
Exploiting Epigenetic Alterations in Prostate Cancer


Abstract
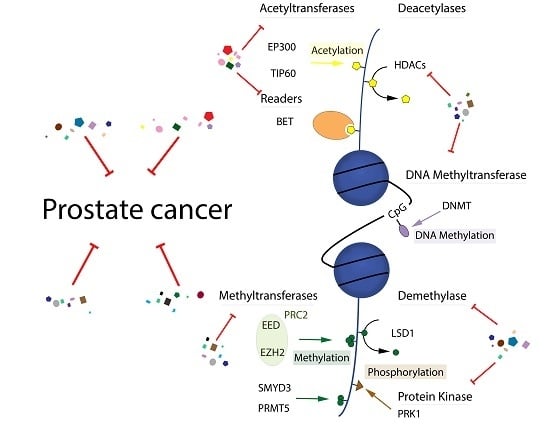
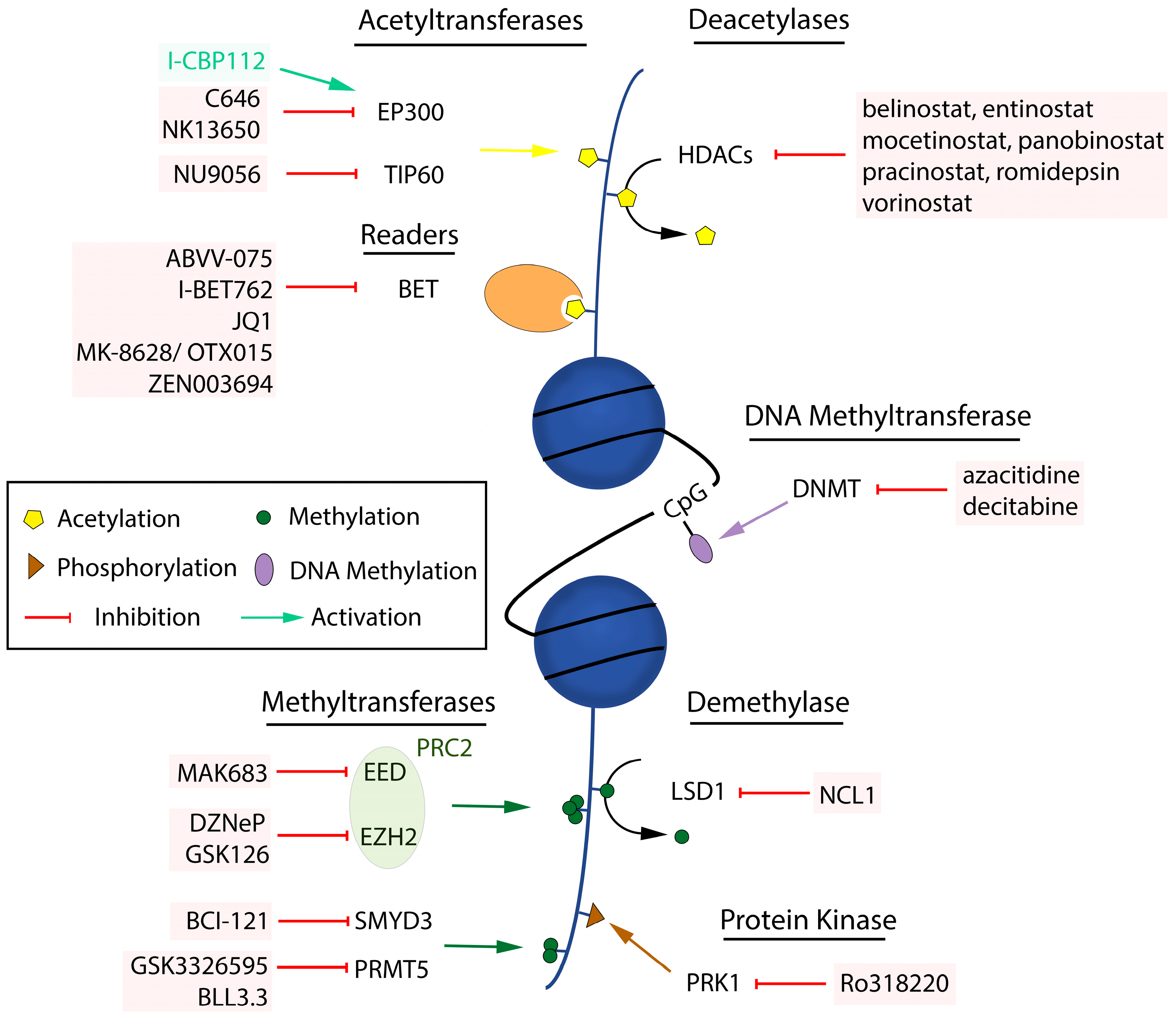
:
1. Introduction
2. Epigenetic Events in Prostate Cancer and Preclinical Efficacy of Inhibitors of Epigenetic Targets
2.1. DNA Methylation
2.2. Histone Acetylation
2.3. Histone Methylation
2.4. Histone Ubiquitylation
2.5. Histone Phosphorylation
3. Clinical Studies in Prostate Cancer Addressing Epigenetic Targets
4. MicroRNAs as Potential Biomarkers for Prostate Cancer
5. Suppressor MicroRNAs as Potential Treatment for Prostate Cancer
6. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dy, G.W.; Gore, J.L.; Forouzanfar, M.H.; Naghavi, M.; Fitzmaurice, C. Global burden of urologic cancers, 1990–2013. Eur. Urol. 2017, 71, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Smith, A.D.; Ferraldeschi, R.; Al-Lazikani, B.; Workman, P.; de Bono, J.S. Drug discovery in advanced prostate cancer: Translating biology into therapy. Nat. Rev. Drug Discov. 2016, 15, 699–718. [Google Scholar] [CrossRef] [PubMed]
- Attard, G.; Parker, C.; Eeles, R.A.; Schroder, F.; Tomlins, S.A.; Tannock, I.; Drake, C.G.; de Bono, J.S. Prostate cancer. Lancet 2016, 387, 70–82. [Google Scholar] [CrossRef]
- Ciccarese, C.; Massari, F.; Iacovelli, R.; Fiorentino, M.; Montironi, R.; di Nunno, V.; Giunchi, F.; Brunelli, M.; Tortora, G. Prostate cancer heterogeneity: Discovering novel molecular targets for therapy. Cancer Treat. Rev. 2017, 54, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, D.N.; Boysen, G.; Sumanasuriya, S.; Seed, G.; Marzo, A.M.; de Bono, J. The molecular underpinnings of prostate cancer: Impacts on management and pathology practice. J. Pathol. 2017, 241, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Squire, J.A.; Park, P.C.; Yoshimoto, M.; Alami, J.; Williams, J.L.; Evans, A.; Joshua, A.M. Prostate cancer as a model system for genetic diversity in tumors. Adv. Cancer Res. 2011, 112, 183–216. [Google Scholar] [PubMed]
- Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell 2015, 163, 1011–1025. [Google Scholar]
- Dellis, A.; Papatsoris, A.G. Phase I and II therapies targeting the androgen receptor for the treatment of castration resistant prostate cancer. Expert Opin. Investig. Drugs 2016, 25, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Bambury, R.M.; Rathkopf, D.E. Novel and next-generation androgen receptor-directed therapies for prostate cancer: Beyond abiraterone and enzalutamide. Urol. Oncol. 2016, 34, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, C.C.; Offit, K.; Nelson, P.S. DNA-repair gene mutations in metastatic prostate cancer. N. Engl. J. Med. 2016, 375, 1804–1805. [Google Scholar] [CrossRef] [PubMed]
- Ta, H.Q.; Gioeli, D. The convergence of DNA damage checkpoint pathways and androgen receptor signaling in prostate cancer. Endocri. Relat. Cancer 2014, 21, R395–407. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.S.; Li, J.; Stockert, J.A.; O’Connor, J.; Herzog, B.; Elaiho, C.; Galsky, M.D.; Tewari, A.K.; Yadav, K.K. Combination effect of therapies targeting the PI3K- and AR-signaling pathways in prostate cancer. Oncotarget 2016, 7, 76181–76196. [Google Scholar] [CrossRef] [PubMed]
- Gundem, G.; Van Loo, P.; Kremeyer, B.; Alexandrov, L.B.; Tubio, J.M.; Papaemmanuil, E.; Brewer, D.S.; Kallio, H.M.; Hognas, G.; Annala, M.; et al. The evolutionary history of lethal metastatic prostate cancer. Nature 2015, 520, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Spratt, D.E.; Zumsteg, Z.S.; Feng, F.Y.; Tomlins, S.A. Translational and clinical implications of the genetic landscape of prostate cancer. Nat. Rev. Clin. Oncol. 2016, 13, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, T.; Neal, D.E. The genomic evolution of human prostate cancer. Br. J. Cancer 2015, 113, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Ngollo, M.; Dagdemir, A.; Karsli-Ceppioglu, S.; Judes, G.; Pajon, A.; Penault-Llorca, F.; Boiteux, J.P.; Bignon, Y.J.; Guy, L.; Bernard-Gallon, D.J. Epigenetic modifications in prostate cancer. Epigenomics 2014, 6, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Cucchiara, V.; Yang, J.C.; Mirone, V.; Gao, A.C.; Rosenfeld, M.G.; Evans, C.P. Epigenomic regulation of androgen receptor signaling: Potential role in prostate cancer therapy. Cancers 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Kgatle, M.M.; Kalla, A.A.; Islam, M.M.; Sathekge, M.; Moorad, R. Prostate cancer: Epigenetic alterations, risk factors, and therapy. Prostate Cancer 2016, 2016, 5653862. [Google Scholar] [CrossRef] [PubMed]
- Gelato, K.A.; Shaikhibrahim, Z.; Ocker, M.; Haendler, B. Targeting epigenetic regulators for cancer therapy: Modulation of bromodomain proteins, methyltransferases, demethylases, and microRNAs. Expert Opin. Ther. Targets 2016, 20, 783–799. [Google Scholar] [CrossRef] [PubMed]
- Graca, I.; Pereira-Silva, E.; Henrique, R.; Packham, G.; Crabb, S.J.; Jeronimo, C. Epigenetic modulators as therapeutic targets in prostate cancer. Clin. Epigenet. 2016, 8, 98. [Google Scholar] [CrossRef] [PubMed]
- Zelic, R.; Fiano, V.; Grasso, C.; Zugna, D.; Pettersson, A.; Gillio-Tos, A.; Merletti, F.; Richiardi, L. Global DNA hypomethylation in prostate cancer development and progression: A systematic review. Prostate Cancer Prostatic Dis. 2015, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Massie, C.E.; Mills, I.G.; Lynch, A.G. The importance of DNA methylation in prostate cancer development. J. Steroid Biochem. Mol. Biol. 2017, 166, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Attar, N.; Kurdistani, S.K. Exploitation of EP300 and CREBBP lysine acetyltransferases by cancer. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Bianco-Miotto, T.; Chiam, K.; Buchanan, G.; Jindal, S.; Day, T.K.; Thomas, M.; Pickering, M.A.; O'Loughlin, M.A.; Ryan, N.K.; Raymond, W.A.; et al. Global levels of specific histone modifications and an epigenetic gene signature predict prostate cancer progression and development. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2611–2622. [Google Scholar] [CrossRef] [PubMed]
