. Biol. Chem. 1997, 272, 30873–30879. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.; Lee, A.S. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene 2013, 32, 805–818. [Google Scholar] [CrossRef] [PubMed]
- Jaattela, M.; Wissing, D. Emerging role of heat shock proteins in biology and medicine. Ann. Med. 1992, 24, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.; Pelham, H.R. An HSP70-like protein in the ER: Identity with the 78 kD glucose-regulated protein and immunoglobulin heavy chain binding protein. Cell 1986, 46, 291–300. [Google Scholar] [CrossRef]
- Hartl, F.U. Molecular chaperones in cellular protein folding. Nature 1996, 381, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, R.J. Stress signaling from the lumen of the endoplasmic reticulum: Coordination of gene transcriptional and translational controls. Genes Dev. 1999, 13, 1211–1233. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.S.; Arvan, P. Endocrinopathies in the family of endoplasmic reticulum (ER) storage diseases: Disorders of protein trafficking and the role of ER molecular chaperones. Endocr. Rev. 1998, 19, 173–202. [Google Scholar] [CrossRef] [PubMed]
- Simmons, D.G.; Kennedy, T.G. Induction of glucose-regulated protein 78 in rat uterine glandular epithelium during uterine sensitization for the decidual cell reaction. Biol. Reprod. 2000, 62, 1168–1176. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Lee, A.S. ER chaperones in mammalian development and human diseases. FEBS Lett. 2007, 581, 3641–3651. [Google Scholar] [CrossRef] [PubMed]
- Pfaffenbach, K.T.; Lee, A.S. The critical role of GRP78 in physiologic and pathologic stress. Curr. Opin. Cell Biol. 2011, 23, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Wiest, D.L.; Burkhardt, J.K.; Hester, S.; Hortsch, M.; Meyer, D.I.; Argon, Y. Membrane biogenesis during B cell differentiation: Most endoplasmic reticulum proteins are expressed coordinately. J. Cell Biol. 1990, 110, 1501–1511. [Google Scholar] [CrossRef] [PubMed]
- Philippova, M.; Ivanov, D.; Joshi, M.B.; Kyriakakis, E.; Rupp, K.; Afonyushkin, T.; Bochkov, V.; Erne, P.; Resink, T.J. Identification of proteins associating with glycosylphosphatidylinositol-anchored T-cadherin on the surface of vascular endothelial cells: Role for GRP78/BiP in T-cadherin-dependent cell survival. Mol. Cell. Biol. 2008, 28, 4004–4017. [Google Scholar] [CrossRef] [PubMed]
- Misra, U.K.; Deedwania, R.; Pizzo, S.V. Binding of activated α2-macroglobulin to its cell surface receptor GRP78 in 1-LN prostate cancer cells regulates PAK-2-dependent activation of LIMK. J. Biol. Chem. 2005, 280, 26278–26286. [Google Scholar] [CrossRef] [PubMed]
- Delpino, A.; Castelli, M. The 78 kDa glucose-regulated protein (GRP78/BiP) is expressed on the cell membrane, is released into cell culture medium and is also present in human peripheral circulation. Biosci. Rep. 2002, 22, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Delpino, A.; Piselli, P.; Vismara, D.; Vendetti, S.; Colizzi, V. Cell surface localization of the 78 kD glucose regulated protein (GRP 78) induced by thapsigargin. Mol. Membr. Biol. 1998, 15, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Corrigall, V.M.; Bodman-Smith, M.D.; Brunst, M.; Cornell, H.; Panayi, G.S. Inhibition of antigen-presenting cell function and stimulation of human peripheral blood mononuclear cells to express an antiinflammatory cytokine profile by the stress protein BiP: Relevance to the treatment of inflammatory arthritis. Arthritis Rheum. 2004, 50, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Delom, F.; Lejeune, P.J.; Vinet, L.; Carayon, P.; Mallet, B. Involvement of oxidative reactions and extracellular protein chaperones in the rescue of misassembled thyroglobulin in the follicular lumen. Biochem. Biophys. Res. Commun. 1999, 255, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, H.; Yoshimori, T.; Yamamoto, A.; Miyata, Y.; Yahara, I.; Inoue, K.; Tashiro, Y. Heavy chain binding protein (BiP/GRP78) and endoplasmin are exported from the endoplasmic reticulum in rat exocrine pancreatic cells, similar to protein disulfide-isomerase. Arch. Biochem. Biophys. 1992, 296, 129–136. [Google Scholar] [CrossRef]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef] [PubMed]
- Beaton, A.; Wilkins, R.J.; Wheeler, T.T. Lactation-associated and prolactin-responsive changes in protein synthesis in mouse mammary cells. Tissue Cell 1997, 29, 509–516. [Google Scholar] [CrossRef]
