
Transposable Elements in Human Cancer: Causes and Consequences of Deregulation


Abstract
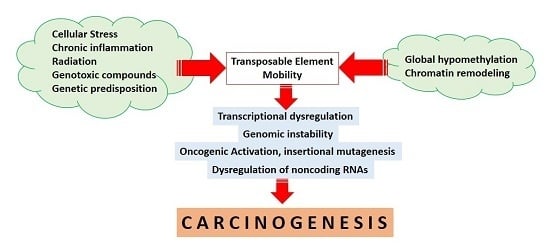
:
1. Introduction
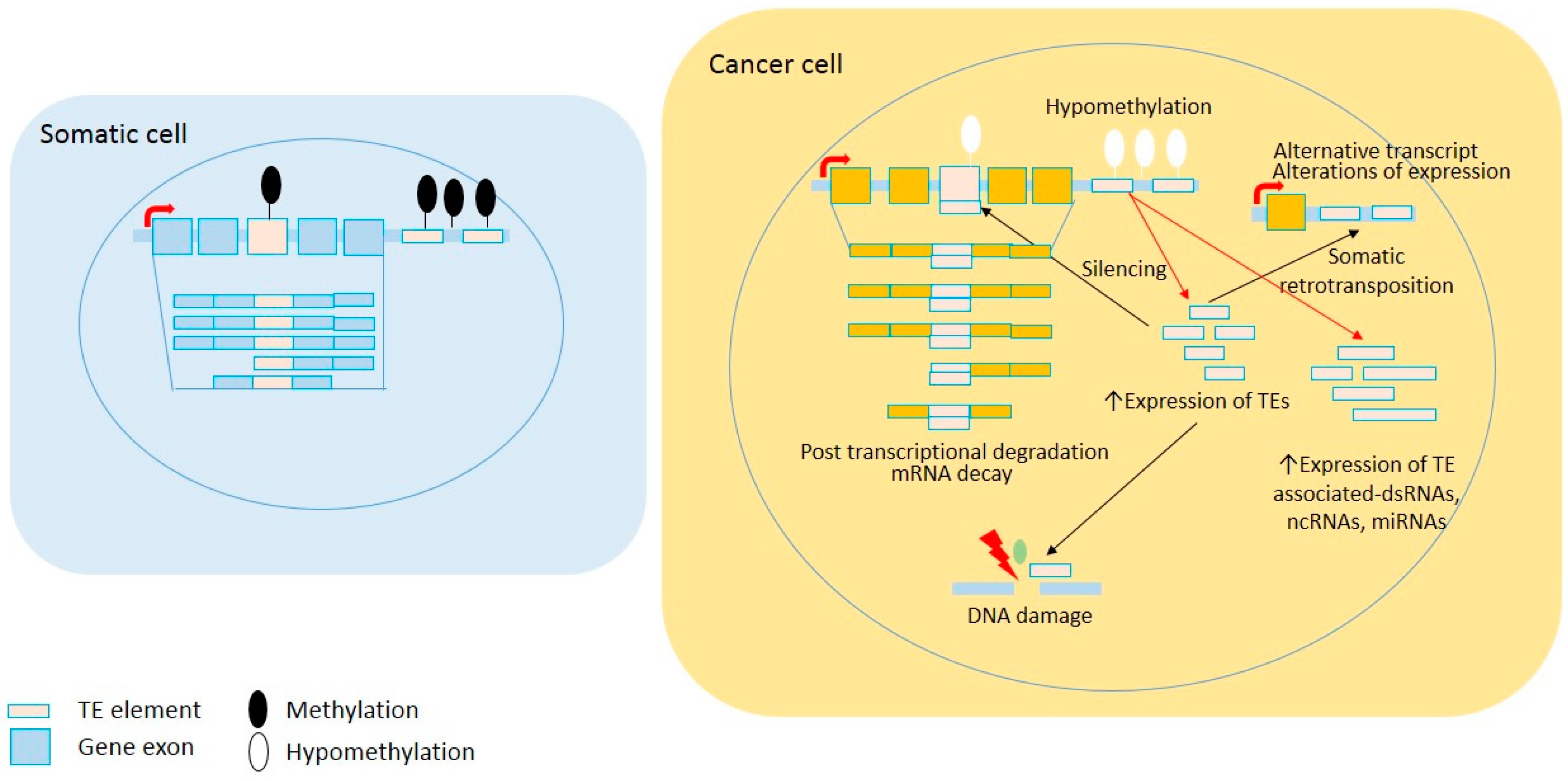
2. Widespread Epigenetic Deregulation of Repetitive Elements in Cancer
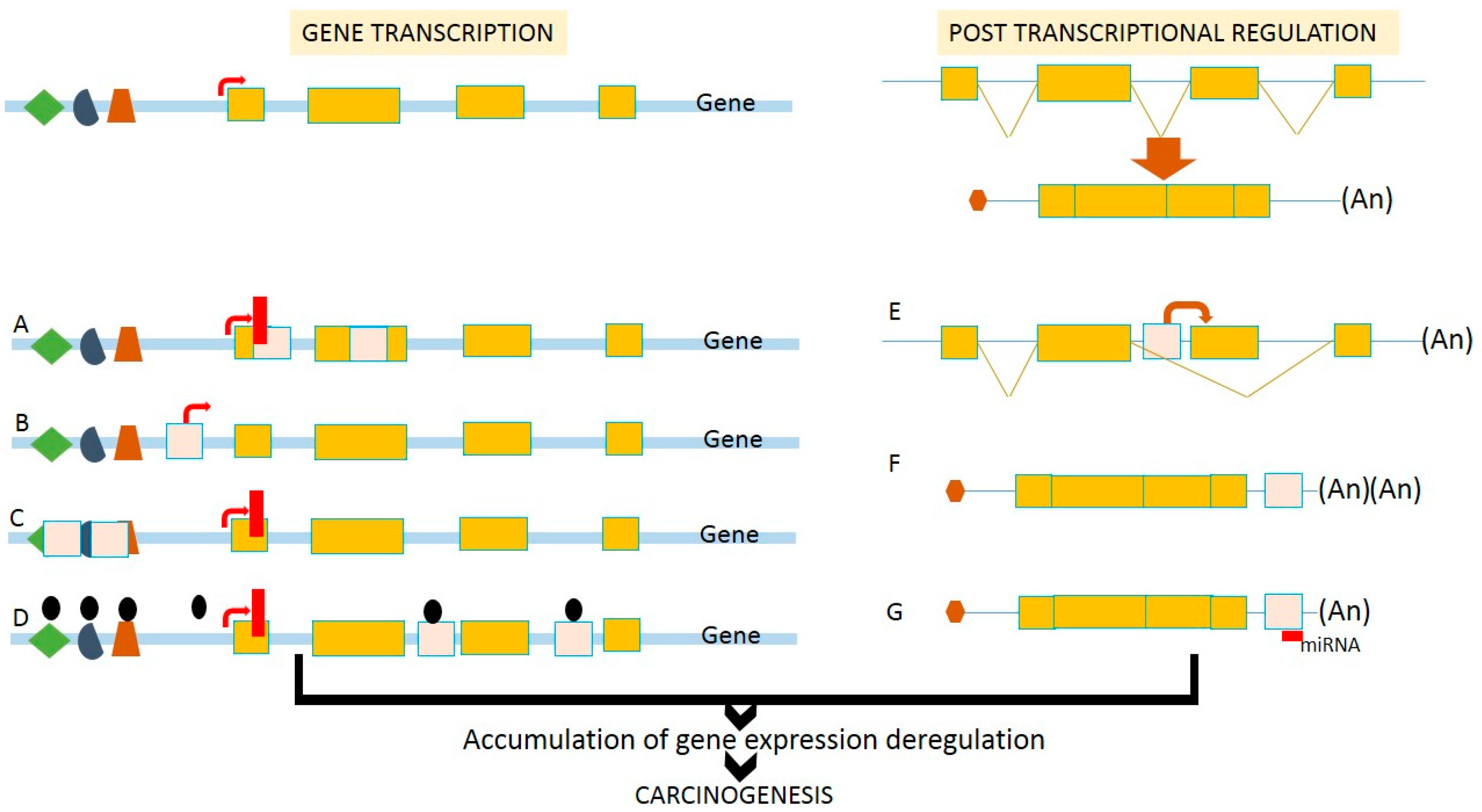
3. Transcriptional Deregulation
4. Genomic Instability and Chromosomal Rearrangements
5. Inactivation of Tumor Suppressor Genes and Activation of Oncogenes
6. Transposable Elements and Non-Coding RNAs
7. Conclusions
Author Contribution
Conflicts of Interest
References
- De Koning, A.P.J.; Gu, W.; Castoe, T.A.; Batzer, M.A.; Pollock, D.D. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011, 7, e1002384. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Iskow, R.; Yang, L.; Gokcumen, O.; Haseley, P.; Luquette, L.J., 3rd; Lohr, J.G.; Harris, C.C.; Ding, L.; Wilson, R.K.; et al. Cancer genome atlas research network landscape of somatic retrotransposition in human cancers. Science 2012, 337, 967–971. [Google Scholar] [CrossRef] [PubMed]
- Rebollo, R.; Romanish, M.T.; Mager, D.L. Transposable elements: An abundant and natural source of regulatory sequences for host genes. Annu. Rev. Genet. 2012, 46, 21–42. [Google Scholar] [CrossRef] [PubMed]
- Kapitonov, V.V.; Jurka, J. A universal classification of eukaryotic transposable elements implemented in Repbase. Nat. Rev. Genet. 2008, 9, 411–412. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.-J.; Xue, H.-Y.; Qi, X.; Xu, J.; Ma, S.-J. LINE-1 in cancer: Multifaceted functions and potential clinical implications. Genet. Med. 2016, 18, 431–439. [Google Scholar]
- Criscione, S.W.; Zhang, Y.; Thompson, W.; Sedivy, J.M.; Neretti, N. Transcriptional landscape of repetitive elements in normal and cancer human cells. BMC Genom. 2014, 15, 583. [Google Scholar] [CrossRef] [PubMed]
- Bodega, B.; Orlando, V. Repetitive elements dynamics in cell identity programming, maintenance and disease. Curr. Opin. Cell Biol. 2014, 31, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Van de Lagemaat, L.N.; Landry, J.R.; Mager, D.L.; Medstrand, P. Transposable elements in mammals promote regulatory variation and diversification of genes with specialized functions. Trends Genet. 2003, 19, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Tress, M.L.; Valencia, A. Alternative splicing and co-option of transposable elements: The case of TMPO/LAP2α and ZNF451 in mammals. Bioinformatics 2015, 31, 2257–2261. [Google Scholar] [CrossRef] [PubMed]
- Roy-Engel, A.M.; El-Sawy, M.; Farooq, L.; Odom, G.L.; Perepelitsa-Belancio, V.; Bruch, H.; Oyeniran, O.O.; Deininger, P.L. Human retroelements may introduce intragenic polyadenylation signals. Cytogenet. Genome Res. 2005, 110, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, T.; Huang, S. 3′-UTR-located inverted Alu repeats facilitate mRNA translational repression and stress granule accumulation. Nucleus 2012, 3, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Teugels, E.; de Brakeleer, S.; Goelen, G.; Lissens, W.; Sermijn, E.; de Grève, J. De novo Alu element insertions targeted to a sequence common to the BRCA1 and BRCA2 genes. Hum. Mutat. 2005, 26, 284. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Vogelstein, B.; Horii, A.; Miyoshi, Y.; Nakamura, Y. Disruption of the APC Gene by a retrotransposal insertion of LI sequence in a colon cancer. Cancer Res. 1992, 52, 643–645. [Google Scholar]
- Rodríguez-Martín, C.; Cidre, F.; Fernández-Teijeiro, A.; Gómez-Mariano, G.; de La Vega, L.; Ramos, P.; Zaballos, A.; Monzón, S.; Alonso, J. Familial retinoblastoma due to intronic LINE-1 insertion causes aberrant and noncanonical mRNA splicing of the RB1 gene. J. Hum. Genet. 2016, 61, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Ade, C.; Roy-Engel, A.M.; Deininger, P.L. Alu elements: An intrinsic source of human genome instability. Curr. Opin. Virol. 2013, 3, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Wolff, E.M.; Byun, H.M.; Han, H.F.; Sharma, S.; Nichols, P.W.; Siegmund, K.D.; Yang, A.S.; Jones, P.A.; Liang, G. Hypomethylation of a LINE-1 promoter activates an alternate transcript of the MET oncogene in bladders with cancer. PLoS Genet. 2010, 6, e1000917. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, T.; Lee, W.; Obenauf, A.C.; Ran, L.; Murali, R.; Zhang, Q.F.; Wong, E.W.P.; Hu, W.; Scott, S.N.; Shah, R.H.; et al. Alternative transcription initiation leads to expression of a novel ALK isoform in cancer. Nature 2015, 526, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Lock, F.E.; Rebollo, R.; Miceli-Royer, K.; Gagnier, L.; Kuah, S.; Babaian, A.; Sistiaga-Poveda, M.; Lai, C.B.; Nemirovsky, O.; Serrano, I.; et al. Distinct isoform of FABP7 revealed by screening for retroelement-activated genes in diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2014, 111, E3534–E3543. [Google Scholar] [CrossRef] [PubMed]
- Scarfò, I.; Pellegrino, E.; Mereu, E.; Kwee, I.; Agnelli, L.; Bergaggio, E.; Garaffo, G.; Vitale, N.; Caputo, M.; Machiorlatti, R.; et al. Identification of a new subclass of ALK-negative ALCL expressing aberrant levels of ERBB4 transcripts. Blood 2016, 127, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, P.; Richardson, S.R.; Mager, D.L.; Faulkner, G.J. Transposable elements in the mammalian embryo: Pioneers surviving through stealth and service. Genome Biol. 2016, 17, 100. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Hong, C.; Zhang, B.; Lowdon, R.F.; Xing, X.; Li, D.; Zhou, X.; Lee, H.J.; Maire, C.L.; Ligon, K.L.; et al. DNA hypomethylation within specific transposable element families associates with tissue-specific enhancer landscape. Nat. Genet. 2013, 45, 836–841. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Paredes, M.; Esteller, M. Cancer epigenetics reaches mainstream oncology. Nat. Med. 2011, 17, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Molecular origins of cancer epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Mukamel, Z.; Tanay, A. Hypomethylation marks enhancers within transposable elements. Nat. Genet. 2013, 45, 717–718. [Google Scholar] [CrossRef] [PubMed]