- Cang, S.; Feng, J.; Konno, S.; Han, L.; Liu, K.; Sharma, S.C.; Choudhury, M.; Chiao, J.W. Deficient histone acetylation and excessive deacetylase activity as epigenomic marks of prostate cancer cells. Int. J. Oncol. 2009, 35, 1417–1422. [Google Scholar] [PubMed]
- Lochrin, S.E.; Price, D.K.; Figg, W.D. BET bromodomain inhibitors—A novel epigenetic approach in castration-resistant prostate cancer. Cancer Biol. Ther. 2014, 15, 1583–1585. [Google Scholar] [CrossRef] [PubMed]
- Shukla, K.K.; Misra, S.; Pareek, P.; Mishra, V.; Singhal, B.; Sharma, P. Recent scenario of microRNA as diagnostic and prognostic biomarkers of prostate cancer. Urol. Oncol. 2017, 35, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Ayub, S.G.; Kaul, D.; Ayub, T. Microdissecting the role of microRNAs in the pathogenesis of prostate cancer. Cancer Genet. 2015, 208, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.L.; Ranieri, G.; Muzi, P.; Marampon, F.; Mancini, A.; Di Pasquale, B.; di Clemente, L.; Dolo, V.; D'Alessandro, A.M.; Festuccia, C. Increased levels of DNA methyltransferases are associated with the tumorigenic capacity of prostate cancer cells. Oncol. Rep. 2013, 29, 1189–1195. [Google Scholar] [PubMed]
- Gravina, G.L.; Marampon, F.; Piccolella, M.; Motta, M.; Ventura, L.; Pomante, R.; Popov, V.M.; Zani, B.M.; Pestell, R.G.; Tombolini, V.; Jannini, E.A.; Festuccia, C. Hormonal therapy promotes hormone-resistant phenotype by increasing DNMT activity and expression in prostate cancer models. Endocrinology 2011, 152, 4550–4561. [Google Scholar] [CrossRef] [PubMed]
- Paziewska, A.; Dabrowska, M.; Goryca, K.; Antoniewicz, A.; Dobruch, J.; Mikula, M.; Jarosz, D.; Zapala, L.; Borowka, A.; Ostrowski, J. DNA methylation status is more reliable than gene expression at detecting cancer in prostate biopsy. Br. J. Cancer 2014, 111, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Brocks, D.; Assenov, Y.; Minner, S.; Bogatyrova, O.; Simon, R.; Koop, C.; Oakes, C.; Zucknick, M.; Lipka, D.B.; Weischenfeldt, J.; et al. Intratumor DNA methylation heterogeneity reflects clonal evolution in aggressive prostate cancer. Cell Rep. 2014, 8, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Festuccia, C.; Gravina, G.L.; D’Alessandro, A.M.; Muzi, P.; Millimaggi, D.; Dolo, V.; Ricevuto, E.; Vicentini, C.; Bologna, M. Azacitidine improves antitumor effects of docetaxel and cisplatin in aggressive prostate cancer models. Endocr. Relat. Cancer 2009, 16, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.L.; Marampon, F.; Di Staso, M.; Bonfili, P.; Vitturini, A.; Jannini, E.A.; Pestell, R.G.; Tombolini, V.; Festuccia, C. 5-Azacitidine restores and amplifies the bicalutamide response on preclinical models of androgen receptor expressing or deficient prostate tumors. Prostate 2010, 70, 1166–1178. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gao, H.; Ren, L.; Gu, J.; Zhang, Y. Demethylation of the miR-146a promoter by 5-aza-2′-deoxycytidine correlates with delayed progression of castration-resistant prostate cancer. BMC Cancer 2014, 14, 308. [Google Scholar] [CrossRef] [PubMed]
- Naldi, I.; Taranta, M.; Gherardini, L.; Pelosi, G.; Viglione, F.; Grimaldi, S.; Pani, L.; Cinti, C. Novel epigenetic target therapy for prostate cancer: A preclinical study. PLoS ONE 2014, 9, e98101. [Google Scholar] [CrossRef] [PubMed]
- McCabe, M.T.; Low, J.A.; Daignault, S.; Imperiale, M.J.; Wojno, K.J.; Day, M.L. Inhibition of DNA methyltransferase activity prevents tumorigenesis in a mouse model of prostate cancer. Cancer Res. 2006, 66, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Rao, A. Connections between TET proteins and aberrant DNA modification in cancer. Trends Genet. 2014, 30, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Spans, L.; Van den Broeck, T.; Smeets, E.; Prekovic, S.; Thienpont, B.; Lambrechts, D.; Karnes, R.J.; Erho, N.; Alshalalfa, M.; Davicioni, E.; et al. Genomic and epigenomic analysis of high-risk prostate cancer reveals changes in hydroxymethylation and TET1. Oncotarget 2016, 7, 24326–24338. [Google Scholar] [CrossRef] [PubMed]
- Ellinger, J.; Kahl, P.; von der Gathen, J.; Rogenhofer, S.; Heukamp, L.C.; Gutgemann, I.; Walter, B.; Hofstadter, F.; Buttner, R.; Muller, S.C.; et al. Global levels of histone modifications predict prostate cancer recurrence. Prostate 2010, 70, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Berman, B.P.; Jariwala, U.; Yan, X.; Cogan, J.P.; Walters, A.; Chen, T.; Buchanan, G.; Frenkel, B.; Coetzee, G.A. Genomic androgen receptor-occupied regions with different functions, defined by histone acetylation, coregulators and transcriptional capacity. PLoS ONE 2008, 3, e3645. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Shen, H.C.; Wantroba, M.; Khalid, O.; Liang, G.; Wang, Q.; Gentzschein, E.; Pinski, J.K.; Stanczyk, F.Z.; Jones, P.A.; et al. Locus-wide chromatin remodeling and enhanced androgen receptor-mediated transcription in recurrent prostate tumor cells. Mol. Cell. Biol. 2006, 26, 7331–7341. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Shrinivas, K.; Young, R.A.; Chakraborty, A.K.; Sharp, P.A. A phase separation model for transcriptional control. Cell 2017, 169, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Mansour, M.R.; Abraham, B.J.; Anders, L.; Berezovskaya, A.; Gutierrez, A.; Durbin, A.D.; Etchin, J.; Lawton, L.; Sallan, S.E.; Silverman, L.B.; et al. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 2014, 346, 1373–1377. [Google Scholar] [CrossRef] [PubMed]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional addiction in cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [PubMed]
- Zuber, V.; Bettella, F.; Witoelar, A.; Consortium, P.; Cruk, G.; Consortium, B.; Consortium, T.; Andreassen, O.A.; Mills, I.G.; Urbanucci, A. Bromodomain protein 4 discriminates tissue-specific super-enhancers containing disease-specific susceptibility loci in prostate and breast cancer. BMC Genom. 2017, 18, 270. [Google Scholar] [CrossRef] [PubMed]
