
Decrease of miR-195 Promotes Chondrocytes Proliferation and Maintenance of Chondrogenic Phenotype via Targeting FGF-18 Pathway

Abstract
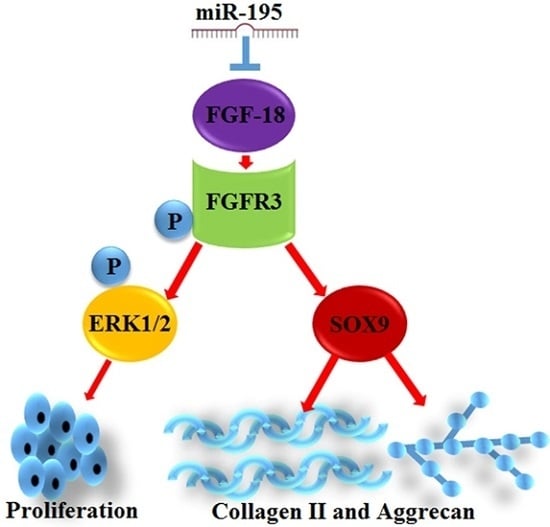
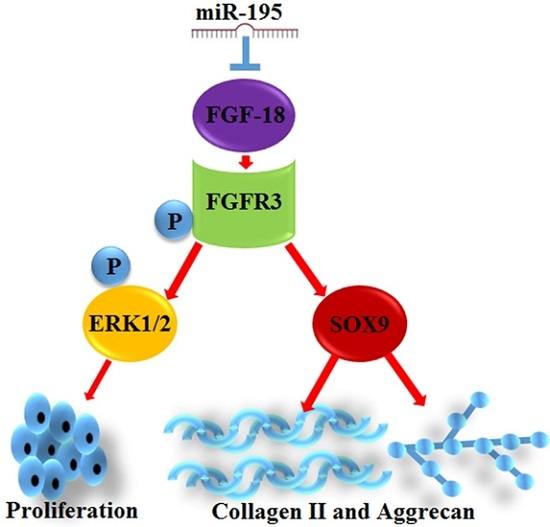
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
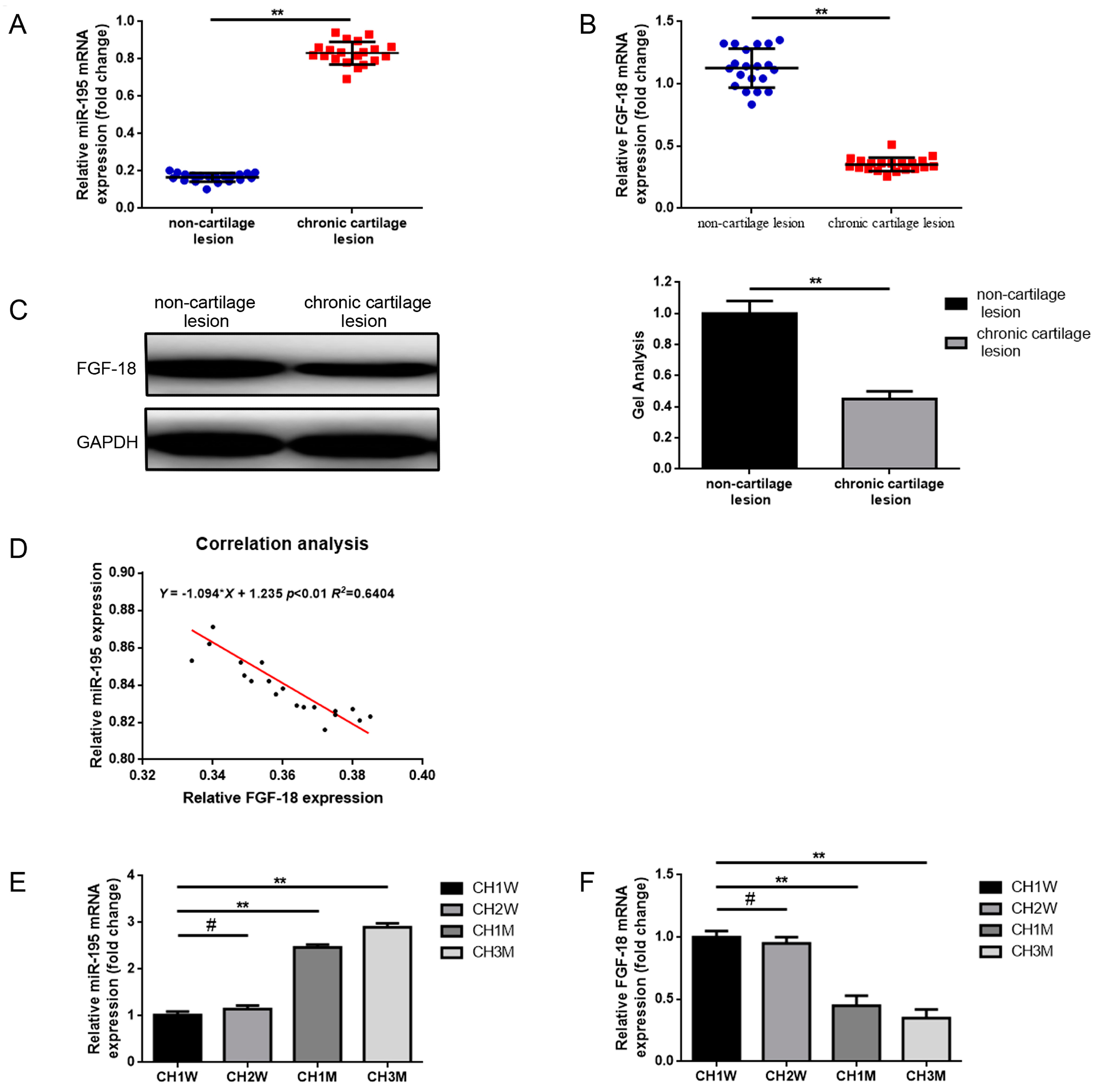