- Hur, K.; Cejas, P.; Feliu, J.; Moreno-Rubio, J.; Burgos, E.; Boland, C.R.; Goel, A. Hypomethylation of long interspersed nuclear element-1 (LINE-1) leads to activation of proto-oncogenes in human colorectal cancer metastasis. Gut 2014, 63, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Swets, M.; Zaalberg, A.; Boot, A.; van Wezel, T.; Frouws, M.; Bastiaannet, E.; Gelderblom, H.; van de Velde, C.; Kuppen, P. Tumor LINE-1 methylation level in association with survival of patients with stage II colon cancer. Int. J. Mol. Sci. 2016, 18, 36. [Google Scholar] [CrossRef] [PubMed]
- Daskalos, A.; Nikolaidis, G.; Xinarianos, G.; Savvari, P.; Cassidy, A.; Zakopoulou, R.; Kotsinas, A.; Gorgoulis, V.; Field, J.K.; Liloglou, T. Hypomethylation of retrotransposable elements correlates with genomic instability in non-small cell lung cancer. Int. J. Cancer 2009, 124, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Nau, M.M.; Zucman-Rossi, J.; Powell, J.I.; Allegra, C.J.; Wright, J.J. LINE-1 element insertion at the t(11;22) translocation breakpoint of a desmoplastic small round cell tumor. Genes Chromosomes Cancer 1997, 18, 232–239. [Google Scholar] [CrossRef]
- Hedges, D.J.; Deininger, P.L. Inviting instability: Transposable elements, double-strand breaks, and the maintenance of genome integrity. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2007, 616, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Ayarpadikannan, S.; Kim, H.-S. The impact of transposable elements in genome evolution and genetic instability and their implications in various diseases. Genom. Inf. 2014, 12, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, R.K.; Martienssen, R. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 2007, 8, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Levin, H.L.; Moran, J.V. Dynamic interactions between transposable elements and their hosts. Nat. Rev. Genet. 2011, 12, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Baba, Y.; Ishimoto, T.; Chikamoto, A.; Kosumi, K.; Hayashi, H.; Nitta, H.; Hashimoto, D.; Beppu, T.; Baba, H. LINE-1 methylation level and patient prognosis in a database of 208 hepatocellular carcinomas. Ann. Surg. Oncol. 2014, 1, 1280–1287. [Google Scholar] [CrossRef] [PubMed]
- Terry, M.B.; Delgado-Cruzata, L.; Vin-Raviv, N.; Wu, H.C.; Santella, R.M. DNA methylation in white blood cells: Association with risk factors in epidemiologic studies. Epigenetics 2011, 6, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.L.; Krech, T.; Hasemeier, B.; Schipper, E.; Schweitzer, N.; Vogel, A.; Kreipe, H.; Lehmann, U. Loss of DNA methylation at imprinted loci is a frequent event in hepatocellular carcinoma and identifies patients with shortened survival. Clin. Epigenet. 2015, 7, 110. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Gammon, M.D.; Hernandez-Vargas, H.; Herceg, Z.; Wetmur, J.G.; Teitelbaum, S.L.; Bradshaw, P.T.; Neugut, A.I.; Santella, R.M.; Chen, J. DNA methylation in peripheral blood measured by LUMA is associated with breast cancer in a population-based study. FASEB J. 2012, 26, 2657–2666. [Google Scholar] [CrossRef] [PubMed]
- Kassiotis, G. Endogenous retroviruses and the development of cancer. J. Immunol. 2014, 192, 1343–1349. [Google Scholar] [CrossRef] [PubMed]
- Penke, T.J.R.; McKay, D.J.; Strahl, B.D.; Gregory Matera, A.; Duronio, R.J. Direct interrogation of the role of H3K9 in metazoan heterochromatin function. Genes Dev. 2016, 30, 1866–1880. [Google Scholar] [CrossRef] [PubMed]
- Fallis, A. An epigenetic switch ensures transponson repressioin upon dynamic loss of DNA methylation in embryonic stem cells. eLife 2013, 53, 1689–1699. [Google Scholar]
- Bulut-Karslioglu, A.; DeLaRosa-Velázquez, I.A.; Ramirez, F.; Barenboim, M.; Onishi-Seebacher, M.; Arand, J.; Galán, C.; Winter, G.E.; Engist, B.; Gerle, B.; et al. Suv39h-dependent H3K9me3 marks intact retrotransposons and silences LINE elements in mouse embryonic stem cells. Mol. Cell 2014, 55, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.-J.; Liu, Z.-P. Histone methylations in heart development, congenital and adult heart diseases. Epigenomics 2015, 7, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Rigal, M.; Mathieu, O. A “mille-feuille” of silencing: Epigenetic control of transposable elements. Biochim. Biophys. Acta Gene Regul. Mech. 2011, 1809, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.; Schwalie, P.C.; Wilson, M.D.; Ballester, B.; Gonalves, Â.; Kutter, C.; Brown, G.D.; Marshall, A.; Flicek, P.; Odom, D.T. Waves of retrotransposon expansion remodel genome organization and CTCF binding in multiple mammalian lineages. Cell 2012, 148, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Pugacheva, E.M.; Teplyakov, E.; Wu, Q.; Li, J.; Chen, C.; Meng, C.; Liu, J.; Robinson, S.; Loukinov, D.; Boukaba, A.; et al. The cancer-associated CTCFL/BORIS protein targets multiple classes of genomic repeats, with a distinct binding and functional preference for humanoid-specific SVA transposable elements. Epigenet. Chromatin 2016, 9, 35. [Google Scholar] [CrossRef] [PubMed]
- Gifford, W.D.; Pfaff, S.L.; MacFarlan, T.S. Transposable elements as genetic regulatory substrates in early development. Trends Cell Biol. 2013, 23, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, A.; Jene-Sanz, A.; Lopez-Bigas, N. The mutational landscape of chromatin regulatory factors across 4623 tumor samples. Genome Biol. 2013, 14, r106. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.A.; Denton, E.L.; Arrowsmith, C.H.; Lupien, M.; Schapira, M. A global assessment of cancer genomic alterations in epigenetic mechanisms. Epigenet. Chromatin 2014, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Hur, S.K.; Park, E.J.; Han, J.E.; Kim, Y.A.; Kim, J.D.; Kang, D.; Kwon, J. Roles of human INO80 chromatin remodeling enzyme in DNA replication and chromosome segregation suppress genome instability. Cell. Mol. Life Sci. 2010, 67, 2283–2296. [Google Scholar] [CrossRef] [PubMed]