- Brewer, J.W.; Cleveland, J.L.; Hendershot, L.M. A pathway distinct from the mammalian unfolded protein response regulates expression of endoplasmic reticulum chaperones in non-stressed cells. EMBO J. 1997, 16, 7207–7216. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Shu, C.W.; Xu, W.; Shiau, C.W.; Grant, D.; Vasile, S.; Cosford, N.D.; Reed, J.C. Chemical biology investigation of cell death pathways activated by endoplasmic reticulum stress reveals cytoprotective modulators of ASK1. J. Biol. Chem. 2009, 284, 1593–1603. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, S.; Miao, L.; Huang, H.; Liang, F.; Teng, X.; Xu, L.; Wang, Q.; Xiao, W.; Ridder, W.H.; et al. Rescue of glaucomatous neurodegeneration by differentially modulating neuronal endoplasmic reticulum stress molecules. J. Neurosci. 2016, 36, 5891–5903. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Arikkath, J.; Yang, L.; Guo, M.L.; Periyasamy, P.; Buch, S. Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders. Autophagy 2016, 12, 225–244. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.C.; Meier, S.E.; Ingram, A.L.; Abisambra, J.F. Perk-opathies: An endoplasmic reticulum stress mechanism underlying neurodegeneration. Curr. Alzheimer Res. 2016, 13, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Penke, B.; Bogar, F.; Fulop, L. Protein folding and misfolding, endoplasmic reticulum stress in neurodegenerative diseases: In trace of novel drug targets. Curr. Protein Pept. Sci. 2016, 17, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, D.T.; Kaufman, R.J. That which does not kill me makes me stronger: Adapting to chronic ER stress. Trends Biochem. Sci. 2007, 32, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Dave, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Kadowaki, H.; Nishitoh, H. Signaling pathways from the endoplasmic reticulum and their roles in disease. Genes 2013, 4, 306–333. [Google Scholar] [CrossRef] [PubMed]
- Michalak, M.; Gye, M.C. Endoplasmic reticulum stress in periimplantation embryos. Clin. Exp. Reprod. Med. 2015, 42, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bifulco, G.; Miele, C.; di Jeso, B.; Beguinot, F.; Nappi, C.; di Carlo, C.; Capuozzo, S.; Terrazzano, G.; Insabato, L.; Ulianich, L. Endoplasmic reticulum stress is activated in endometrial adenocarcinoma. Gynecol. Oncol. 2012, 125, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Schroder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res. 2005, 569, 29–63. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mrna is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Liu, J.; Ibi, D.; Taniguchi, K.; Lee, J.; Herrema, H.; Akosman, B.; Mucka, P.; Salazar Hernandez, M.A.; Uyar, M.F.; Park, S.W.; et al. Inflammation improves glucose homeostasis through IKKβ-XBP1s interaction. Cell 2016, 167, 1052–1066. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Zhou, Y.; Lee, J.; Lu, A.; Sun, C.; Chung, J.; Ueki, K.; Ozcan, U. The regulatory subunits of PI3K, p85α and p85β, interact with XBP-1 and increase its nuclear translocation. Nat. Med. 2010, 16, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Heath-Engel, H.M.; Chang, N.C.; Shore, G.C. The endoplasmic reticulum in apoptosis and autophagy: Role of the BCL-2 protein family. Oncogene 2008, 27, 6419–6433. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.V.; Peel, A.; Logvinova, A.; del Rio, G.; Hermel, E.; Yokota, T.; Goldsmith, P.C.; Ellerby, L.M.; Ellerby, H.M.; Bredesen, D.E. Coupling endoplasmic reticulum stress to the cell death program: Role of the ER chaperone GRP78. FEBS Lett. 2002, 514, 122–128. [Google Scholar] [CrossRef]
- Kayisli, U.A.; Mahutte, N.G.; Arici, A. Uterine chemokines in reproductive physiology and pathology. Am. J. Reprod. Immunol. 2002, 47, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.M.; Couse, J.F.; Korach, K.S. The multifaceted mechanisms of estradiol and estrogen receptor signaling. J. Biol. Chem. 2001, 276, 36869–36872. [Google Scholar] [CrossRef] [PubMed]
- Truss, M.; Beato, M. Steroid hormone receptors: Interaction with deoxyribonucleic acid and transcription factors. Endocr. Rev. 1993, 14, 459–479. [Google Scholar] [PubMed]
- Levin, E.R. Plasma membrane estrogen receptors. Trends Endocrinol. Metab. 2009, 20, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Seval, Y.; Cakmak, H.; Kayisli, U.A.; Arici, A. Estrogen-mediated regulation of p38 mitogen-activated protein kinase in human endometrium. J. Clin. Endocrinol. Metab. 2006, 91, 2349–2357. [Google Scholar] [CrossRef] [PubMed]
- Tabibzadeh, S.; Kong, Q.F.; Satyaswaroop, P.G.; Babaknia, A. Heat shock proteins in human endometrium throughout the menstrual cycle. Hum. Reprod. 1996, 11, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Koshiyama, M.; Konishi, I.; Nanbu, K.; Nanbu, Y.; Mandai, M.; Komatsu, T.; Yamamoto, S.; Mori, T.; Fujii, S. Immunohistochemical localization of heat shock proteins HSP70 and HSP90 in the human endometrium: Correlation with sex steroid receptors and Ki-67 antigen expression. J. Clin. Endocrinol. Metab. 1995, 80, 1106–1112. [Google Scholar] [CrossRef] [PubMed]