- Culig, Z.; Comuzzi, B.; Steiner, H.; Bartsch, G.; Hobisch, A. Expression and function of androgen receptor coactivators in prostate cancer. J. Steroid Biochem. Mol. Biol. 2004, 92, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Coffey, K.; Robson, C.N. Regulation of the androgen receptor by post-translational modifications. J. Endocrinol. 2012, 215, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Faus, H.; Haendler, B. Androgen receptor acetylation sites differentially regulate gene control. J. Cell Biochem. 2008, 104, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Lavery, D.N.; Bevan, C.L. Androgen receptor signalling in prostate cancer: The functional consequences of acetylation. J. Biomed. Biotechnol. 2011, 2011, 862125. [Google Scholar] [CrossRef] [PubMed]
- Ianculescu, I.; Wu, D.Y.; Siegmund, K.D.; Stallcup, M.R. Selective roles for cAMP response element-binding protein binding protein and p300 protein as coregulators for androgen-regulated gene expression in advanced prostate cancer cells. J. Biol. Chem. 2012, 287, 4000–4013. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Sunkel, B.; Chen, Z.; Liu, X.; Ye, Z.; Li, Q.; Grenade, C.; Ke, J.; Zhang, C.; Chen, H.; et al. Three-tiered role of the pioneer factor GATA2 in promoting androgen-dependent gene expression in prostate cancer. Nucleic Acids Res. 2014, 42, 3607–3622. [Google Scholar] [CrossRef] [PubMed]
- Culig, Z. Androgen receptor coactivators in regulation of growth and differentiation in prostate cancer. J. Cell. Physiol. 2016, 231, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Zucconi, B.E.; Luef, B.; Xu, W.; Henry, R.A.; Nodelman, I.M.; Bowman, G.D.; Andrews, A.J.; Cole, P.A. Modulation of p300/CBP acetylation of nucleosomes by bromodomain ligand I-CBP112. Biochemistry 2016, 55, 3727–3734. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, A.; Chaurasia, P.; Xiao, G.Q.; Philizaire, M.; Lv, X.; Yao, S.; Burnstein, K.L.; Liu, D.P.; Levine, A.C.; Mujtaba, S. Coactivator MYST1 regulates nuclear factor-κB and androgen receptor functions during proliferation of prostate cancer cells. Mol. Endocrinol. 2014, 28, 872–885. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Yu, J.; Abdulkadir, S.A.; Chakravarti, D. KAT8 regulates androgen signaling in prostate cancer cells. Mol. Endocrinol. 2016, 30, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Halkidou, K.; Gnanapragasam, V.J.; Mehta, P.B.; Logan, I.R.; Brady, M.E.; Cook, S.; Leung, H.Y.; Neal, D.E.; Robson, C.N. Expression of Tip60, an androgen receptor coactivator, and its role in prostate cancer development. Oncogene 2003, 22, 2466–2477. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Yokomizo, A.; Masubuchi, D.; Tada, Y.; Inokuchi, J.; Eto, M.; Uchiumi, T.; Fujimoto, N.; Naito, S. Tip60 promotes prostate cancer cell proliferation by translocation of androgen receptor into the nucleus. Prostate 2010, 70, 540–554. [Google Scholar] [CrossRef] [PubMed]
- Richters, A.; Koehler, A.N. Epigenetic modulation using small molecules—Targeting histone acetyltransferases in disease. Curr. Med. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Santer, F.R.; Hoschele, P.P.; Oh, S.J.; Erb, H.H.; Bouchal, J.; Cavarretta, I.T.; Parson, W.; Meyers, D.J.; Cole, P.A.; Culig, Z. Inhibition of the acetyltransferases p300 and CBP reveals a targetable function for p300 in the survival and invasion pathways of prostate cancer cell lines. Mol. Cancer Ther. 2011, 10, 1644–1655. [Google Scholar] [CrossRef] [PubMed]
- Tohyama, S.; Tomura, A.; Ikeda, N.; Hatano, M.; Odanaka, J.; Kubota, Y.; Umekita, M.; Igarashi, M.; Sawa, R.; Morino, T. Discovery and characterization of NK13650s, naturally occurring p300-selective histone acetyltransferase inhibitors. J. Org. Chem. 2012, 77, 9044–9052. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Bourke, E.; Eriksson, L.A.; Kerin, M.J. Targeting cancer using KAT inhibitors to mimic lethal knockouts. Biochem. Soc. Trans. 2016, 44, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Coffey, K.; Blackburn, T.J.; Cook, S.; Golding, B.T.; Griffin, R.J.; Hardcastle, I.R.; Hewitt, L.; Huberman, K.; McNeill, H.V.; Newell, D.R.; et al. Characterisation of a Tip60 specific inhibitor, NU9056, in prostate cancer. PLoS ONE 2012, 7, e45539. [Google Scholar] [CrossRef] [PubMed]
- Waltregny, D.; North, B.; Van Mellaert, F.; de Leval, J.; Verdin, E.; Castronovo, V. Screening of histone deacetylases (HDAC) expression in human prostate cancer reveals distinct class I HDAC profiles between epithelial and stromal cells. Eur. J. Histochem. 2004, 48, 273–290. [Google Scholar] [PubMed]
- Weichert, W.; Roske, A.; Gekeler, V.; Beckers, T.; Stephan, C.; Jung, K.; Fritzsche, F.R.; Niesporek, S.; Denkert, C.; Dietel, M.; et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br. J. Cancer 2008, 98, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Gaughan, L.; Logan, I.R.; Neal, D.E.; Robson, C.N. Regulation of androgen receptor and histone deacetylase 1 by Mdm2-mediated ubiquitylation. Nucleic Acids Res. 2005, 33, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Welsbie, D.S.; Xu, J.; Chen, Y.; Borsu, L.; Scher, H.I.; Rosen, N.; Sawyers, C.L. Histone deacetylases are required for androgen receptor function in hormone-sensitive and castrate-resistant prostate cancer. Cancer Res. 2009, 69, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Olzscha, H.; Bekheet, M.E.; Sheikh, S.; La Thangue, N.B. HDAC inhibitors. Methods Mol. Biol. 2016, 1436, 281–303. [Google Scholar] [PubMed]
- Bruzzese, F.; Pucci, B.; Milone, M.R.; Ciardiello, C.; Franco, R.; Chianese, M.I.; Rocco, M.; Di Gennaro, E.; Leone, A.; Luciano, A.; et al. Panobinostat synergizes with zoledronic acid in prostate cancer and multiple myeloma models by increasing ROS and modulating mevalonate and p38-MAPK pathways. Cell Death Dis. 2013, 4, e878. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.L.; Marampon, F.; Giusti, I.; Carosa, E.; Di Sante, S.; Ricevuto, E.; Dolo, V.; Tombolini, V.; Jannini, E.A.; Festuccia, C. Differential effects of PXD101 (belinostat) on androgen-dependent and androgen-independent prostate cancer models. Int. J. Oncol. 2012, 40, 711–720. [Google Scholar] [PubMed]
- Qian, D.Z.; Wei, Y.F.; Wang, X.; Kato, Y.; Cheng, L.; Pili, R. Antitumor activity of the histone deacetylase inhibitor MS-275 in prostate cancer models. Prostate 2007, 67, 1182–1193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Sun, M.; Zhou, S.; Guo, B. Class I HDAC inhibitor mocetinostat induces apoptosis by activation of miR-31 expression and suppression of E2F6. Cell Death Discov. 2016, 2, 16036. [Google Scholar] [CrossRef] [PubMed]