- Papamichos-Chronakis, M.; Peterson, C.L. Chromatin and the genome integrity network. Nat. Rev. Genet. 2013, 14, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Lev-Maor, G.; Sorek, R.; Shomron, N.; Ast, G. The birth of an alternatively spliced exon: 3’ Splice-site selection in Alu exons. Science 2003, 300, 1288–1291. [Google Scholar] [CrossRef] [PubMed]
- Lev-Maor, G.; Ram, O.; Kim, E.; Sela, N.; Goren, A.; Levanon, E.Y.; Ast, G. Intronic Alus influence alternative splicing. PLoS Genet. 2008, 4, e1000204. [Google Scholar] [CrossRef] [PubMed]
- Sela, N.; Mersch, B.; Hotz-Wagenblatt, A.; Ast, G. Characteristics of transposable element exonization within human and mouse. PLoS ONE 2010, 5, e10907. [Google Scholar] [CrossRef] [PubMed]
- Sorek, R.; Ast, G.; Graur, D. Alu-containing exons are alternatively spliced. Genome Res. 2002, 12, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.H.; Allton, K.; Duncan, S.A.; Shen, L.; Barton, M.C. Foxa1 functions as a pioneer transcription factor at transposable elements to activate Afp during differentiation of embryonic stem cells. J. Biol. Chem. 2010, 285, 16135–16144. [Google Scholar] [CrossRef] [PubMed]
- Javier Piedrafita, F.; Molander, R.B.; Vansant, G.; Orlova, E.A.; Pfahl, M.; Reynolds, W.F. An Alu element in the myeloperoxidase promoter contains a composite SP1-thyroid hormone-retinoic acid response element. J. Biol. Chem. 1996, 271, 14412–14420. [Google Scholar]
- Hambor, J.E.; Mennone, J.; Coon, M.E.; Hanke, J.H.; Kavathas, P. Identification and characterization of an Alu-containing, T-cell-specific enhancer located in the last intron of the human CD8α gene. Mol. Cell. Biol. 1993, 13, 7056–7070. [Google Scholar] [CrossRef] [PubMed]
- Cui, F.; Sirotin, M.V.; Zhurkin, V.B. Impact of Alu repeats on the evolution of human p53 binding sites. Biol. Direct 2011, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Vansant, G.; Reynolds, W.F. The consensus sequence of a major Alu subfamily contains a functional retinoic acid response element. Proc. Natl. Acad. Sci. USA 1995, 92, 8229–8233. [Google Scholar] [CrossRef] [PubMed]
- Menendez, D.; Inga, A.; Resnick, M.A. The expanding universe of p53 targets. Nat. Rev. Cancer 2009, 9, 724–737. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.B.; Wang, H.Y.; Lu, H.Y.; Xiong, J.; Li, H.H.; Qiu, X.H.; Liu, H.Q. Increased level of polymerase III transcribed Alu RNA in hepatocellular carcinoma tissue. Mol. Carcinog. 2005, 42, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Wylie, A.; Jones, A.E.; D’Brot, A.; Lu, W.J.; Kurtz, P.; Moran, J.V.; Rakheja, D.; Chen, K.S.; Hammer, R.E.; Comerford, S.A.; et al. p53 genes function to restrain mobile elements. Genes Dev. 2016, 30, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, V.; Cheng, Y.; Ma, Z.; Li, D.; Xing, X.; Edge, P.; Snyder, M.P.; Wang, T. Widespread contribution of transposable elements to the innovation of gene regulatory networks. Genome Res. 2014, 24, 1963–1976. [Google Scholar] [CrossRef] [PubMed]
- Van Bortle, K.; Corces, V.G. The role of chromatin insulators in nuclear architecture and genome function. Curr. Opin. Genet. Dev. 2013, 23, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Rebollo, R.; Farivar, S.; Mager, D.L. C-GATE—Catalogue of genes affected by transposable elements. Mob. DNA 2012, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Valles, I.; Pajares, M.J.; Segura, V.; Guruceaga, E.; Gomez-Roman, J.; Blanco, D.; Tamura, A.; Montuenga, L.M.; Pio, R. Identification of novel deregulated RNA metabolism-related genes in non-small cell lung cancer. PLoS ONE 2012, 7, e42086. [Google Scholar] [CrossRef] [PubMed]
- Paz, N.; Levanon, E.Y.; Amariglio, N.; Heimberger, A.B.; Ram, Z.; Constantini, S.; Barbash, Z.S.; Adamsky, K.; Safran, M.; Hirschberg, A.; et al. Altered adenosine-to-inosine RNA editing in human cancer Altered adenosine-to-inosine RNA editing in human cancer. Genome Res. 2007, 17, 1586–1595. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Chang, J.W.O.; Park, J.K.U.; Hwang, S.G. Increased aldehyde reductase expression mediates acquired radioresistance of laryngeal cancer cells via modulating p53. Cancer Biol. Ther. 2012, 13, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Wang, M.; Zhong, D.; Tong, N.; Chu, H.; Sheng, X.; Zhang, Z. ADH1C Ile350Val polymorphism and cancer risk: Evidence from 35 case-control studies. PLoS ONE 2012, 7, e37227. [Google Scholar] [CrossRef] [PubMed]
- Zamanian-Daryoush, M.; di Donato, J.A. Apolipoprotein A–I and cancer. Front. Pharmacol. 2015, 6, 265. [Google Scholar] [CrossRef] [PubMed]
- Flor, I.; Bullerdiek, J. The dark side of a success story: MicroRNAs of the C19MC cluster in human tumours. J. Pathol. 2012, 227, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Bekku, S.; Mochizuki, H.; Yamamoto, T.; Ueno, H.; Takayama, E.; Tadakuma, T. Expression of carbonic anhydrase I or II and correlation to clinical aspects of colorectal cancer. Hepatogastroenterology 2000, 47, 998–1001. [Google Scholar] [PubMed]
- Dorr, C.; Janik, C.; Weg, M.; Been, R.A.; Bader, J.; Kang, R.; Ng, B.; Foran, L.; Landman, S.R.; O’Sullivan, M.G.; et al. Transposon mutagenesis screen identifies potential lung cancer drivers and CUL3 as a tumor suppressor. Mol. Cancer Res. 2015, 13, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Rhee, I.; Jair, K.W.; Yen, R.W.; Lengauer, C.; Herman, J.G.; Kinzler, K.W.; Vogelstein, B.; Baylin, S.B.; Schuebel, K.E. CpG methylation is maintained in human cancer cells lacking DNMT1. Nature 2000, 404, 1003–1007. [Google Scholar] [PubMed]
- McAtee, C.O.; Barycki, J.J.; Simpson, M.A. Emerging roles for hyaluronidase in cancer metastasis and therapy. Adv. Cancer Res. 2014, 123, 1–34. [Google Scholar] [PubMed]