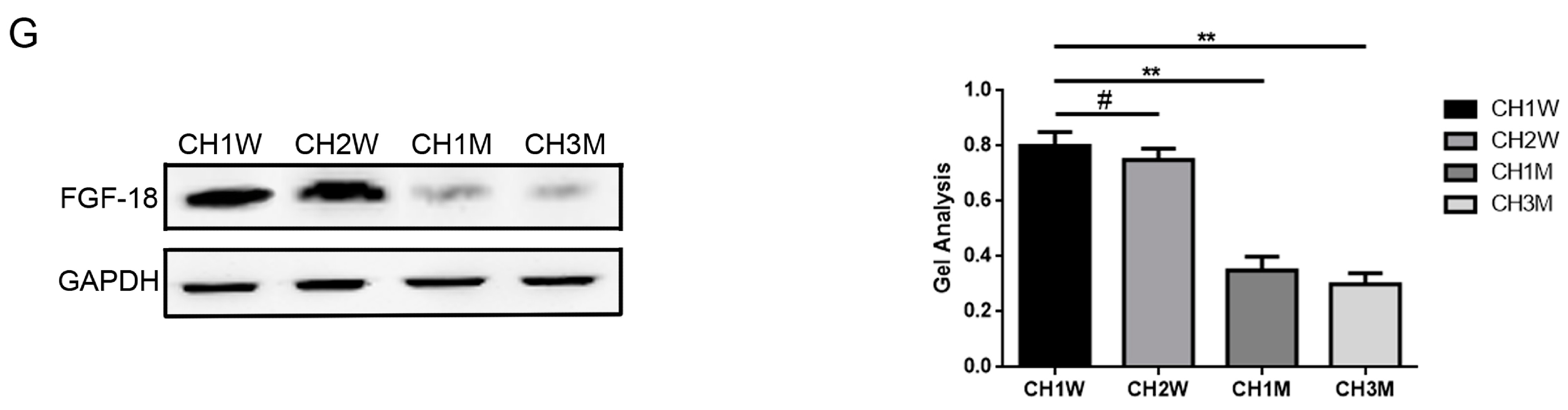
2.1. Expression of miR-195 and FGF-18 in the Joint Fluid of Patients with Chronic Cartilage Lesions and with Respect to