- Ruscetti, M.; Dadashian, E.L.; Guo, W.; Quach, B.; Mulholland, D.J.; Park, J.W.; Tran, L.M.; Kobayashi, N.; Bianchi-Frias, D.; Xing, Y.; et al. HDAC inhibition impedes epithelial-mesenchymal plasticity and suppresses metastatic, castration-resistant prostate cancer. Oncogene 2016, 35, 3781–3795. [Google Scholar] [CrossRef] [PubMed]
- Bjorkman, M.; Iljin, K.; Halonen, P.; Sara, H.; Kaivanto, E.; Nees, M.; Kallioniemi, O.P. Defining the molecular action of HDAC inhibitors and synergism with androgen deprivation in ERG-positive prostate cancer. Int. J. Cancer 2008, 123, 2774–2781. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Ngo, D.; Forman, L.W.; Qin, D.C.; Jacob, J.; Faller, D.V. Sirtuin 1 is required for antagonist-induced transcriptional repression of androgen-responsive genes by the androgen receptor. Mol. Endocrinol. 2007, 21, 1807–1821. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Vakoc, C.R. Targeting cancer cells with BET bromodomain inhibitors. Cold Spring Harb. Perspect. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Sahai, V.; Redig, A.J.; Collier, K.A.; Eckerdt, F.D.; Munshi, H.G. Targeting BET bromodomain proteins in solid tumors. Oncotarget 2016, 7, 53997–54009. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Gelato, K.A.; Fernandez-Montalvan, A.; Siegel, S.; Haendler, B. Targeting BET bromodomains for cancer treatment. Epigenomics 2015, 7, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, Y. The bromodomain and extra-terminal domain (BET) family: Functional anatomy of BET paralogous proteins. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Wyce, A.; Degenhardt, Y.; Bai, Y.; Le, B.; Korenchuk, S.; Crouthame, M.C.; McHugh, C.F.; Vessella, R.; Creasy, C.L.; Tummino, P.J.; et al. Inhibition of BET bromodomain proteins as a therapeutic approach in prostate cancer. Oncotarget 2013, 4, 2419–2429. [Google Scholar] [CrossRef] [PubMed]
- Asangani, I.A.; Dommeti, V.L.; Wang, X.; Malik, R.; Cieslik, M.; Yang, R.; Escara-Wilke, J.; Wilder-Romans, K.; Dhanireddy, S.; Engelke, C.; et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014, 510, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Asangani, I.A.; Wilder-Romans, K.; Dommeti, V.L.; Krishnamurthy, P.M.; Apel, I.J.; Escara-Wilke, J.; Plymate, S.R.; Navone, N.M.; Wang, S.; Feng, F.Y.; et al. BET bromodomain inhibitors enhance efficacy and disrupt resistance to AR antagonists in the treatment of prostate cancer. Mol. Cancer Res. 2016, 14, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.C.; Selth, L.A.; Li, Y.; Nyquist, M.D.; Miao, L.; Bradner, J.E.; Raj, G.V.; Tilley, W.D.; Dehm, S.M. Targeting chromatin binding regulation of constitutively active AR variants to overcome prostate cancer resistance to endocrine-based therapies. Nucleic Acids Res. 2015, 43, 5880–5897. [Google Scholar] [CrossRef] [PubMed]
- Faivre, E.J.; Wilcox, D.; Lin, X.; Hessler, P.; Torrent, M.; He, W.; Uziel, T.; Albert, D.H.; McDaniel, K.; Kati, W.; et al. Exploitation of castration-resistant prostate cancer transcription factor dependencies by the novel BET inhibitor ABBV-075. Mol. Cancer Res. 2017, 15, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Coleman, D.J.; Van Hook, K.; King, C.J.; Schwartzman, J.; Lisac, R.; Urrutia, J.; Sehrawat, A.; Woodward, J.; Wang, N.J.; Gulati, R.; et al. Cellular androgen content influences enzalutamide agonism of F877L mutant androgen receptor. Oncotarget 2016, 7, 40690–40703. [Google Scholar] [CrossRef] [PubMed]
- Blee, A.M.; Liu, S.; Wang, L.; Huang, H. BET bromodomain-mediated interaction between ERG and BRD4 promotes prostate cancer cell invasion. Oncotarget 2016, 7, 38319–38332. [Google Scholar] [CrossRef] [PubMed]
- Neklesa, T.K.; Winkler, J.D.; Crews, C.M. Targeted protein degradation by PROTACs. Pharmacol. Ther. 2017. [Google Scholar] [CrossRef] [PubMed]
- Raina, K.; Lu, J.; Qian, Y.; Altieri, M.; Gordon, D.; Rossi, A.M.; Wang, J.; Chen, X.; Dong, H.; Siu, K.; et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 7124–7129. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.X.; Guo, L.; Revenko, A.S.; Tepper, C.G.; Gemo, A.T.; Kung, H.J.; Chen, H.W. Androgen-induced coactivator ANCCA mediates specific androgen receptor signaling in prostate cancer. Cancer Res. 2009, 69, 3339–3346. [Google Scholar] [CrossRef] [PubMed]
- Theurillat, J.P.; Udeshi, N.D.; Errington, W.J.; Svinkina, T.; Baca, S.C.; Pop, M.; Wild, P.J.; Blattner, M.; Groner, A.C.; Rubin, M.A.; et al. Prostate cancer. Ubiquitylome analysis identifies dysregulation of effector substrates in SPOP-mutant prostate cancer. Science 2014, 346, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Groner, A.C.; Cato, L.; de Tribolet-Hardy, J.; Bernasocchi, T.; Janouskova, H.; Melchers, D.; Houtman, R.; Cato, A.C.; Tschopp, P.; Gu, L.; et al. TRIM24 is an oncogenic transcriptional activator in prostate cancer. Cancer Cell 2016, 29, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Tavassoli, P.; Wafa, L.A.; Cheng, H.; Zoubeidi, A.; Fazli, L.; Gleave, M.; Snoek, R.; Rennie, P.S. TAF1 differentially enhances androgen receptor transcriptional activity via its N-terminal kinase and ubiquitin-activating and -conjugating domains. Mol. Endocrinol. 2010, 24, 696–708. [Google Scholar] [CrossRef] [PubMed]
- Bouché, L.; Christ, C.D.; Siegel, S.; Fernández-Montalván, A.E.; Holton, S.J.; Fedorov, O.; Ter Laak, A.; Sugawara, T.; Stöckigt, D.; Tallant, C.; et al. Benzoisoquinolinediones as potent and selective inhibitors of BRPF2 and TAF1/TAF1L bromodomains. J. Med. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.; Fedorov, O.; Tallant, C.; Monteiro, O.; Meier, J.; Gamble, V.; Savitsky, P.; Nunez-Alonso, G.A.; Haendler, B.; Rogers, C.; et al. Discovery of a chemical tool inhibitor targeting the bromodomains of TRIM24 and BRPF. J. Med. Chem. 2016, 59, 1642–1647. [Google Scholar] [CrossRef] [PubMed]