- Zhang, Y.; Romanish, M.T.; Mager, D.L. Distributions of transposable elements reveal hazardous zones in mammalian introns. PLoS Comput. Biol. 2011, 7, e1002046. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Takimoto, K. Selective expression of HERG and Kv2 channels influences proliferation of uterine cancer cells. Int. J. Oncol. 2004, 25, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Romanish, M.T.; Nakamura, H.; Lai, B.C.; Wang, Y.; Mager, D.L. A novel protein isoform of the multicopy human NAIP gene derives from intragenic Alu SINE promoters. PLoS ONE 2009, 4, e5761. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.L.; Krech, T.; Hasemeier, B.; Schipper, E.; Schweitzer, N.; Vogel, A.; Kreipe, H.; Lehmann, U. Deregulation of RB1 expression by loss of imprinting in human hepatocellular carcinoma. J. Pathol. 2014, 233, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, E.; Sugioka-Sugiyama, R.; Mizuguchi, T.; Mehta, S.; Cui, B.; Grewal, S.I.S. A homolog of male sex-determining factor SRY cooperates with a transposon-derived CENP-B protein to control sex-specific directed recombination. Proc. Natl. Acad. Sci. USA 2011, 108, 18754–18759. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa-Watanabe, Y.; Inoue, J.-I.; Semba, K. Transcriptional activity of testis-determining factor SRY is modulated by the Wilms’ tumor 1 gene product, WT1. Oncogene 2003, 22, 7900–7904. [Google Scholar] [CrossRef] [PubMed]
- Pelkonen, M.; Luostari, K.; Tengstrom, M.; Ahonen, H.; Berdel, B.; Kataja, V.; Soini, Y.; Kosma, V.-M.; Mannermaa, A. Low expression levels of hepsin and TMPRSS3 are associated with poor breast cancer survival. BMC Cancer 2015, 15, 431. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, K.; Wang, Z.; Bachvarova, M.; Gregoire, J.; Renaud, M.C.; Plante, M.; Bachvarov, D. A novel genome-based approach correlates TMPRSS3 overexpression in ovarian cancer with DNA hypomethylation. Gynecol. Oncol. 2012, 125, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Cappadocia, L.; Pichler, A.; Lima, C.D. Structural basis for catalytic activation by the human ZNF451 SUMO E3 ligase. Nat. Struct. Mol. Biol. 2015, 22, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Rebollo, R.; Karimi, M.M.; Bilenky, M.; Gagnier, L.; Miceli-Royer, K.; Zhang, Y.; Goyal, P.; Keane, T.M.; Jones, S.; Hirst, M.; et al. Retrotransposon-induced heterochromatin spreading in the mouse revealed by insertional polymorphisms. PLoS Genet. 2011, 7, e1002301. [Google Scholar] [CrossRef] [PubMed]
- Mateo, L.; Ullastres, A.; González, J. A Transposable element insertion confers xenobiotic resistance in Drosophila. PLoS Genet. 2014, 10, e1004560. [Google Scholar] [CrossRef] [PubMed]
- Wallace, M.R.; Andersen, L.B.; Saulino, A.M.; Gregory, P.E.; Glover, T.W.; Collins, F.S. A de novo Alu insertion results in neurofibromatosis type. Nature 1991, 353, 864–866. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.M.; Stenson, P.D.; Cooper, D.N.; Férec, C. A systematic analysis of LINE-1 endonuclease-dependent retrotranspositional events causing human genetic disease. Hum. Genet. 2005, 117, 411–427. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Edwards, A.; Fan, W.; Deininger, P.; Zhang, K. Alu distribution and mutation types of cancer genes. BMC Genom. 2011, 12, 157. [Google Scholar] [CrossRef] [PubMed]
- Voineagu, I.; Narayanan, V.; Lobachev, K.S.; Mirkin, S.M. Replication stalling at unstable inverted repeats: Interplay between DNA hairpins and fork stabilizing proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 9936–9941. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Wang, G.; Bacolla, A.; Zhao, J.; Spitser, S.; Vasquez, K.M. Short inverted repeats are hotspots for genetic instability: Relevance to cancer genomes. Cell Rep. 2015, 10, 1674–1680. [Google Scholar] [CrossRef] [PubMed]
- Lobachev, K.S.; Stenger, J.E.; Kozyreva, O.G.; Jurka, J.; Gordenin, D.A.; Resnick, M.A. Inverted Alu repeats unstable in yeast are excluded from the human genome. EMBO J. 2000, 19, 3822–3830. [Google Scholar] [CrossRef] [PubMed]
- Cordaux, R.; Hedges, D.J.; Herke, S.W.; Batzer, M.A. Estimating the retrotransposition rate of human Alu elements. Gene 2006, 373, 134–137. [Google Scholar] [CrossRef] [PubMed]
- De Biase, I.; Rasmussen, A.; Monticelli, A.; Al-Mahdawi, S.; Pook, M.; Cocozza, S.; Bidichandani, S.I. Somatic instability of the expanded GAA triplet-repeat sequence in Friedreich ataxia progresses throughout life. Genomics 2007, 90, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Conti, A.; Carnevali, D.; Bollati, V.; Fustinoni, S.; Pellegrini, M.; Dieci, G. Identification of RNA polymerase III-transcribed Alu loci by computational screening of RNA-Seq data. Nucleic Acids Res. 2015, 43, 817–835. [Google Scholar] [CrossRef] [PubMed]
- Valeri, N.; Gasparini, P.; Fabbri, M.; Braconi, C.; Veronese, A.; Lovat, F.; Adair, B.; Vannini, I.; Fanini, F.; Bottoni, A.; et al. Modulation of mismatch repair and genomic stability by miR-155. Proc. Natl. Acad. Sci. USA 2010, 107, 6982–6987. [Google Scholar] [CrossRef] [PubMed]
- Lissens, W.; Teugels, E.; Brakeleer, S.D.; Gr, J.D. Systematic detection of pathogenic Alu element insertions in NGS-based diagnostic screens: The BRCA1/BRCA2 example. Hum. Mutat. 2013, 34, 785–791. [Google Scholar]
- Halling, K.C.; Lazzaro, C.R.; Honchel, R.; Bufill, J.A.; Powell, S.M.; Arndt, C.A.; Lindor, N.M. Hereditary desmoid disease in a family with a germline Alu I repeat mutation of the APC gene. Hum. Hered. 1999, 49, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Belgnaoui, S.M.; Gosden, R.G.; Semmes, O.J.; Haoudi, A. Human LINE-1 retrotransposon induces DNA damage and apoptosis in cancer cells. Cancer Cell Int. 2006, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Gasior, S.L.; Wakeman, T.P.; Xu, B.; Deininger, P.L. The human LINE-1 retrotransposon creates DNA double-strand breaks. J. Mol. Biol. 2006, 357, 1383–1393. [Google Scholar] [CrossRef] [PubMed]