- Reese, J.; Das, S.K.; Paria, B.C.; Lim, H.; Song, H.; Matsumoto, H.; Knudtson, K.L.; DuBois, R.N.; Dey, S.K. Global gene expression analysis to identify molecular markers of uterine receptivity and embryo implantation. J. Biol. Chem. 2001, 276, 44137–44145. [Google Scholar] [CrossRef] [PubMed]
- Guzel, E.; Basar, M.; Ocak, N.; Arici, A.; Kayisli, U.A. Bidirectional interaction between unfolded-protein-response key protein HSPA5 and estrogen signaling in human endometrium. Biol. Reprod. 2011, 85, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Jacquier-Sarlin, M.R.; Fuller, K.; Dinh-Xuan, A.T.; Richard, M.J.; Polla, B.S. Protective effects of HSP70 in inflammation. Experientia 1994, 50, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, H.N.; Kelly, R.W.; Fraser, H.M.; Critchley, H.O. Endocrine regulation of menstruation. Endocr. Rev. 2006, 27, 17–46. [Google Scholar] [CrossRef] [PubMed]
- Kayisli, U.A.; Guzeloglu-Kayisli, O.; Arici, A. Endocrine-immune interactions in human endometrium. Ann. N. Y. Acad. Sci. 2004, 1034, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Bulmer, J.N.; Morrison, L.; Longfellow, M.; Ritson, A.; Pace, D. Granulated lymphocytes in human endometrium: Histochemical and immunohistochemical studies. Hum. Reprod. 1991, 6, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Kamat, B.R.; Isaacson, P.G. The immunocytochemical distribution of leukocytic subpopulations in human endometrium. Am. J. Pathol. 1987, 127, 66–73. [Google Scholar] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Tan, J.; Raja, S.; Halder, J.; Paria, B.C.; Dey, S.K. Estrogen targets genes involved in protein processing, calcium homeostasis, and wnt signaling in the mouse uterus independent of estrogen receptor-α and -β. J. Biol. Chem. 2000, 275, 28834–28842. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Hou, X.; Zhou, H.E.; Wang, H.; Das, S.K. BiP is a molecular link between the phase I and phase II estrogenic responses in uterus. Mol. Endocrinol. 2006, 20, 1825–1837. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, K.; Anchoori, R.; Iizuka, Y.; Meints, J.; MacNeill, L.; Vogel, R.I.; Orlowski, R.Z.; Lee, M.K.; Roden, R.B.; Bazzaro, M. Small-molecule RA-9 inhibits proteasome-associated DUBs and ovarian cancer in vitro and in vivo via exacerbating unfolded protein responses. Clin. Cancer Res. 2014, 20, 3174–3186. [Google Scholar] [CrossRef] [PubMed]
- Littlefield, B.A.; Gurpide, E.; Markiewicz, L.; McKinley, B.; Hochberg, R.B. A simple and sensitive microtiter plate estrogen bioassay based on stimulation of alkaline phosphatase in Ishikawa cells: Estrogenic action of Δ5 adrenal steroids. Endocrinology 1990, 127, 2757–2762. [Google Scholar] [CrossRef] [PubMed]
- Ocak, N.S.; Guzel, E.; Bozkurt, I.; Bagriyanik, A.; Arici, A.; Kayisli, U.A. Endoplasmic reticulum (ER) homeostasis is cycle-dependent and the inflammatory cytokines TNF-α and ILI-1β induce ER stress by regulating BiP expression in human endometrial endothelial cells. Fertil. Steril. 2011, 96, S27. [Google Scholar] [CrossRef]
- Cakmak, H.; Guzeloglu-Kayisli, O.; Kayisli, U.A.; Arici, A. Immune-endocrine interactions in endometriosis. Front. Biosci. 2009, 1, 429–443. [Google Scholar]
- Giudice, L.C. Clinical practice. Endometriosis. N. Engl. J. Med. 2010, 362, 2389–2398. [Google Scholar] [CrossRef] [PubMed]
- Giudice, L.C.; Kao, L.C. Endometriosis. Lancet 2004, 364, 1789–1799. [Google Scholar] [CrossRef]
- Sampson, J.A. Metastatic or embolic endometriosis, due to the menstrual dissemination of endometrial tissue into the venous circulation. Am. J. Pathol. 1927, 3, 93–110. [Google Scholar] [PubMed]
- Mott, J.D.; Werb, Z. Regulation of matrix biology by matrix metalloproteinases. Curr. Opin. Cell Biol. 2004, 16, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhao, F.; Lin, F.; Chen, J.; Huang, Y. Lipoxin A4 inhibits the development of endometriosis in mice: The role of anti-inflammation and anti-angiogenesis. Am. J. Reprod. Immunol. 2012, 67, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, S.; Darcha, C. Epithelial to mesenchymal transition-like and mesenchymal to epithelial transition-like processes might be involved in the pathogenesis of pelvic endometriosis. Hum. Reprod. 2012, 27, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Van Langendonckt, A.; Casanas-Roux, F.; Donnez, J. Oxidative stress and peritoneal endometriosis. Fertil. Steril. 2002, 77, 861–870. [Google Scholar] [CrossRef]
- Guzel, E.; Ocak, N.S.; Basar, M.; Bozkurt, I.; Arici, A.; Kayisli, U.A. Bidirectional regulation of unfolding protein response in human endometrium and endometriosis. Fertil. Steril. 2010, 94, S216. [Google Scholar] [CrossRef]