- Palmer, W.S.; Poncet-Montange, G.; Liu, G.; Petrocchi, A.; Reyna, N.; Subramanian, G.; Theroff, J.; Yau, A.; Kost-Alimova, M.; Bardenhagen, J.P.; et al. Structure-guided design of IACS-9571, a selective high-affinity dual TRIM24-BRPF1 bromodomain inhibitor. J. Med. Chem. 2016, 59, 1440–1454. [Google Scholar] [CrossRef] [PubMed]
- Seligson, D.B.; Horvath, S.; Shi, T.; Yu, H.; Tze, S.; Grunstein, M.; Kurdistani, S.K. Global histone modification patterns predict risk of prostate cancer recurrence. Nature 2005, 435, 1262–1266. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.; Yang, Y.W.; Huang, Y.; Yang, J.; Zhang, H.; Chen, R.; Dong, L.; Huang, Y.; Wang, D.; Liu, J.; et al. P110β inhibition reduces histone H3K4 di-methylation in prostate cancer. Prostate 2017, 77, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Behbahani, T.E.; Kahl, P.; von der Gathen, J.; Heukamp, L.C.; Baumann, C.; Gutgemann, I.; Walter, B.; Hofstadter, F.; Bastian, P.J.; von Ruecker, A.; et al. Alterations of global histone H4K20 methylation during prostate carcinogenesis. BMC Urol. 2012, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Yuan, X.; Balk, S.P. Androgen receptor epigenetics. Transl. Androl. Urol. 2013, 2, 148–157. [Google Scholar] [PubMed]
- Yang, Y.A.; Yu, J. EZH2, an epigenetic driver of prostate cancer. Protein Cell 2013, 4, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Melling, N.; Thomsen, E.; Tsourlakis, M.C.; Kluth, M.; Hube-Magg, C.; Minner, S.; Koop, C.; Graefen, M.; Heinzer, H.; Wittmer, C.; et al. Overexpression of enhancer of zeste homolog 2 (EZH2) characterizes an aggressive subset of prostate cancers and predicts patient prognosis independently from pre- and postoperatively assessed clinicopathological parameters. Carcinogenesis 2015, 36, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Yu, J.; Mani, R.S.; Cao, Q.; Brenner, C.J.; Cao, X.; Wang, X.; Wu, L.; Li, J.; Hu, M.; et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell 2010, 17, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Crea, F. EZH2 and cancer stem cells: Fact or fiction? Epigenomics 2011, 3, 127–128. [Google Scholar] [CrossRef] [PubMed]
- Volkel, P.; Dupret, B.; Le Bourhis, X.; Angrand, P.O. Diverse involvement of EZH2 in cancer epigenetics. Am. J. Transl. Res. 2015, 7, 175–193. [Google Scholar] [PubMed]
- Deb, G.; Thakur, V.S.; Gupta, S. Multifaceted role of EZH2 in breast and prostate tumorigenesis: Epigenetics and beyond. Epigenetics 2013, 8, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Crea, F.; Hurt, E.M.; Mathews, L.A.; Cabarcas, S.M.; Sun, L.; Marquez, V.E.; Danesi, R.; Farrar, W.L. Pharmacologic disruption of Polycomb Repressive Complex 2 inhibits tumorigenicity and tumor progression in prostate cancer. Mol. Cancer 2011, 10, 40. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Jin, X.; Yang, J.; Yang, Y.; He, Y.; Ding, L.; Pan, Y.; Chen, S.; Jiang, J.; Huang, H. Inhibition of EZH2 by chemo- and radiotherapy agents and small molecule inhibitors induces cell death in castration-resistant prostate cancer. Oncotarget 2016, 7, 3440–3452. [Google Scholar] [PubMed]
- He, Y.; Selvaraju, S.; Curtin, M.L.; Jakob, C.G.; Zhu, H.; Comess, K.M.; Shaw, B.; The, J.; Lima-Fernandes, E.; Szewczyk, M.M.; et al. The EED protein-protein interaction inhibitor A-395 inactivates the PRC2 complex. Nat. Chem. Biol 2017, 13, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Zhao, K.; Gu, J.; Huang, Y.; Wang, Y.; Zhang, H.; Zhang, M.; Zhang, J.; Yu, Z.; Li, L.; et al. An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat. Chem. Biol. 2017, 13, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhang, J.; Yu, Z.; Zhang, H.; Wang, Y.; Lingel, A.; Qi, W.; Gu, J.; Zhao, K.; Shultz, M.D.; et al. Discovery of first-in-class, potent, and orally bioavailable Embryonic Ectoderm Development (EED) inhibitor with robust anticancer efficacy. J. Med. Chem. 2017, 60, 2215–2226. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, H.; Zhang, M.; Zhao, M.; Feng, L.; Luo, X.; Gao, Z.; Huang, Y.; Ardayfio, O.; Zhang, J.H.; Lin, Y.; et al. Discovery and molecular basis of a diverse set of Polycomb Repressive Complex 2 inhibitors recognition by EED. PLoS ONE 2017, 12, e0169855. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Xu, A.M. SET and MYND domain containing protein 3 in cancer. Am. J. Transl. Res. 2017, 9, 1–14. [Google Scholar] [PubMed]
- Liu, C.; Wang, C.; Wang, K.; Liu, L.; Shen, Q.; Yan, K.; Sun, X.; Chen, J.; Liu, J.; Ren, H.; et al. SMYD3 as an oncogenic driver in prostate cancer by stimulation of androgen receptor transcription. J. Natl. Cancer Inst. 2013, 105, 1719–1728. [Google Scholar] [CrossRef] [PubMed]
- Vieira, F.Q.; Costa-Pinheiro, P.; Almeida-Rios, D.; Graca, I.; Monteiro-Reis, S.; Simoes-Sousa, S.; Carneiro, I.; Sousa, E.J.; Godinho, M.I.; Baltazar, F.; et al. SMYD3 contributes to a more aggressive phenotype of prostate cancer and targets Cyclin D2 through H4K20me3. Oncotarget 2015, 6, 13644–13657. [Google Scholar] [CrossRef] [PubMed]
- Peserico, A.; Germani, A.; Sanese, P.; Barbosa, A.J.; di Virgilio, V.; Fittipaldi, R.; Fabini, E.; Bertucci, C.; Varchi, G.; Moyer, M.P.; et al. A SMYD3 small-molecule inhibitor impairing cancer cell growth. J. Cell. Physiol. 2015, 230, 2447–2460. [Google Scholar] [CrossRef] [PubMed]
- Stopa, N.; Krebs, J.E.; Shechter, D. The PRMT5 arginine methyltransferase: Many roles in development, cancer and beyond. Cell. Mol. Life Sci. 2015, 72, 2041–2059. [Google Scholar] [CrossRef] [PubMed]
- Mounir, Z.; Korn, J.M.; Westerling, T.; Lin, F.; Kirby, C.A.; Schirle, M.; McAllister, G.; Hoffman, G.; Ramadan, N.; Hartung, A.; et al. ERG signaling in prostate cancer is driven through PRMT5-dependent methylation of the Androgen Receptor. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Shao, G.; Zhang, H.T.; Li, C.; Zhang, D.; Cheng, L.; Elzey, B.D.; Pili, R.; Ratliff, T.L.; Huang, J.; et al. Protein arginine methyltransferase 5 functions as an epigenetic activator of the androgen receptor to promote prostate cancer cell growth. Oncogene 2017, 36, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Kahl, P.; Gullotti, L.; Heukamp, L.C.; Wolf, S.; Friedrichs, N.; Vorreuther, R.; Solleder, G.; Bastian, P.J.; Ellinger, J.; Metzger, E.; et al. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res. 2006, 66, 11341–11347. [Google Scholar] [CrossRef] [PubMed]