- Hoang, M.L.; Tan, F.J.; Lai, D.C.; Celniker, S.E.; Hoskins, R.A.; Dunham, M.J.; Zheng, Y.; Koshland, D. Competitive repair by naturally dispersed repetitive DNA during non-allelic homologous recombination. PLoS Genet. 2010, 6, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Luzhna, L.; Ilnytskyy, Y.; Kovalchuk, O. Mobilization of LINE-1 in irradiated mammary gland tissue may potentially contribute to low dose radiation-induced genomic instability. Genes Cancer 2015, 6, 71–81. [Google Scholar] [PubMed]
- Walsh, T.; Casadei, S.; Coats, K.H.; Swisher, E.; Stray, S.M.; Higgins, J.; Roach, K.C.; Mandell, J.; Lee, M.K.; Ciernikova, S.; et al. Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. JAMA 2006, 295, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, I.; Salomon, D.; Erne, B.; Schaeren-Wiemers, N.; Peles, E. Caspr3 and caspr4, two novel members of the caspr family are expressed in the nervous system and interact with PDZ domains. Mol. Cell. Neurosci. 2002, 20, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Cybulski, C.; Wokołorczyk, D.; Huzarski, T.; Byrski, T.; Gronwald, J.; Górski, B.; Debniak, T.; Masojć, B.; Jakubowska, A.; Gliniewicz, B.; et al. A large germline deletion in the Chek2 kinase gene is associated with an increased risk of prostate cancer. J. Med. Genet. 2006, 43, 863–866. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.; Reynolds, L.E.; Robinson, S.D.; Lees, D.M.; Parsons, M.; Elia, G.; Hodivala-Dilke, K. Stromal claudin14-heterozygosity, but not deletion, increases tumour blood leakage without affecting tumour growth. PLoS ONE 2013, 8, e62516. [Google Scholar] [CrossRef] [PubMed]
- Knower, K.C.; To, S.Q.; Simpson, E.R.; Clyne, C.D. Epigenetic mechanisms regulating CYP19 transcription in human breast adipose fibroblasts. Mol. Cell. Endocrinol. 2010, 321, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Bonnefoy, N.; Bastid, J.; Alberici, G.; Bensussan, A.; Eliaou, J.-F. CD39: A complementary target to immune checkpoints to counteract tumor-mediated immunosuppression. Oncoimmunology 2015, 4, e1003015. [Google Scholar] [CrossRef] [PubMed]
- Padró, M.; Cobler, L.; Garrido, M.; De Bolós, C. Down-regulation of FUT3 and FUT5 by shRNA alters Lewis antigens expression and reduces the adhesion capacities of gastric cancer cells. Biochim. Biophys. Acta Gen. Subj. 2011, 1810, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Setiawan, V.W.; Hankinson, S.E.; Colditz, G.A.; Hunter, D.J.; De Vivo, I. HSD17B1 gene polymorphisms and risk of endometrial and breast cancer. Cancer Epidemiol. Biomark. Prev. 2004, 13, 213–219. [Google Scholar] [CrossRef]
- Ihle, J.N.; Smith-White, B.; Sisson, B.; Parker, D.; Blair, D.G.; Schultz, A.; Kozak, C.; Lunsford, R.D.; Askew, D.; Weinstein, Y.; et al. Activation of the c-H-ras proto-oncogene by retrovirus insertion and chromosomal rearrangement in a Moloney leukemia virus-induced T-cell leukemia. J. Virol. 1989, 63, 2959–2966. [Google Scholar] [PubMed]
- Li, L.; McVety, S.; Younan, R.; Liang, P.; Du Sart, D.; Gordon, P.H.; Hutter, P.; Hogervorst, F.B.L.; Chong, G.; Foulkes, W.D. Distinct patterns of germ-line deletions in MLH1 and MSH2: The implication of Alu repetitive element in the genetic etiology of Lynch syndrome (HNPCC). Hum. Mutat. 2006, 27, 388. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Yamazaki, T. Nucleic acids research. Nucleic Acids Res. 1989, 17, 1–2. [Google Scholar]
- Katsanis, N.; Beales, P.L.; Woods, M.O.; Lewis, R.A.; Green, J.S.; Parfrey, P.S.; Ansley, S.J.; Davidson, W.S.; Lupski, J.R. Mutations in MKKS cause obesity, retinal dystrophy and renal malformations associated with Bardet-Biedl syndrome. Nat. Genet. 2000, 26, 67–70. [Google Scholar] [PubMed]
- Morse, B.; Rotherg, P.G.; South, V.J.; Spandorfer, J.M.; Astrin, S.M. Insertional mutagenesis of the myc locus by a LINE-1 sequence in a human breast carcinoma. Nature 1988, 333, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Pastan, I.; Hassan, R. Discovery of mesothelin and exploiting it as a target for immunotherapy. Cancer Res. 2014, 74, 2907–2912. [Google Scholar] [CrossRef] [PubMed]
- Rodić, N.; Zampella, J.G.; Cornish, T.C.; Wheelan, S.J.; Burns, K.H. Translocation junctions in TCF3-PBX1 acute lymphoblastic leukemia/lymphoma cluster near transposable elements. Mob. DNA 2013, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; He, Z.; Zhou, K.; Cheng, J.; Yao, H.; Lu, D.; Cai, R.; Jin, Y.; Dong, B.; Xu, Y.; Wang, Y. Essential role of trpc6 channels in G2/M phase transition and development of human glioma. J. Natl. Cancer Inst. 2010, 102, 1052–1068. [Google Scholar] [CrossRef] [PubMed]
- Elliott, B.; Richardson, C.; Jasin, M. Chromosomal translocation mechanisms at intronic Alu elements in mammalian cells. Mol. Cell 2005, 17, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Beck, C.R.; Collier, P.; Macfarlane, C.; Malig, M.; Kidd, J.M.; Eichler, E.E.; Badge, R.M.; Moran, J.V. LINE-1 retrotransposition activity in human genomes. Cell 2010, 141, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, S.-Y.; Chen, W.-Y.; Yeh, T.-S.; Sheen, I.-S.; Huang, S.-F. High-frequency Alu-mediated genomic recombination/deletion within the caspase-activated DNase gene in human hepatoma. Oncogene 2005, 24, 6584–6589. [Google Scholar] [CrossRef] [PubMed]
- Morrish, T.A.; Gilbert, N.; Myers, J.S.; Vincent, B.J.; Stamato, T.D.; Taccioli, G.E.; Batzer, M.A.; Moran, J.V. DNA repair mediated by endonuclease-independent LINE-1 retrotransposition. Nat. Genet. 2002, 31, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.A.; Liu, G.; Eichler, E.E. An Alu transposition model for the origin and expansion of human segmental duplications. Am. J. Hum. Genet. 2003, 73, 823–834. [Google Scholar] [CrossRef] [PubMed]