the Different Ages of Chondrocytes
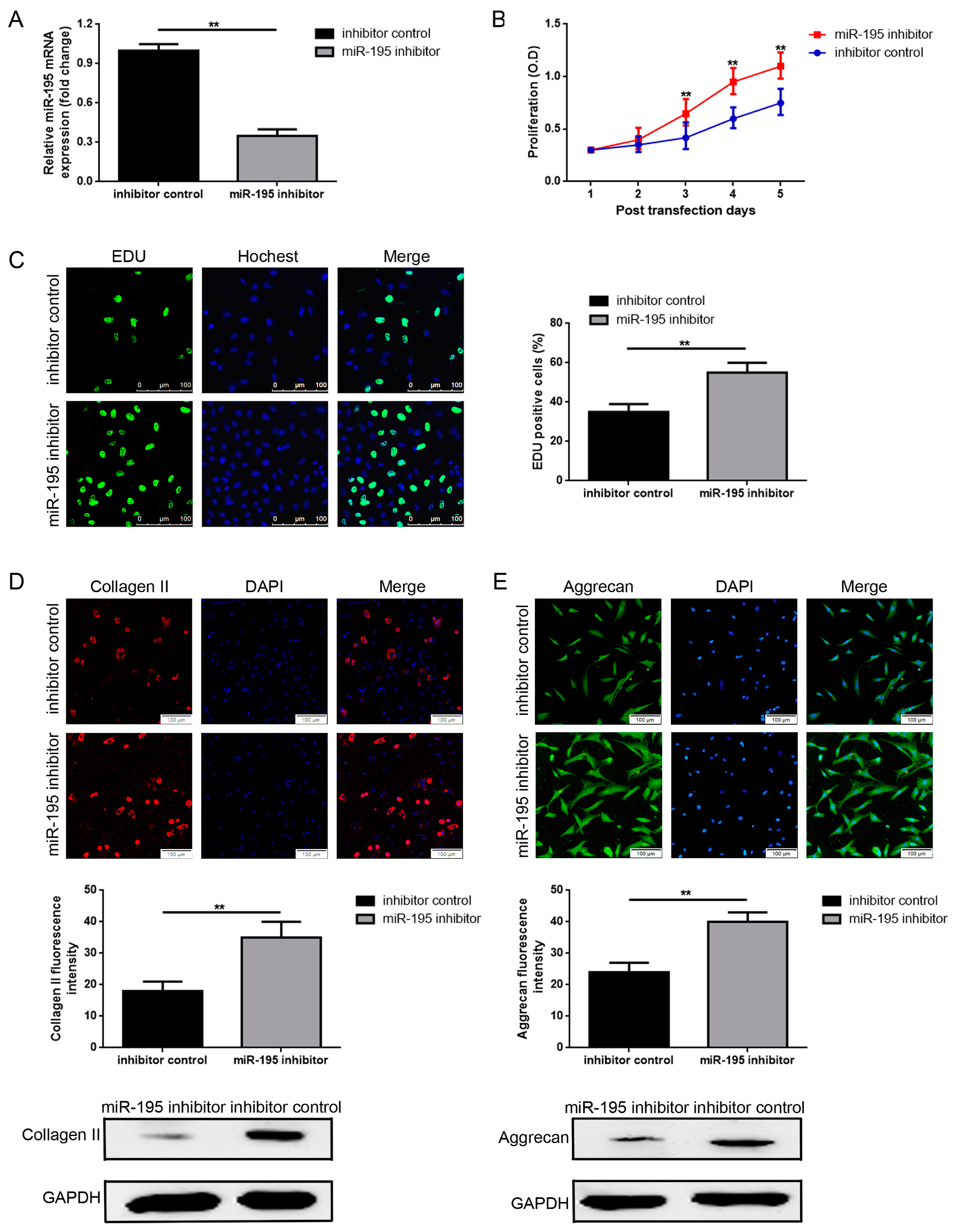
2.2. Decrease of miR-195 Promotes Chondrocytes Proliferation and Col2a1/Aggrecan Expression