- Taylor, R.N.; Yu, J.; Torres, P.B.; Schickedanz, A.C.; Park, J.K.; Mueller, M.D.; Sidell, N. Mechanistic and therapeutic implications of angiogenesis in endometriosis. Reprod. Sci. 2009, 16, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Cali, G.; Insabato, L.; Conza, D.; Bifulco, G.; Parrillo, L.; Mirra, P.; Fiory, F.; Miele, C.; Raciti, G.A.; di Jeso, B.; et al. GRP78 mediates cell growth and invasiveness in endometrial cancer. J. Cell. Physiol. 2014, 229, 1417–1426. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.H.; Bae, J.S.; Kim, K.M.; Park, H.S.; Cho, D.H.; Jang, K.Y.; Moon, W.S.; Kang, M.J.; Lee, D.G.; Chung, M.J. Expression of CHOP in squamous tumor of the uterine cervix. Korean J. Pathol. 2012, 46, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Lopez, I.; Tournillon, A.S.; Nylander, K.; Fahraeus, R. p53-mediated control of gene expression via mRNA translation during endoplasmic reticulum stress. Cell Cycle 2015, 14, 3373–3378. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.Y.; Ryan, K.M. Autophagy and cancer—Issues we need to digest. J. Cell Sci. 2012, 125, 2349–2358. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Sinha, S.; Kroemer, G. Bcl-2 family members: Dual regulators of apoptosis and autophagy. Autophagy 2008, 4, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Liu, J.H.; Zhang, J.; Zhang, N.; Wang, Z.H. Blockade of autophagy aggravates endoplasmic reticulum stress and improves Paclitaxel cytotoxicity in human cervical cancer cells. Cancer Res. Treat. 2015, 47, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Wen, N.; Xia, M.; Zhang, Y.U.; Liu, W.; Xu, Y.E.; Sun, L. Calcium efflux from the endoplasmic reticulum regulates cisplatin-induced apoptosis in human cervical cancer Hela cells. Oncol. Lett. 2016, 11, 2411–2419. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Yu, H.; Qin, H.; Kang, J.; Yu, C.; Zhong, J.; Su, J.; Li, H.; Sun, L. Inhibition of autophagy enhances cisplatin cytotoxicity through endoplasmic reticulum stress in human cervical cancer cells. Cancer Lett. 2012, 314, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Kim, H.S.; Jung, E.J.; Lee, J.Y.; B, K.T.; Lim, J.M.; Song, Y.S. Curcumin induces ER stress-mediated apoptosis through selective generation of reactive oxygen species in cervical cancer cells. Mol. Carcinog. 2016, 55, 918–928. [Google Scholar] [CrossRef] [PubMed]
- Gwak, H.; Kim, S.; Dhanasekaran, D.N.; Song, Y.S. Resveratrol triggers ER stress-mediated apoptosis by disrupting N-linked glycosylation of proteins in ovarian cancer cells. Cancer Lett. 2016, 371, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Yanagimachi, R. Fertility of mammalian spermatozoa: Its development and relativity. Zygote 1994, 2, 371–372. [Google Scholar] [CrossRef] [PubMed]
- Austin, C.R. The capacitation of the mammalian sperm. Nature 1952, 170, 326. [Google Scholar] [CrossRef] [PubMed]
- Naz, R.K.; Rajesh, P.B. Role of tyrosine phosphorylation in sperm capacitation/acrosome reaction. Reprod. Biol. Endocrinol. 2004, 2, 75. [Google Scholar] [CrossRef] [PubMed]
- Visconti, P.E.; Stewart-Savage, J.; Blasco, A.; Battaglia, L.; Miranda, P.; Kopf, G.S.; Tezon, J.G. Roles of bicarbonate, camp, and protein tyrosine phosphorylation on capacitation and the spontaneous acrosome reaction of hamster sperm. Biol. Reprod. 1999, 61, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Naz, R.K.; Ahmad, K.; Kumar, R. Role of membrane phosphotyrosine proteins in human spermatozoal function. J. Cell Sci. 1991, 99, 157–165. [Google Scholar] [PubMed]
- Leyton, L.; Saling, P. 95 kD sperm proteins bind ZP3 and serve as tyrosine kinase substrates in response to zona binding. Cell 1989, 57, 1123–1130. [Google Scholar] [CrossRef]
- Lachance, C.; Bailey, J.L.; Leclerc, P. Expression of HSP60 and GRP78 in the human endometrium and oviduct, and their effect on sperm functions. Hum. Reprod. 2007, 22, 2606–2614. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Chen, H.Q.; Cui, Z.H.; Yin, L.; Zhang, W.L.; Liu, W.B.; Han, F.; Ao, L.; Cao, J.; Liu, J.Y. Low-dose and combined effects of oral exposure to bisphenol A and diethylstilbestrol on the male reproductive system in adult Sprague-Dawley rats. Environ. Toxicol. Pharmacol. 2016, 43, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.L.; Wang, H.; Zhao, X.F.; Wang, Q.; Zhang, C.; Zhang, Y.; Zhao, M.; Chen, Y.H.; Meng, X.H.; Xu, D.X. Crosstalk between endoplasmic reticulum stress and mitochondrial pathway mediates cadmium-induced germ cell apoptosis in testes. Toxicol. Sci. 2011, 124, 446–459. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Park, S.J.; Kim, T.S.; Park, H.J.; Park, J.; Kim, B.K.; Kim, G.R.; Kim, J.M.; Huang, S.M.; Chae, J.I.; et al. Testicular hyperthermia induces unfolded protein response signaling activation in spermatocyte. Biochem. Biophys. Res. Commun. 2013, 434, 861–866. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.Y.; Avila, F.W.; Clark, A.G.; Wolfner, M.F. Induction of excessive endoplasmic reticulum stress in the drosophila male accessory gland results in infertility. PLoS ONE 2015, 10, e0119386. [Google Scholar] [CrossRef] [PubMed]