- Alkan, C.; Kidd, J.M.; Marques-Bonet, T.; Aksay, G.; Antonacci, F.; Hormozdiari, F.; Kitzman, J.O.; Baker, C.; Malig, M.; Mutlu, O.; et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat. Genet. 2009, 41, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Jeffs, A.R.; Benjes, S.M.; Smith, T.L.; Sowerby, S.J.; Morris, C.M. The BCR gene recombines preferentially with Alu elements in complex BCR-ABL translocations of chronic myeloid leukaemia. Hum. Mol. Genet. 1998, 7, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Montagna, M.; Santacatterina, M.; Torri, A.; Menin, C.; Zullato, D.; Chieco-Bianchi, L.; D’andrea, E. Identification of a 3 kb Alu-mediated BRCA1 gene rearrangement in two breast/ovarian cancer families. Oncogene 1999, 18, 4160–4165. [Google Scholar] [CrossRef] [PubMed]
- Sluiter, M.D.; van Rensburg, E.J. Large genomic rearrangements of the BRCA1 and BRCA2 genes: Review of the literature and report of a novel BRCA1 mutation. Breast Cancer Res. Treat. 2011, 125, 325–349. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.; Senz, J.; Kaurah, P.; Pinheiro, H.; Sanges, R.; Haegert, A.; Corso, G.; Schouten, J.; Fitzgerald, R.; Vogelsang, H.; et al. Germline CDH1 deletions in hereditary diffuse gastric cancer families. Hum. Mol. Genet. 2009, 18, 1545–1555. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhang, X.; Liu, T.; Liu, X.; Geng, J.; He, X.; Liu, Y.; Pang, D. Increased expression of mitotic arrest deficient-like 1 (MAD1L1) is associated with poor prognosis and insensitive to taxol treatment in breast cancer. Breast Cancer Res. Treat. 2013, 140, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Schichman, S.A.; Caligiuri, M.A.; Strout, M.P.; Carter, S.L.; Gu, Y.; Canaani, E.; Bloomfield, C.D.; Croce, C.M. ALL-1 Tandem duplication in acute myeloid leukemia with a normal karyotype involves homologous recombination between Alu elements. Cancer Res. 1994, 54, 4277–4280. [Google Scholar] [PubMed]
- O’Neil, J.; Tchinda, J.; Gutierrez, A.; Moreau, L.; Maser, R.S.; Wong, K.-K.; Li, W.; McKenna, K.; Liu, X.S.; Feng, B.; et al. Alu elements mediate MYB gene tandem duplication in human T-ALL. J. Exp. Med. 2007, 204, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Franke, G.; Bausch, B.; Hoffmann, M.M.; Cybulla, M.; Wilhelm, C.; Kohlhase, J.; Scherer, G.; Neumann, H.P.H. Alu-Alu recombination underlies the vast majority of large VHL germline deletions: Molecular characterization and genotype-phenotype correlations in VHL patients. Hum. Mutat. 2009, 30, 776–786. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Lin, L.; Cai, J.J.; Jiang, P.; Kenkel, E.J.; Stroik, M.R.; Sato, S.; Davidson, B.L.; Xing, Y. Widespread establishment and regulatory impact of Alu exons in human genes. Proc. Natl. Acad. Sci. USA 2011, 108, 2837–2842. [Google Scholar] [CrossRef] [PubMed]
- Strout, M.P.; Marcucci, G.; Bloomfield, C.D.; Caligiuri, M.A. The partial tandem duplication of ALL1 (MLL) is consistently generated by Alu-mediated homologous recombination in acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 1998, 95, 2390–2395. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Zhao, H.; Yi, Y.; Nakata, Y.; Kalota, A.; Gewirtz, A.M. c-Myb binds MLL through menin in human leukemia cells and is an important driver of MLL-associated leukemogenesis. J. Clin. Investig. 2010, 120, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.V.; Croce, C.M. MicroRNA dysregulation in cancer: Diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol. Med. 2012, 4, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Croce, C.M. Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet. 2009, 10, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Smalheiser, N.R.; Torvik, V.I. Mammalian microRNAs derived from genomic repeats. Trends Genet. 2005, 21, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Piriyapongsa, J.; Mariño-Ramírez, L.; Jordan, I.K. Origin and evolution of human microRNAs from transposable elements. Genetics 2007, 176, 1323–1337. [Google Scholar] [CrossRef] [PubMed]
- Borchert, G.M.; Holton, N.W.; Williams, J.D.; Hernan, W.L.; Bishop, I.P.; Dembosky, J.A.; Elste, J.E.; Gregoire, N.S.; Kim, J.-A.; Koehler, W.W.; et al. Comprehensive analysis of microRNA genomic loci identifies pervasive repetitive-element origins. Mob. Genet. Elements 2011, 1, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.T.; Cooper, E.A.; Favreau, C.J.; Howell, J.S.; Lane, L.G.; Mills, J.E.; Newman, D.C.; Perry, T.J.; Russell, M.E.; Wallace, B.M.; et al. Continuing analysis of microRNA origins: Formation from transposable element insertions and noncoding RNA mutations. Mob. Genet. Elements 2013, 3, e27755. [Google Scholar] [CrossRef] [PubMed]
- Piriyapongsa, J.; Jordan, I.K. A family of human microRNA genes from miniature inverted-repeat transposable elements. PLoS ONE 2007, 2, e203. [Google Scholar] [CrossRef] [PubMed]
- Spizzo, R.; Almeida, M.I.; Colombatti, A.; Calin, G.A. Long non-coding RNAs and cancer: A new frontier of translational research? Oncogene 2012, 31, 4577–4587. [Google Scholar] [CrossRef] [PubMed]
- Dhamija, S.; Diederichs, S. From junk to master regulators of invasion: LncRNA functions in migration, EMT and metastasis. Int. J. Cancer 2016, 139, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Maquat, L.E. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature 2011, 470, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.R.; Rinn, J.L. Transposable elements reveal a stem cell specific class of long noncoding RNAs. Genome Biol. 2012, 13, R107. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.O.; Wang, H.B.; Zhang, Y.; Lu, X.; Chen, L.L.; Yang, L. Complementary sequence-mediated exon circularization. Cell 2014, 159, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Hadjiargyrou, M.; Delihas, N. The intertwining of transposable elements and non-coding RNAs. Int. J. Mol. Sci. 2013, 14, 13307–13328. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.L.; Krech, T.; Hasemeier, B.; Schipper, E.; Schweitzer, N.; Vogel, A.; Kreipe, H.; Lehmann, U. Loss of imprinting and allelic switching at the DLK1-MEG3 locus in human hepatocellular carcinoma. PLoS ONE 2012, 7, e49462. [Google Scholar] [CrossRef] [PubMed]