- Bjorkman, M.; Ostling, P.; Harma, V.; Virtanen, J.; Mpindi, J.P.; Rantala, J.; Mirtti, T.; Vesterinen, T.; Lundin, M.; Sankila, A.; et al. Systematic knockdown of epigenetic enzymes identifies a novel histone demethylase PHF8 overexpressed in prostate cancer with an impact on cell proliferation, migration and invasion. Oncogene 2012, 31, 3444–3456. [Google Scholar] [CrossRef] [PubMed]
- Coffey, K.; Rogerson, L.; Ryan-Munden, C.; Alkharaif, D.; Stockley, J.; Heer, R.; Sahadevan, K.; O'Neill, D.; Jones, D.; Darby, S.; et al. The lysine demethylase, KDM4B, is a key molecule in androgen receptor signalling and turnover. Nucleic Acids Res. 2013, 41, 4433–4446. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, V.; Ahmad, S.; Nilsson, E.M.; Helczynski, L.; Kenna, S.; Persson, J.L.; Gudas, L.J.; Mongan, N.P. The lysine specific demethylase-1 (LSD1/KDM1A) regulates VEGF-A expression in prostate cancer. Mol. Oncol. 2013, 7, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Xu, W.; Cheng, P.; Jin, H.; Wang, X. Histone demethylase lysine demethylase 5B in development and cancer. Oncotarget 2017, 8, 8980–8991. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, A.; Haendler, B. Histone demethylation and steroid receptor function in cancer. Mol. Cell. Endocrinol. 2012, 348, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Metzger, E.; Wissmann, M.; Yin, N.; Muller, J.M.; Schneider, R.; Peters, A.H.; Gunther, T.; Buettner, R.; Schule, R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Wissmann, M.; Yin, N.; Muller, J.M.; Greschik, H.; Fodor, B.D.; Jenuwein, T.; Vogler, C.; Schneider, R.; Gunther, T.; Buettner, R.; et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat. Cell Biol. 2007, 9, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; He, H.H.; Chen, S.; Coleman, I.; Wang, H.; Fang, Z.; Chen, S.; Nelson, P.S.; Liu, X.S.; Brown, M.; et al. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell 2011, 20, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Etani, T.; Suzuki, T.; Naiki, T.; Naiki-Ito, A.; Ando, R.; Iida, K.; Kawai, N.; Tozawa, K.; Miyata, N.; Kohri, K.; et al. NCL1, a highly selective lysine-specific demethylase 1 inhibitor, suppresses prostate cancer without adverse effect. Oncotarget 2015, 6, 2865–2878. [Google Scholar] [CrossRef] [PubMed]
- Tong, D.; Liu, Q.; Liu, G.; Yuan, W.; Wang, L.; Guo, Y.; Lan, W.; Zhang, D.; Dong, S.; Wang, Y.; et al. The HIF/PHF8/AR axis promotes prostate cancer progression. Oncogenesis 2016, 5, e283. [Google Scholar] [CrossRef] [PubMed]
- Maina, P.K.; Shao, P.; Liu, Q.; Fazli, L.; Tyler, S.; Nasir, M.; Dong, X.; Qi, H.H. c-MYC drives histone demethylase PHF8 during neuroendocrine differentiation and in castration-resistant prostate cancer. Oncotarget 2016, 7, 75585–75602. [Google Scholar] [CrossRef] [PubMed]
- Ackloo, S.; Brown, P.J.; Muller, S. Chemical probes targeting epigenetic proteins: Applications beyond oncology. Epigenetics 2017, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Gulzar, Z.G.; Salari, K.; Lapointe, J.; Brooks, J.D.; Pollack, J.R. Recurrent deletion of CHD1 in prostate cancer with relevance to cell invasiveness. Oncogene 2012, 31, 4164–4170. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, L.U.; Rider, L.; Nieto, C.; Romero, L.; Karimpour-Fard, A.; Loda, M.; Lucia, M.S.; Wu, M.; Shi, L.; Cimic, A.; et al. Coordinate loss of MAP3K7 and CHD1 promotes aggressive prostate cancer. Cancer Res. 2015, 75, 1021–1034. [Google Scholar] [CrossRef] [PubMed]
- Kari, V.; Mansour, W.Y.; Raul, S.K.; Baumgart, S.J.; Mund, A.; Grade, M.; Sirma, H.; Simon, R.; Will, H.; Dobbelstein, M.; et al. Loss of CHD1 causes DNA repair defects and enhances prostate cancer therapeutic responsiveness. EMBO Rep. 2016, 17, 1609–1623. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Lu, X.; Wang, G.; Lan, Z.; Liao, W.; Li, J.; Liang, X.; Chen, J.R.; Shah, S.; Shang, X.; et al. Synthetic essentiality of chromatin remodelling factor CHD1 in PTEN-deficient cancer. Nature 2017, 542, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Du, H.N. Transcription, DNA damage and beyond: The roles of histone ubiquitination and deubiquitination. Current Prot. Pept. Sci. 2012, 13, 447–466. [Google Scholar] [CrossRef]
- Johnsen, S.A. The enigmatic role of H2Bub1 in cancer. FEBS Lett. 2012, 586, 1592–1601. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Yan, Q. Histone ubiquitination and deubiquitination in transcription, DNA damage response, and cancer. Front. Oncol. 2012, 2, 26. [Google Scholar] [CrossRef] [PubMed]
- Jaaskelainen, T.; Makkonen, H.; Visakorpi, T.; Kim, J.; Roeder, R.G.; Palvimo, J.J. Histone H2B ubiquitin ligases RNF20 and RNF40 in androgen signaling and prostate cancer cell growth. Mol. Cell. Endocrinol. 2012, 350, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Draker, R.; Sarcinella, E.; Cheung, P. USP10 deubiquitylates the histone variant H2A.Z and both are required for androgen receptor-mediated gene activation. Nucleic Acids Res. 2011, 39, 3529–3542. [Google Scholar] [CrossRef] [PubMed]
- Faus, H.; Meyer, H.A.; Huber, M.; Bahr, I.; Haendler, B. The ubiquitin-specific protease USP10 modulates androgen receptor function. Mol. Cell. Endocrinol. 2005, 245, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.; Rajapakshe, K.; Shah, S.S.; Shou, J.; Eedunuri, V.K.; Foley, C.; Fiskus, W.; Rajendran, M.; Chew, S.A.; Zimmermann, M.; et al. Androgen receptor is the key transcriptional mediator of the tumor suppressor SPOP in prostate cancer. Cancer Res. 2014, 74, 5631–5643. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.T.; Okada, M.; Nakato, R.; Izumi, K.; Bando, M.; Shirahige, K. The deubiquitinating enzyme USP7 regulates androgen receptor activity by modulating its binding to chromatin. J. Biol. Chem. 2015, 290, 21713–21723. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Liu, N.; Hua, X.; Cai, J.; Xia, X.; Wang, X.; Huang, H.; Liu, J. Proteasome-associated deubiquitinase ubiquitin-specific protease 14 regulates prostate cancer proliferation by deubiquitinating and stabilizing androgen receptor. Cell Death Dis. 2017, 8, e2585. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Blattner, M.; Liu, D.; Robinson, B.D.; Huang, D.; Poliakov, A.; Gao, D.; Nataraj, S.; Deonarine, L.D.; Augello, M.A.; Sailer, V.; et al. SPOP mutation drives prostate tumorigenesis in vivo through coordinate regulation of PI3K/mTOR and AR signaling. Cancer Cell 2017, 31, 436–451. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Fan, L.; Hussain, A. Implications of ubiquitin ligases in castration-resistant prostate cancer. Curr. Opin. Oncol. 2015, 27, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Faus, H.; Haendler, B. Post-translational modifications of steroid receptors. Biomed. Pharmacother. 2006, 60, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Banerjee, T.; Vinckevicius, A.; Luo, Q.; Parker, J.B.; Baker, M.R.; Radhakrishnan, I.; Wei, J.J.; Barish, G.D.; Chakravarti, D. A role for WDR5 in integrating threonine 11 phosphorylation to lysine 4 methylation on histone H3 during androgen signaling and in prostate cancer. Mol. Cell 2014, 54, 613–625. [Google Scholar] [CrossRef] [PubMed]
- Kohler, J.; Erlenkamp, G.; Eberlin, A.; Rumpf, T.; Slynko, I.; Metzger, E.; Schule, R.; Sippl, W.; Jung, M. Lestaurtinib inhibits histone phosphorylation and androgen-dependent gene expression in prostate cancer cells. PLoS ONE 2012, 7, e34973. [Google Scholar] [CrossRef] [PubMed]
- Thibault, A.; Figg, W.D.; Bergan, R.C.; Lush, R.M.; Myers, C.E.; Tompkins, A.; Reed, E.; Samid, D. A phase II study of 5-aza-2′deoxycytidine (decitabine) in hormone independent metastatic (D2) prostate cancer. Tumori 1998, 84, 87–89. [Google Scholar] [PubMed]
- Singal, R.; Ramachandran, K.; Gordian, E.; Quintero, C.; Zhao, W.; Reis, I.M. Phase I/II study of azacitidine, docetaxel, and prednisone in patients with metastatic castration-resistant prostate cancer previously treated with docetaxel-based therapy. Clin. Genitourin. Cancer 2015, 13, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Bradley, D.; Rathkopf, D.; Dunn, R.; Stadler, W.M.; Liu, G.; Smith, D.C.; Pili, R.; Zwiebel, J.; Scher, H.; Hussain, M. Vorinostat in advanced prostate cancer patients progressing on prior chemotherapy (National Cancer Institute Trial 6862): Trial results and interleukin-6 analysis: A study by the Department of Defense Prostate Cancer Clinical Trial Consortium and University of Chicago Phase 2 Consortium. Cancer 2009, 115, 5541–5549. [Google Scholar] [PubMed]
- Molife, L.R.; Attard, G.; Fong, P.C.; Karavasilis, V.; Reid, A.H.; Patterson, S.; Riggs, C.E., Jr.; Higano, C.; Stadler, W.M.; McCulloch, W.; et al. Phase II, two-stage, single-arm trial of the histone deacetylase inhibitor (HDACi) romidepsin in metastatic castration-resistant prostate cancer (CRPC). Ann. Oncol. 2010, 21, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Eigl, B.J.; North, S.; Winquist, E.; Finch, D.; Wood, L.; Sridhar, S.S.; Powers, J.; Good, J.; Sharma, M.; Squire, J.A.; et al. A phase II study of the HDAC inhibitor SB939 in patients with castration resistant prostate cancer: NCIC clinical trials group study IND195. Investig. New Drugs 2015, 33, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Rathkopf, D.E.; Picus, J.; Hussain, A.; Ellard, S.; Chi, K.N.; Nydam, T.; Allen-Freda, E.; Mishra, K.K.; Porro, M.G.; Scher, H.I.; et al. A phase 2 study of intravenous panobinostat in patients with castration-resistant prostate cancer. Cancer Chemother. Pharmacol. 2013, 72, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Rathkopf, D.; Carducci, M.A.; Morris, M.J.; Slovin, S.F.; Eisenberger, M.A.; Pili, R.; Denmeade, S.R.; Kelsen, M.; Curley, T.; Halter, M.; et al. Phase II trial of docetaxel with rapid androgen cycling for progressive noncastrate prostate cancer. J. Clin. Oncol. 2008, 26, 2959–2965. [Google Scholar] [CrossRef] [PubMed]
- Munster, P.N.; Marchion, D.; Thomas, S.; Egorin, M.; Minton, S.; Springett, G.; Lee, J.H.; Simon, G.; Chiappori, A.; Sullivan, D.; et al. Phase I trial of vorinostat and doxorubicin in solid tumours: Histone deacetylase 2 expression as a predictive marker. Br. J. Cancer 2009, 101, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, D.; Vashistha, V.; Isharwal, S.; Sediqe, S.A.; Lin, M.F. Histone deacetylase inhibitors in castration-resistant prostate cancer: Molecular mechanism of action and recent clinical trials. Ther. Adv. Urol. 2015, 7, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Tsujikawa, L.; Norek, K.; Calosing, C.; Attwell, S.; Gilham, D.; Sharma, N.; Tobin, J.; Haager, M.; Jahagirdar, R.; Lakhotia, S.; et al. Preclinical development and clinical validation of a whole blood pharmacodynamic marker assay for the BET bromodomain inhibitor ZEN-3694 in metastatic castration-resistant prostate cancer (mCRPC) patients. In Proceedings of the AACR Annual Meeting 2017, Washington, DC, USA, 1–5 April 2017. [Google Scholar]
- Bertoli, G.; Cava, C.; Castiglioni, I. MicroRNAs as niomarkers for diagnosis, prognosis and theranostics in prostate cancer. Int. J. Mol. Sci. 2016, 17, 421. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [PubMed]
- Kojima, S.; Goto, Y.; Naya, Y. The roles of microRNAs in the progression of castration-resistant prostate cancer. J. Hum. Genet. 2017, 62, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Ostling, P.; Leivonen, S.K.; Aakula, A.; Kohonen, P.; Makela, R.; Hagman, Z.; Edsjo, A.; Kangaspeska, S.; Edgren, H.; Nicorici, D.; et al. Systematic analysis of microRNAs targeting the androgen receptor in prostate cancer cells. Cancer Res. 2011, 71, 1956–1967. [Google Scholar] [CrossRef] [PubMed]
- Cannistraci, A.; Federici, G.; Addario, A.; Di Pace, A.L.; Grassi, L.; Muto, G.; Collura, D.; Signore, M.; De Salvo, L.; Sentinelli, S.; et al. C-Met/miR-130b axis as novel mechanism and biomarker for castration resistance state acquisition. Oncogene 2017. [Google Scholar] [CrossRef] [PubMed]
- Hao, Z.; Fan, W.; Hao, J.; Wu, X.; Zeng, G.Q.; Zhang, L.J.; Nie, S.F.; Wang, X.D. Efficient delivery of micro RNA to bone-metastatic prostate tumors by using aptamer-conjugated atelocollagen in vitro and in vivo. Drug Deliv. 2016, 23, 874–881. [Google Scholar] [PubMed]