- Sebkova, N.; Ded, L.; Vesela, K.; Dvorakova-Hortova, K. Progress of sperm izumo1 relocation during spontaneous acrosome reaction. Reproduction 2014, 147, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Fujiwara, E.; Kakiuchi, Y.; Okabe, M.; Satouh, Y.; Baba, S.A.; Chiba, K.; Hirohashi, N. Most fertilizing mouse spermatozoa begin their acrosome reaction before contact with the zona pellucida during in vitro fertilization. Proc. Natl. Acad. Sci. USA 2011, 108, 4892–4896. [Google Scholar] [CrossRef] [PubMed]
- Marin-Briggiler, C.I.; Gonzalez-Echeverria, M.F.; Munuce, M.J.; Ghersevich, S.; Caille, A.M.; Hellman, U.; Corrigall, V.M.; Vazquez-Levin, M.H. Glucose-regulated protein 78 (GRP78/BiP) is secreted by human oviduct epithelial cells and the recombinant protein modulates sperm-zona pellucida binding. Fertil. Steril. 2010, 93, 1574–1584. [Google Scholar] [CrossRef] [PubMed]
- Galgani, M.; Insabato, L.; Cali, G.; Della Gatta, A.N.; Mirra, P.; Papaccio, F.; Santopaolo, M.; Alviggi, C.; Mollo, A.; Strina, I.; et al. Regulatory T cells, inflammation, and endoplasmic reticulum stress in women with defective endometrial receptivity. Fertil. Steril. 2015, 103, 1579–1586. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.T.; Bonneau, A.R.; Giraldez, A.J. Zygotic genome activation during the maternal-to-zygotic transition. Annu. Rev. Cell Dev. Biol. 2014, 30, 581–613. [Google Scholar] [CrossRef] [PubMed]
- Barckmann, B.; Simonelig, M. Control of maternal mRNA stability in germ cells and early embryos. Biochim. Biophys. Acta 2013, 1829, 714–724. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zheng, P.; Dean, J. Maternal control of early mouse development. Development 2010, 137, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Gosden, R.G. Oogenesis as a foundation for embryogenesis. Mol. Cell. Endocrinol. 2002, 186, 149–153. [Google Scholar] [CrossRef]
- Tatsuta, T.; Hosono, M.; Miura, Y.; Sugawara, S.; Kariya, Y.; Hakomori, S.; Nitta, K. Involvement of ER stress in apoptosis induced by sialic acid-binding lectin (leczyme) from bullfrog eggs. Int. J. Oncol. 2013, 43, 1799–1808. [Google Scholar] [PubMed]
- Wu, L.L.; Russell, D.L.; Norman, R.J.; Robker, R.L. Endoplasmic reticulum (ER) stress in cumulus-oocyte complexes impairs pentraxin-3 secretion, mitochondrial membrane potential (ΔΨm), and embryo development. Mol. Endocrinol. 2012, 26, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.L.; Russell, D.L.; Wong, S.L.; Chen, M.; Tsai, T.S.; St John, J.C.; Norman, R.J.; Febbraio, M.A.; Carroll, J.; Robker, R.L. Mitochondrial dysfunction in oocytes of obese mothers: Transmission to offspring and reversal by pharmacological endoplasmic reticulum stress inhibitors. Development 2015, 142, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Harada, M.; Nose, E.; Takahashi, N.; Hirota, Y.; Hirata, T.; Yoshino, O.; Koga, K.; Fujii, T.; Osuga, Y. Evidence of the activation of unfolded protein response in granulosa and cumulus cells during follicular growth and maturation. Gynecol. Endocrinol. 2015, 31, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Schultz, R.M. RNA transcript profiling during zygotic gene activation in the preimplantation mouse embryo. Dev. Biol. 2005, 283, 40–57. [Google Scholar] [CrossRef] [PubMed]
- Abraham, T.; Pin, C.L.; Watson, A.J. Embryo collection induces transient activation of XBP1 arm of the ER stress response while embryo vitrification does not. Mol. Hum. Reprod. 2012, 18, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Lane, M.; Gardner, D.K. Understanding cellular disruptions during early embryo development that perturb viability and fetal development. Reprod. Fertil. Dev. 2005, 17, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Diao, Y.F.; Kim, H.R.; Jin, D.I. Inhibition of endoplasmic reticulum stress improves mouse embryo development. PLoS ONE 2012, 7, e40433. [Google Scholar] [CrossRef] [PubMed]