- Carrieri, C.; Cimatti, L.; Biagioli, M.; Beugnet, A.; Zucchelli, S.; Fedele, S.; Pesce, E.; Ferrer, I.; Collavin, L.; Santoro, C.; et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature 2012, 491, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Khanduja, J.S.; Calvo, I.A.; Joh, R.I.; Hill, I.T.; Motamedi, M. Nuclear noncoding RNAs and genome stability. Mol. Cell 2016, 63, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Villeneuve, L.; Morris, K.; Rossi, J. Argonaute-1 directs siRNA-mediated transcriptional gene silencing in human cells. Nat. Struct. Mol. Biol. 2006, 13, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.V.; Chan, S.W.-L.; Jacobsen, S.E.; Looney, D.J. Small interfering RNA-induced transcriptional gene silencing in human cells. Science 2004, 305, 1289–1292. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, Y.; Pilpel, Y.; Oren, M. MicroRNAs and Alu elements in the p53–Mdm2–Mdm4 regulatory network. J. Mol. Cell Biol. 2014, 6, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Elkahloun, A.G.; Candotti, F.; Grajkowski, A.; Beaucage, S.L.; Petricoin, E.F.; Calvert, V.; Juhl, H.; Mills, F.; Mason, K.; et al. A novel function of RNAs arising from the long terminal repeat of human endogenous retrovirus 9 in cell cycle arrest. J. Virol. 2013, 87, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Ponicsan, S.L.; Kugel, J.F.; Goodrich, J.A. Genomic gems: SINE RNAs regulate mRNA production. Curr. Opin. Genet. Dev. 2010, 20, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, B.; Lopez, M.F.; Wang, J.; Krastins, B.; Sarracino, D.; Tollervey, J.R.; Dobke, M.; Jordan, I.K.; Lunyak, V.V. Protein interactions with piALU RNA indicates putative participation of retroRNA in the cell cycle, DNA repair and chromatin assembly. Mob. Genet. Elements 2012, 2, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Bamezai, S.; Rawat, V.P.S.; Buske, C. Concise review: The PIWI–piRNA axis: Pivotal beyond transposon silencing. Stem Cells 2012, 30, 2603–2611. [Google Scholar] [CrossRef] [PubMed]
- Kim, V.N.; Han, J.; Siomi, M.C. Biogenesis of small RNAs in animals. Nat. Rev. Mol. Cell Biol. 2009, 10, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Le Thomas, A.; Rogers, A.K.; Webster, A.; Marinov, G.K.; Liao, S.E.; Perkins, E.M.; Hur, J.K.; Aravin, A.A.; Tóth, K.F. Piwi induces piRNA-guided transcriptional silencing and establishment of a repressive chromatin state. Genes Dev. 2013, 27, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Aravin, A.A.; Hannon, G.J.; Brennecke, J. The Piwi-piRNA pathway provides an adaptive defense in the transposon arms race. Science 2007, 318, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Simonelig, M. Developmental functions of piRNAs and transposable elements: A Drosophila point-of-view. RNA Biol. 2011, 8, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Malone, C.D.; Hannon, G.J. Small RNAs as guardians of the genome. Cell 2009, 136, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Tomizawa, S.; Mitsuya, K.; Totoki, Y.; Yamamoto, Y.; Kuramochi-Miyagawa, S.; Iida, N.; Hoki, Y.; Murphy, P.J.; Toyoda, A.; et al. Role for piRNAs and noncoding RNA in de novo DNA methylation of the imprinted mouse Rasgrf1 locus. Science 2011, 332, 848–852. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus or Genes | TE(s) | Mechanisms | Associated Cancers | References |
|---|---|---|---|---|
| ADARB1 | Alu | Alternative splicing | Lung, brain cancer | [65,66,67] |
| AKR1A1 | Alu | Alternative splicing | Head and neck cancer | [65,68] |
| ALS2CR8 | LINE1 | silencing | Colorectal cancer | [2] |
| ANKS1B_ | Alu | Deletion, silencing | Colorectal cancer | [2] |
| ANO9 | LINE1 | Alternative splicing | Colorectal cancer | [2] |
| ADH1C | LTR | Primary promoter | Cancers | [65,69] |
| ALK | LINE1, LTR | Alternative promoter | Melanoma, cancers | [17] |
| APOA | LINE1 | Enhancer | Cancers | [65,70] |
| APOC | LTR | Alternative promoter | Gastric cancer | [65] |
| ARNT | Alu | Alternative splicing | Lung cancer, metastasis of different cancers | [65] |
| ARHGEF12 | LINE1 | Alternative splicing | Ovarian cancer | [2] |
| ASMT | LINE1 | Alternative splicing | Glioma | [65] |
| B3GALNT2 | Alu | Alternative splicing | Breast cancer | [65] |
| B3GALT5 | Alu | Alternative promoter | Breast cancer | [65] |
| BAAT | LTR | Primary promoter | Lung cancer | [65] |
| BAHCC1 | Alu | Alternative splicing | Colorectal cancer | [65] |
| BBS7 | LINE1 | Primary promoter | Prostate cancer | [2] |
| BLVRA | Alu | Alternative splicing | Breast cancer | [65] |
| C19MC miRNA | Alu | POL III promoter | Hepatocellular, human cancers | [35,65,71] |
| CA1 | LTR | Primary promoter | Colorectal cancer | [8,72] |
| CASPR4 | LTR | Alternative promoter | Breast cancer, nasopharyngeal cancer | [65] |
| CD8A | LTR | Enhancer | Colorectal, pancreatic cancer | [65] |
| CDH12, CDH20 | LINE1 | Alternative splicing | Colorectal cancer | [2] |
| CHRM3 | LINE1 | Alternative promoter | Endometrial cancer | [65] |
| CHRNA1 | Alu | Alternative splicing | Testicular cancer | [65] |
| CLDN14 | LTR | Alternative promoter | Hepatocellular carcinoma | [65] |
| COL11A1, COL9A1 | LINE1 | Alternative splicing | Colorectal, prostate cancer | [2] |
| COX7B2 | LINE1 | Alternative splicing | Prostate cancer | [2] |
| CTNNA2 | LINE1 | Alternative splicing | Colorectal cancer | [2] |
| CWF19L1 | Alu | Alternative splicing | Breast cancer | [65] |
| CYB561D1 | Alu | Alternative splicing | Lung cancer | [65] |
| CYP19A1 | LTR | Alternative promoter | Breast cancer | [65] |
| CUL3 | LTR | Alternative splicing | Lung cancer | [73] |
| DAPK1 | ERV1 | Alternative splicing | Multiple myeloma | [2] |