- Yao, C.; Liu, J.; Wu, X.; Tai, Z.; Gao, Y.; Zhu, Q.; Li, J.; Zhang, L.; Hu, C.; Gu, F.; Gao, J.; Gao, S. Reducible self-assembling cationic polypeptide-based micelles mediate co-delivery of doxorubicin and microRNA-34a for androgen-independent prostate cancer therapy. J. Control. Release 2016, 232, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.B.; Ma, A.H.; Xue, L.; Li, M.; Nguyen, H.G.; Yang, J.C.; Tepper, C.G.; Gandour-Edwards, R.; Evans, C.P.; Kung, H.J.; et al. miR-124 and androgen receptor signaling inhibitors repress prostate cancer growth by downregulating androgen receptor splice variants, EZH2, and Src. Cancer Res. 2015, 75, 5309–5317. [Google Scholar] [CrossRef] [PubMed]
- Larne, O.; Hagman, Z.; Lilja, H.; Bjartell, A.; Edsjo, A.; Ceder, Y. miR-145 suppress the androgen receptor in prostate cancer cells and correlates to prostate cancer prognosis. Carcinogenesis 2015, 36, 858–866. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yan, M.; Yun, Y.; Zhang, J.; Zhang, R.; Li, Y.; Wu, X.; Liu, Q.; Miao, W.; Jiang, H. MicroRNA-455–3p functions as a tumor suppressor by targeting eIF4E in prostate cancer. Oncol. Rep. 2017. [Google Scholar] [CrossRef] [PubMed]
- Nip, H.; Dar, A.A.; Saini, S.; Colden, M.; Varahram, S.; Chowdhary, H.; Yamamura, S.; Mitsui, Y.; Tanaka, Y.; Kato, T.; et al. Oncogenic microRNA-4534 regulates PTEN pathway in prostate cancer. Oncotarget 2016, 7, 68371–68384. [Google Scholar] [CrossRef] [PubMed]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Rekoske, B.T.; McNeel, D.G. Immunotherapy for prostate cancer: False promises or true hope? Cancer 2016, 122, 3598–3607. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Target | Compound | Combination | Indication | Phase | Identifier | Status |
|---|---|---|---|---|---|---|
| DNMT | Azacitidine | Phenylbutirate | Prostate cancer | 2 | NCT00006019 | Completed |
| DNMT | Azacitidine | Prostate cancer | 2 | NCT00384839 | Completed | |
| DNMT | Azacitidine | Docetaxel, prednisone | mCRPC Post-chemotherapy | 1/2 | NCT00503984 | Terminated |
| HDAC | Vorinostat | Includes prostate cancer | 1 | NCT00005634 | Completed | |
| HDAC | Vorinostat | Includes prostate cancer | 1 | NCT00045006 | Completed | |
| HDAC | Vorinostat | Advanced CRPC Post-chemotherapy | 2 | NCT00330161 | Completed | |
| HDAC | Vorinostat | Docetaxel | Includes prostate cancer | 1 | NCT00565227 | Terminated |
| HDAC | Vorinostat | Androgen deprivation | Localized prostate cancer | 2 | NCT00589472 | Completed |
| HDAC | Vorinostat | Temsirolimus | mCRPC | 1 | NCT01174199 | Terminated |
| HDAC | Entinostat | Includes prostate cancer | 1 | NCT00020579 | Completed | |
| HDAC | Romidpesin | Prostatic neoplasms | 2 | NCT00106301 | Completed | |
| HDAC | Romidepsin | mCRPC | 2 | NCT00106418 | Completed | |
| HDAC | Romidepsin | CRPC | 1 | NCT01638533 | Recruiting | |
| HDAC | Belinostat | Includes prostate cancer | 1 | NCT00413075 | Completed | |
| HDAC | Belinostat | 5-fluorouracil | Includes prostate cancer | 1 | NCT00413322 | Completed |
| HDAC | Panobinostat | Docetaxel, prednisone | CRPC | 1 | NCT00419536 | Terminated |
| HDAC | Panobinostat | Docetaxel, prednisone | CRPC | 1 | NCT00493766 | Terminated |
| HDAC | Panobinostat | Docetaxel, prednisone | CRPC | 1 | NCT00663832 | Completed |
| HDAC | Panobinostat | mCRPC | 2 | NCT00667862 | Completed | |
| HDAC | Panobinostat | External beam radiotherapy | Includes prostate cancer | 1 | NCT00670553 | Completed |
| HDAC | Panobinostat | Bicalutamide | Recurrent CRPC | 1/2 | NCT00878436 | Completed |
| HDAC | Mocetinostat | Docetaxel | Includes prostate cancer | 1 | NCT00511576 | Terminated |
| HDAC | Valproic acid | CRPC | 2 | NCT00670046 | Unknown | |
| HDAC | Pracinostat | mCRPC | 2 | NCT01075308 | Completed |
| Target | Compound | Combination | Indication | Phase | Identifier | Status |
|---|---|---|---|---|---|---|
| BET | GSK525762 | CRPC | 1 | NCT01587703 | Recruiting | |
| BET | OTX105 MK-8628 | CRPC | 1 | NCT02259114 | Active, not recruting | |
| BET | OTX105 MK-8628 | CRPC | 1 | NCT02698176 | Active, not recruting | |
| BET | INCB054329 | CRPC | 1/2 | NCT02431260 | Recruiting | |
| BET | INCB057643 | CRPC | 1/2 | NCT02711137 | Recruitng | |
| BET | ZEN003694 | mCRPC | 1 | NCT02705469 | Recuiting | |
| BET | ZEN003694 | Enzalutamide | mCRPC | 1b | NCT02711956 | Recruiting |
| EED | MAK683 | Includes prostate cancer | 1 | NCT02900651 | Recruiting | |
| PRMT5 | GSK3326595 | Includes prostate cancer | 1 | NCT02783300 | Recruiting |
| Outcome Measure | Treatment | Indication | Identifier | Status |
|---|---|---|---|---|
| miRNA profiling | Prostate cancer | NCT01220427 | Terminated | |
| miRNA profiling using Nano-string technology | Prostate cancer | NCT02964351 | Not yet recruiting | |
| Serum exosomal miRNA profiling using next-generation sequencing | Androgen deprivation | Prostate cancer | NCT02366494 | Recruiting |
| Preselected miRNA profiling | Enzalutamide | mCRPC | NCT02471469 | Recruiting |
| miRNA profiling | Radiotherapy | Prostate cancer | NCT02745587 | Recruiting |
| Preselected miRNA profiling | Abiraterone acetate | mCRPC | NCT01503229 | Ongoing, not recruiting |
| miRNA-141, -375 levels | Focal brachytherapy | Low-risk prostate cancer | NCT02391051 | Recruiting |
| miRNA profiling | Androgen deprivation + cixutumumab | Metastatic prostate cancer | NCT01120236 | Ongoing, not recruiting |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baumgart, S.J.; Haendler, B. Exploiting Epigenetic Alterations in Prostate Cancer. Int. J. Mol. Sci. 2017, 18, 1017. https://doi.org/10.3390/ijms18051017
Baumgart SJ, Haendler B. Exploiting Epigenetic Alterations in Prostate Cancer. International Journal of Molecular Sciences. 2017; 18(5):1017. https://doi.org/10.3390/ijms18051017
Chicago/Turabian StyleBaumgart, Simon J., and Bernard Haendler. 2017. "Exploiting Epigenetic Alterations in Prostate Cancer" International Journal of Molecular Sciences 18, no. 5: 1017. https://doi.org/10.3390/ijms18051017
APA StyleBaumgart, S. J., & Haendler, B. (2017). Exploiting Epigenetic Alterations in Prostate Cancer. International Journal of Molecular Sciences, 18(5), 1017. https://doi.org/10.3390/ijms18051017