- Basar, M.; Bozkurt, I.; Guzeloglu-Kayisli, O.; Sozen, B.; Tekmen, I.; Schatz, F.; Arici, A.; Lockwood, C.J.; Kayisli, U.A. Unfolded protein response prevents blastocyst formation during preimplantation embryo development in vitro. Fertil. Steril. 2014, 102, 1777–1784. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Song, B.S.; Lee, K.S.; Kim, D.H.; Kim, S.U.; Choo, Y.K.; Chang, K.T.; Koo, D.B. Tauroursodeoxycholic acid enhances the pre-implantation embryo development by reducing apoptosis in pigs. Reprod. Domest. Anim. 2012, 47, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Diao, Y.F.; Kang, J.W.; Lee, J.E.; Kim, D.K.; Jin, D.I. Tauroursodeoxycholic acid improves the implantation and live-birth rates of mouse embryos. Reprod. Biol. 2015, 15, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Agrawal, H.; Mullani, N.; Sandhu, A.; Singh, M.K.; Chauhan, M.S.; Singla, S.K.; Palta, P.; Manik, R.S. Supplementation of tauroursodeoxycholic acid during IVC did not enhance in vitro development and quality of buffalo IVF embryos but combated endoplasmic reticulum stress. Theriogenology 2015, 84, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Kaufman, R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.; Almanza, G.; Burns, J.C.; Wheeler, M.; Zanetti, M. Endoplasmic reticulum stress drives a regulatory phenotype in human T-cell clones. Cell. Immunol. 2010, 266, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Brosens, J.J.; Salker, M.S.; Teklenburg, G.; Nautiyal, J.; Salter, S.; Lucas, E.S.; Steel, J.H.; Christian, M.; Chan, Y.W.; Boomsma, C.M.; et al. Uterine selection of human embryos at implantation. Sci. Rep. 2014, 4, 3894. [Google Scholar] [CrossRef] [PubMed]
- Groom, K.M.; McCowan, L.M.; Mackay, L.K.; Lee, A.C.; Said, J.M.; Kane, S.C.; Walker, S.P.; van Mens, T.E.; Hannan, N.J.; Tong, S.; et al. Enoxaparin for the prevention of preeclampsia and intrauterine growth restriction in women with a history: A randomized trial. Am. J. Obstet. Gynecol. 2017, 216, 296. [Google Scholar] [CrossRef] [PubMed]
- Pardi, G.; Marconi, A.M.; Cetin, I. Placental-fetal interrelationship in IUGR fetuses—A review. Placenta 2002, 23, S136–S141. [Google Scholar] [CrossRef] [PubMed]
- Cetin, I.; Antonazzo, P. The role of the placenta in intrauterine growth restriction (IUGR). Z. Geburtshilfe Neonatol. 2009, 213, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Lockwood, C.J.; Basar, M.; Kayisli, U.A.; Guzeloglu-Kayisli, O.; Murk, W.; Wang, J.; de Paz, N.; Shapiro, J.P.; Masch, R.J.; Semerci, N.; et al. Interferon-γ protects first-trimester decidual cells against aberrant matrix metalloproteinases 1, 3, and 9 expression in preeclampsia. Am. J. Pathol. 2014, 184, 2549–2559. [Google Scholar] [CrossRef] [PubMed]
- Lian, I.A.; Loset, M.; Mundal, S.B.; Fenstad, M.H.; Johnson, M.P.; Eide, I.P.; Bjorge, L.; Freed, K.A.; Moses, E.K.; Austgulen, R. Increased endoplasmic reticulum stress in decidual tissue from pregnancies complicated by fetal growth restriction with and without pre-eclampsia. Placenta 2011, 32, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Hubel, C.A. Oxidative stress in the pathogenesis of preeclampsia. Proc. Soc. Exp. Biol. Med. 1999, 222, 222–235. [Google Scholar] [CrossRef] [PubMed]
- Yung, H.W.; Alnaes-Katjavivi, P.; Jones, C.J.; El-Bacha, T.; Golic, M.; Staff, A.C.; Burton, G.J. Placental endoplasmic reticulum stress in gestational diabetes: The potential for therapeutic intervention with chemical chaperones and antioxidants. Diabetologia 2016, 59, 2240–2250. [Google Scholar] [CrossRef] [PubMed]
- Sacks, D.A.; Hadden, D.R.; Maresh, M.; Deerochanawong, C.; Dyer, A.R.; Metzger, B.E.; Lowe, L.P.; Coustan, D.R.; Hod, M.; Oats, J.J.; et al. Frequency of gestational diabetes mellitus at collaborating centers based on IADPSG consensus panel-recommended criteria: The hyperglycemia and adverse pregnancy outcome (hapo) study. Diabetes Care 2012, 35, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Bryson, C.L.; Ioannou, G.N.; Rulyak, S.J.; Critchlow, C. Association between gestational diabetes and pregnancy-induced hypertension. Am. J. Epidemiol. 2003, 158, 1148–1153. [Google Scholar] [CrossRef] [PubMed]
- Lockwood, C.J. Predicting premature delivery—No easy task. N. Engl. J. Med. 2002, 346, 282–284. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Mazor, M.; Wu, Y.K.; Sirtori, M.; Oyarzun, E.; Mitchell, M.D.; Hobbins, J.C. Infection in the pathogenesis of preterm labor. Semin. Perinatol. 1988, 12, 262–279. [Google Scholar] [PubMed]
- Shim, S.S.; Romero, R.; Hong, J.S.; Park, C.W.; Jun, J.K.; Kim, B.I.; Yoon, B.H. Clinical significance of intra-amniotic inflammation in patients with preterm premature rupture of membranes. Am. J. Obstet. Gynecol. 2004, 191, 1339–1345. [Google Scholar] [CrossRef] [PubMed]
- Witt, A.; Berger, A.; Gruber, C.J.; Petricevic, L.; Apfalter, P.; Husslein, P. IL-8 concentrations in maternal serum, amniotic fluid and cord blood in relation to different pathogens within the amniotic cavity. J. Perinat Med. 2005, 33, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Saji, F.; Samejima, Y.; Kamiura, S.; Sawai, K.; Shimoya, K.; Kimura, T. Cytokine production in chorioamnionitis. J. Reprod. Immunol. 2000, 47, 185–196. [Google Scholar] [CrossRef]