| DBC1 | LINE1 | Deletion, Alternative promoter | Colorectal cancer | [2] |
| DHRS2 | LTR | Alternative promoter | Breast cancer | [65] |
| DNMT1 | Alu | Alternative splicing | Different cancers | [65,74] |
| DSCR4, DSCR8 | LTR | Primary promoter | Colorectal cancer | [65] |
| EPHA6 | LINE1 | Alternative transcript | Colorectal cancer | [2] |
| ERBB4 | LTR | Alternative promoter | Lymphoma, colorectal cancer | [2,19] |
| EBR | LTR | Alternative promoter | Bladder cancer | [65] |
| FABP7 | LTR | Alternative promoter | Lymphoma | [18] |
| FLT4/VEGFR3 | LTR | Alternative splicing | Different cancers | [65] |
| FMO1 | LINE1 | Silencer | Lung cancer | [65] |
| FOXP2 | LINE1 | Alternative promoter | Ovarian cancer | [2] |
| FUT5 | LINE1, Alu | Alternative splicing | Colorectal cancer | [65] |
| GABRG3 | LINE1 | Alternative splicing | Colorectal cancer | [2] |
| GBP5 | LTR | Primary promoter | Breast cancer | [65] |
| HHLA2, HHLA3 | LTR | Polyadenylation signal | Bladder cancer | [65] |
| HINFP/ZNF743 | Alu | Alternative splicing | Lung cancer | [65] |
| HYAL-4 | LINE1, Alu | Primary promoter | Cancers | [8,75] |
| IFNγ | Alu | Binding sites | Cancers | [65] |
| KCNH6 | Alu, LTR | Alternative splicing | Endometrial cancer | [76,77] |
| KDR | LINE1 | Alternative promoter | Colorectal cancer | [2] |
| MCTP2 | LINE1 | Deletion, Alternative promoter | Colorectal cancer | [2] |
| MET | LINE1 | Alternative splicing | Bladder cancer | [16] |
| MKKS | LTR | Alternative promoter | Colorectal cancer | [65] |
| MSLN | LTR | Primary promoter | Pancreatic cancer | [65] |
| NAIP | Alu | Alternative promoter | Cancer | [78] |
| NFKBID | Alu | Alternative splicing | Colorectal cancer | [65] |
| NOS3, NOSIP | LR | Alternative promoter | Different cancers | [65] |
| NPAS3 | LINE1 | Alternative promoter | Colorectal cancer | [2] |
| NRXN3 | LINE1 | Alternative splicing | Colorectal cancer | [2] |
| RB1 | LTR | Alternative promoter | Hepatocellular carcinoma, retinoblastoma | [14,79] |
| ROBO2 | LINE1 | Alternative splicing | Colorectal cancer | [2] |
| SLCC44A5, SLC35F1 | LINE1 | Alternative promoter, slencing | Colorectal cancer | [2] |
| SRY | LTR | Alternative transcript | Wilm’s tumor | [80,81] |
| STXBP5L | LINE1 | Alternative promoter | Colorectal cancer | [2] |
| TMED7 | Alu | Alternative promoter | Colorectal cancer | [2] |
| TMEM16J, TMEM56 | Alu | Alternative splicing, silencing | Colorectal cancer | [2] |
| TMPRSS3 | Alu, LTR | Alternative transcript | Breast, ovarian cancer | [8,82,83] |
| TP53 | Alu | Binding sites | Cancers, Pancreatic cancer | [58] |
| P63 | LTR | Primary promoter | Breast cancer | [65] |
| PDZK1 | Alu | Alternative splicing | Lung cancer | [65] |
| PODXL | Alu | Alternative splicing | Pancreatic cancer | [65] |
| PTN | LTR | Alternative promoter | Different cancers | [65] |
| ZNF451 | LTR | Alternative splicing | Hematological cancer | [9,84] |
| ZNF177, ZNF257, ZNF418 | LINE1, Alu | Alternative splicing | Different cancers | [65] |
| Locus or Genes | TE(s) | Associated Cancers | References |
|---|---|---|---|
| APC | Alu | Colon cancer | [13,99] |
| BRCA1 | Alu | Breast, ovarian cancer | [12,98,104] |
| BRCA2 | Alu | Breast, ovarian cancer | [12,98,104] |
| CASPR4 | LTR | Brain cancers | [8,105] |
| CHEK2 | Alu | Breast, ovarian, prostate cancer | [104,106] |
| CLDN14 | LTR | Melanoma | [8,107] |
| CYP19 | LTR | Breast cancer | [8,108] |
| ENTPD1 | LTR | Melanoma | [8,109] |
| FUT5 | LINE1, Alu | Gastric cancer | [8,110] |
| HSD17B1 | LTR | Breast, endometrial cancers | [111] |
| HRAS | LTR | T-cell leukemia | [112] |
| MLH1 | Alu | Colorectal cancer | [2,113] |
| MLVI2 | Alu | Leukemia | [114] |
| MKKS | LINE2, LTR | Embryonic cancers | [8,115] |
| MLH2 | Alu | Colorectal cancer | [113] |
| MYC | LINE1 | Breast cancer | [116] |
| NF1 | Alu | Neurofibromatosis type I | [87] |
| MSLN | LTR | Human cancers | [8,117] |
| RB1 | Alu, LTR | Retinoblastoma, hepatocellular cancer | [14,79] |
| TCF3-PBX1 | LTR | ALL | [118] |
| TRPC6 | LTR | Breast, prostate, gastric cancers, glioma | [76,119] |
| Locus of Genes | TE(s) | Chromosomal Defects | Associated Cancer | References |
|---|---|---|---|---|
| BCR-ABL | Alu | Chromosomal translocation | CML | [126] |
| BRCA1 | Alu | Chromosomal deletion, duplication, insertion | Breast, ovarian cancer | [127] |
| BRCA2 | Alu | Chromosomal deletion, duplication, insertion | Breast, ovarian cancer | [12,128] |
| CAD | Alu | Chromosomal deletion | Hepatocellular carcinoma | [122] |
| CDH1 | Alu | Chromosomal deletion | Diffuse gastric cancer | [129] |
| EWSR1-ETV | Alu, LINE1 | Chromosomal translocation | Ewing sarcoma | [28] |
| MAD1L1 | LTR | Chromosomal instability | Breast cancer | [8,130] |
| MLL1 | Alu | Chromosomal duplication | AML | [131] |
| MYB | Alu | Chromosomal duplication | T-ALL | [132] |
| VHL | Alu | Chromosomal deletion | von Hippel Lindau disease | [133] |
| WT1 | LINE1 | Chromosomal translocation | Sarcoma | [28] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anwar, S.L.; Wulaningsih, W.; Lehmann, U. Transposable Elements in Human Cancer: Causes and Consequences of Deregulation. Int. J. Mol. Sci. 2017, 18, 974. https://doi.org/10.3390/ijms18050974
Anwar SL, Wulaningsih W, Lehmann U. Transposable Elements in Human Cancer: Causes and Consequences of Deregulation. International Journal of Molecular Sciences. 2017; 18(5):974. https://doi.org/10.3390/ijms18050974
Chicago/Turabian StyleAnwar, Sumadi Lukman, Wahyu Wulaningsih, and Ulrich Lehmann. 2017. "Transposable Elements in Human Cancer: Causes and Consequences of Deregulation" International Journal of Molecular Sciences 18, no. 5: 974. https://doi.org/10.3390/ijms18050974
APA StyleAnwar, S. L., Wulaningsih, W., & Lehmann, U. (2017). Transposable Elements in Human Cancer: Causes and Consequences of Deregulation. International Journal of Molecular Sciences, 18(5), 974. https://doi.org/10.3390/ijms18050974