- Atkins, P.C.; Schwartz, L.B.; Adkinson, N.F.; von Allmen, C.; Valenzano, M.; Zweiman, B. In vivo antigen-induced cutaneous mediator release: Simultaneous comparisons of histamine, tryptase, and prostaglandin D2 release and the effect of oral corticosteroid administration. J. Allergy Clin. Immunol. 1990, 86, 360–370. [Google Scholar] [CrossRef]
- Zaga, V.; Estrada-Gutierrez, G.; Beltran-Montoya, J.; Maida-Claros, R.; Lopez-Vancell, R.; Vadillo-Ortega, F. Secretions of interleukin-1β and tumor necrosis factor α by whole fetal membranes depend on initial interactions of amnion or choriodecidua with lipopolysaccharides or group B streptococci. Biol. Reprod. 2004, 71, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Hardy, D.B.; Janowski, B.A.; Corey, D.R.; Mendelson, C.R. Progesterone receptor plays a major antiinflammatory role in human myometrial cells by antagonism of nuclear factor-κB activation of cyclooxygenase 2 expression. Mol. Endocrinol. 2006, 20, 2724–2733. [Google Scholar] [CrossRef] [PubMed]
- Lockwood, C.J.; Murk, W.K.; Kayisli, U.A.; Buchwalder, L.F.; Huang, S.J.; Arcuri, F.; Li, M.; Gopinath, A.; Schatz, F. Regulation of interleukin-6 expression in human decidual cells and its potential role in chorioamnionitis. Am. J. Pathol. 2010, 177, 1755–1764. [Google Scholar] [CrossRef] [PubMed]
- Oner, C.; Schatz, F.; Kizilay, G.; Murk, W.; Buchwalder, L.F.; Kayisli, U.A.; Arici, A.; Lockwood, C.J. Progestin-inflammatory cytokine interactions affect matrix metalloproteinase-1 and -3 expression in term decidual cells: Implications for treatment of chorioamnionitis-induced preterm delivery. J. Clin. Endocrinol. Metab. 2008, 93, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Yung, H.W.; Hemberger, M.; Watson, E.D.; Senner, C.E.; Jones, C.P.; Kaufman, R.J.; Charnock-Jones, D.S.; Burton, G.J. Endoplasmic reticulum stress disrupts placental morphogenesis: Implications for human intrauterine growth restriction. J. Pathol. 2012, 228, 554–564. [Google Scholar] [CrossRef] [PubMed]
- Veerbeek, J.H.; Tissot van Patot, M.C.; Burton, G.J.; Yung, H.W. Endoplasmic reticulum stress is induced in the human placenta during labour. Placenta 2015, 36, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Liong, S.; Lappas, M. Endoplasmic reticulum stress is increased after spontaneous labor in human fetal membranes and myometrium where it regulates the expression of prolabor mediators. Biol. Reprod. 2014, 91, 70. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wong, H.N.; Song, B.; Miller, C.N.; Scheuner, D.; Kaufman, R.J. The unfolded protein response sensor IRE1α is required at 2 distinct steps in B cell lymphopoiesis. J. Clin. Investig. 2005, 115, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Iwawaki, T.; Akai, R.; Yamanaka, S.; Kohno, K. Function of IRE1α in the placenta is essential for placental development and embryonic viability. Proc. Natl. Acad. Sci. USA 2009, 106, 16657–16662. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.X.; He, W.H.; Yin, L.J.; Lv, P.P.; Zhang, Y.; Sheng, J.Z.; Leung, P.C.; Huang, H.F. Sustained endoplasmic reticulum stress as a cofactor of oxidative stress in decidual cells from patients with early pregnancy loss. J. Clin. Endocrinol. Metab. 2011, 96, E493–E497. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.J.; Zhu, Y.M.; He, W.H.; Liu, A.X.; Dong, M.Y.; Jin, M.; Sheng, J.Z.; Huang, H.F. Endoplasmic reticulum stress induced by oxidative stress in decidual cells: A possible mechanism of early pregnancy loss. Mol. Biol. Rep. 2012, 39, 9179–9186. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.K.; Nicholson, C.J.; Holloway, A.C.; Hardy, D.B. Maternal nicotine exposure leads to impaired disulfide bond formation and augmented endoplasmic reticulum stress in the rat placenta. PLoS ONE 2015, 10, e0122295. [Google Scholar] [CrossRef] [PubMed]
- Yung, H.W.; Calabrese, S.; Hynx, D.; Hemmings, B.A.; Cetin, I.; Charnock-Jones, D.S.; Burton, G.J. Evidence of placental translation inhibition and endoplasmic reticulum stress in the etiology of human intrauterine growth restriction. Am. J. Pathol. 2008, 173, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; He, F.; Kuang, L.; Tang, W.; Li, Y.; Chen, D. eNOS/iNOS and endoplasmic reticulum stress-induced apoptosis in the placentas of patients with preeclampsia. J. Hum. Hypertens. 2017, 31, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Mizuuchi, M.; Cindrova-Davies, T.; Olovsson, M.; Charnock-Jones, D.S.; Burton, G.J.; Yung, H.W. Placental endoplasmic reticulum stress negatively regulates transcription of placental growth factor via ATF4 and ATF6β: Implications for the pathophysiology of human pregnancy complications. J. Pathol. 2016, 238, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Yung, H.W.; Atkinson, D.; Campion-Smith, T.; Olovsson, M.; Charnock-Jones, D.S.; Burton, G.J. Differential activation of placental unfolded protein response pathways implies heterogeneity in causation of early- and late-onset pre-eclampsia. J. Pathol. 2014, 234, 262–276. [Google Scholar] [CrossRef] [PubMed]
- Sobrevia, L.; Salsoso, R.; Fuenzalida, B.; Barros, E.; Toledo, L.; Silva, L.; Pizarro, C.; Subiabre, M.; Villalobos, R.; Araos, J.; et al. Insulin is a key modulator of fetoplacental endothelium metabolic disturbances in gestational diabetes mellitus. Front. Physiol. 2016, 7, 119. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ER Stress Molecules | Alteration/Sources | Action/Significance/Association | Pregnancy Stage/Groups |
|---|---|---|---|
| HSC70 | Increased secretion from blastocyst | Paracrine action for proper folding of newly translated and misfolded proteins in decidual cells | Implantation window [133] |
| GRP78 | Increased in endometrial stromal cells | Recurrent miscarriage | Implantation window [114] |
| IRE1α | Knock-out mouse Smaller placenta and embryo sizes | A reduced VEGF-A levels in the placenta as well as severe dysfunction of the labyrinth placenta | Placentation in mouse [157] |
| GRP78 and VCP | Down-regulation in decidual cells | Acts with oxidative stress as cofactor for molecular induction of early pregnancy loss | Specimens from Early pregnancy loss [159] |
| GRP78, P-eIF2α and XBP-1 | Increased levels in syncytiotrophoblasts | Increased ER stress during normal labor | Labor vs. Non-labor placentas [154] |
| GRP78, IRE1 and XBP-1s | -In fetal membranes and myometrium -LPS mediated increase in explant cultures of fetal membranes and myometrium | - Increased ER stress in preterm and term labor - Infection may induce ER stress | Term and spontaneous pre-term labor vs. non-labor placenta specimens. Fetal membranes and myometrium from non-laboring women at the time of term Cesarean section [155] |
| GRP78, P-eIF2α, ATF4, and CHOP | Increased in placenta | Elevated ER stress and deregulation of proper protein folding during pregnancy | During pregnancy in rat [160] |
| P-eIF2α | Increased in placenta | Increased ER stress that reduce placental protein synthesis | FGR (GA weeks 28–38) vs. term control (GA weeks 39–40) [161] |
| GRP78 and 94, P-PERK, eIF2a, P-eIF2a, XBP1, CHOP, IRE1, P-IRE1 | Elevated levels in placentas | Exaggerated ER stress in preeclampsia | Preeclamptic (mean GA weeks 33.6) vs. control placentas (mean GA weeks 39.2) [162] |
| UPR transcription factors ATF4, ATF6α and ATF6β | Increased nuclear localization in the syncytiotrophoblasts | Increased ER stress and contributes to reduced PlGF protein levels | Preeclamptic placentas (GA < 34 weeks ) vs. term control [163] |
| PERK-induced p-eIF2α, ATF6 and XBP1u | Increased levels in extra-villous trophoblasts, decidual cells and macrophages | Increased ER stress may impair placental growth associated with FGR and FGR + pre-eclampsia | Decidual tissues from FGR (mean GA weeks 31.9) or FGR with pre-eclampsia (mean GA weeks 30.3) vs. term control (mean GA weeks 38.7) [138] |
| P-IRE1α, ATF6, XBP-1, GRP78 and GRP94 | Increased in placental lysates | Impaired ER stress may cause placental dysfunction that triggers preeclampsia | Early-onset (<34 weeks) pre-eclampsia vs. late-onset pre-eclampsia and normotensive controls [164] |
| P-eIF2α, eIF2α, XBP-1 and GRP78 | Increased in placental lysates | Association between increased ER stress and preterm labor | Spontaneous pre-term placentas (due to acute chorioamnionitis and other conditions) vs. term controls [164] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guzel, E.; Arlier, S.; Guzeloglu-Kayisli, O.; Tabak, M.S.; Ekiz, T.; Semerci, N.; Larsen, K.; Schatz, F.; Lockwood, C.J.; Kayisli, U.A. Endoplasmic Reticulum Stress and Homeostasis in Reproductive Physiology and Pathology. Int. J. Mol. Sci. 2017, 18, 792. https://doi.org/10.3390/ijms18040792
Guzel E, Arlier S, Guzeloglu-Kayisli O, Tabak MS, Ekiz T, Semerci N, Larsen K, Schatz F, Lockwood CJ, Kayisli UA. Endoplasmic Reticulum Stress and Homeostasis in Reproductive Physiology and Pathology. International Journal of Molecular Sciences. 2017; 18(4):792. https://doi.org/10.3390/ijms18040792
Chicago/Turabian StyleGuzel, Elif, Sefa Arlier, Ozlem Guzeloglu-Kayisli, Mehmet Selcuk Tabak, Tugba Ekiz, Nihan Semerci, Kellie Larsen, Frederick Schatz, Charles Joseph Lockwood, and Umit Ali Kayisli. 2017. "Endoplasmic Reticulum Stress and Homeostasis in Reproductive Physiology and Pathology" International Journal of Molecular Sciences 18, no. 4: 792. https://doi.org/10.3390/ijms18040792
APA StyleGuzel, E., Arlier, S., Guzeloglu-Kayisli, O., Tabak, M. S., Ekiz, T., Semerci, N., Larsen, K., Schatz, F., Lockwood, C. J., & Kayisli, U. A. (2017). Endoplasmic Reticulum Stress and Homeostasis in Reproductive Physiology and Pathology. International Journal of Molecular Sciences, 18(4), 792. https://doi.org/10.3390/ijms18040792