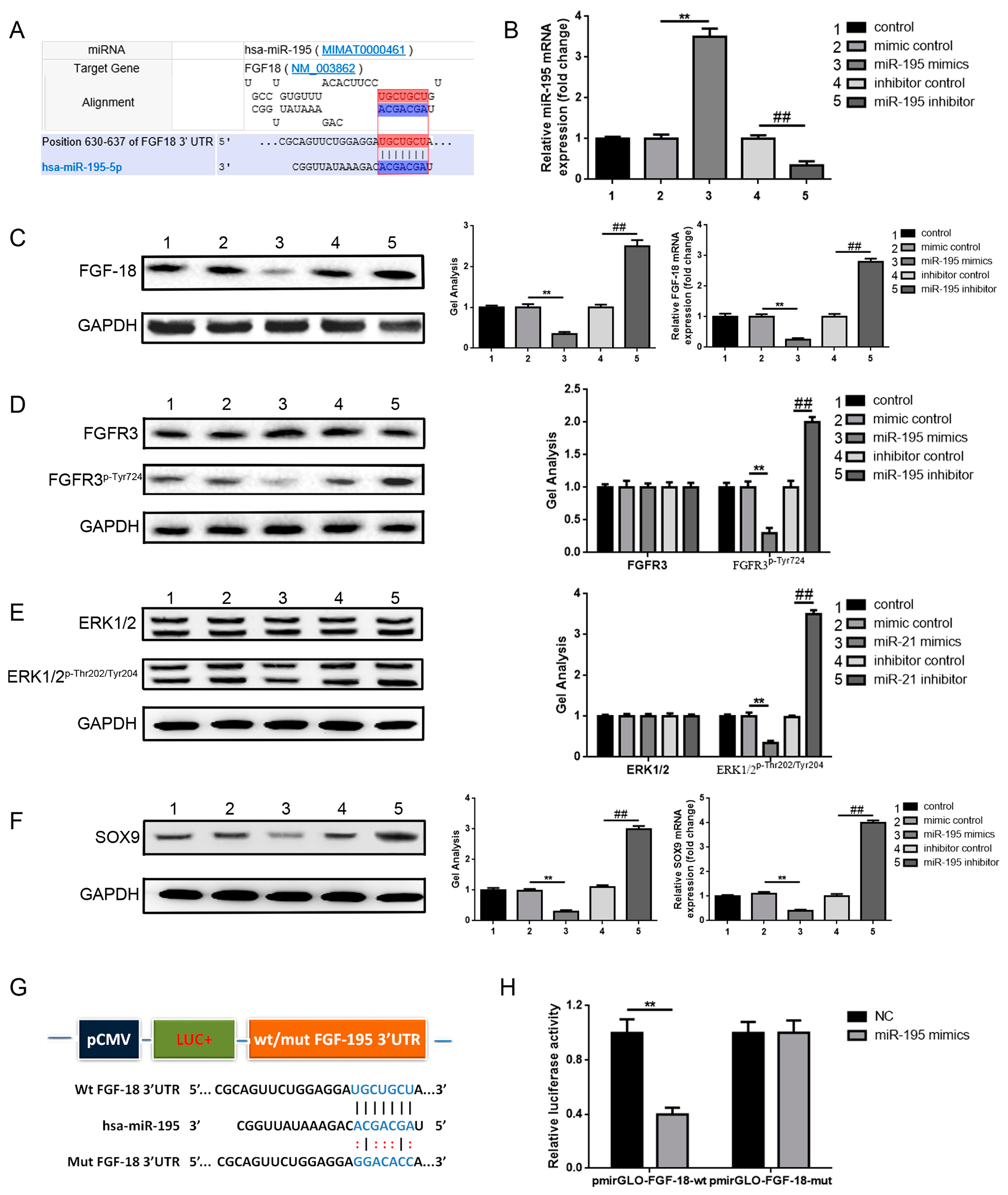
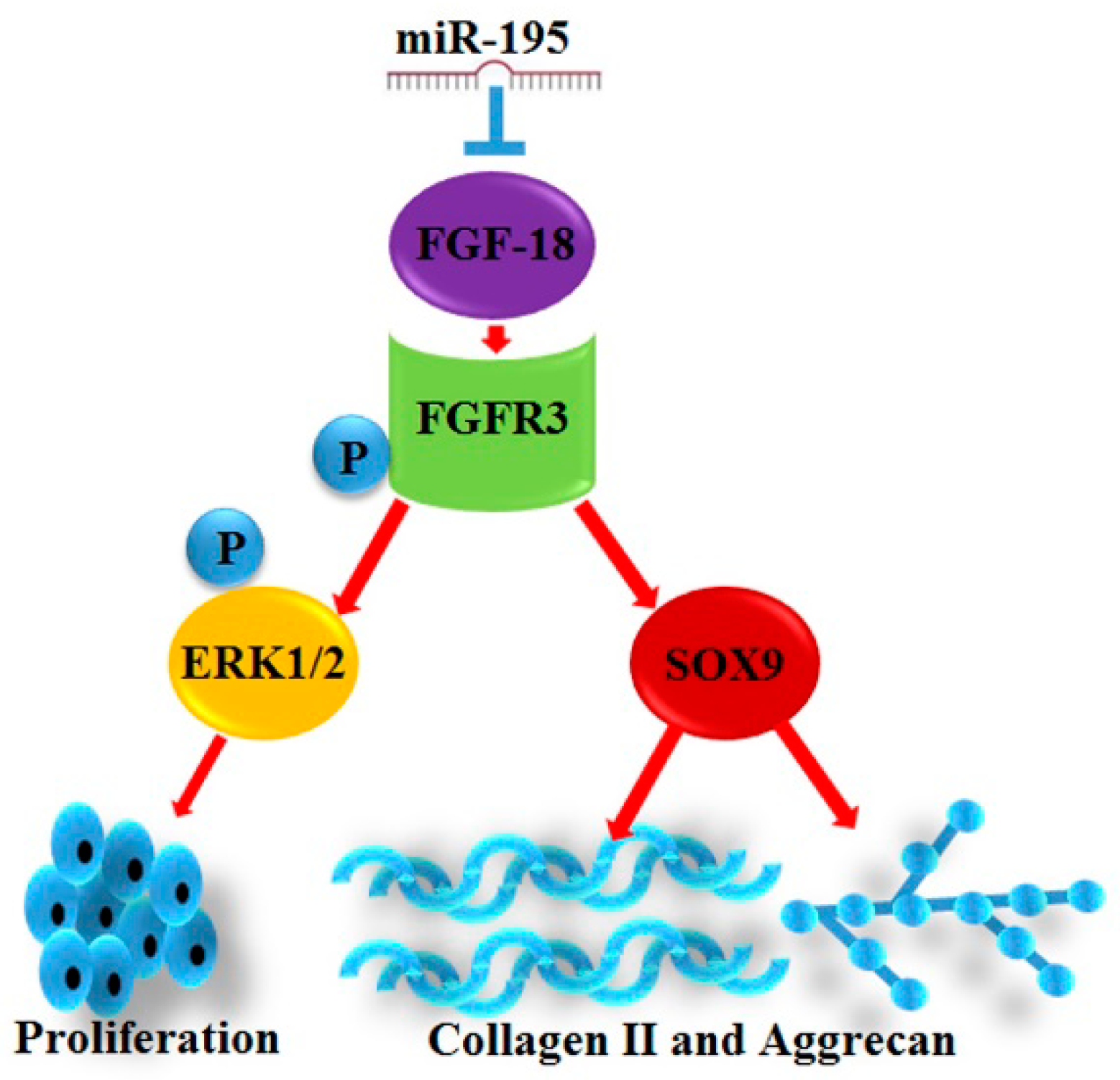
2.3. miR-195 Targets FGF-18 and Regulates Its Downstream Pathway
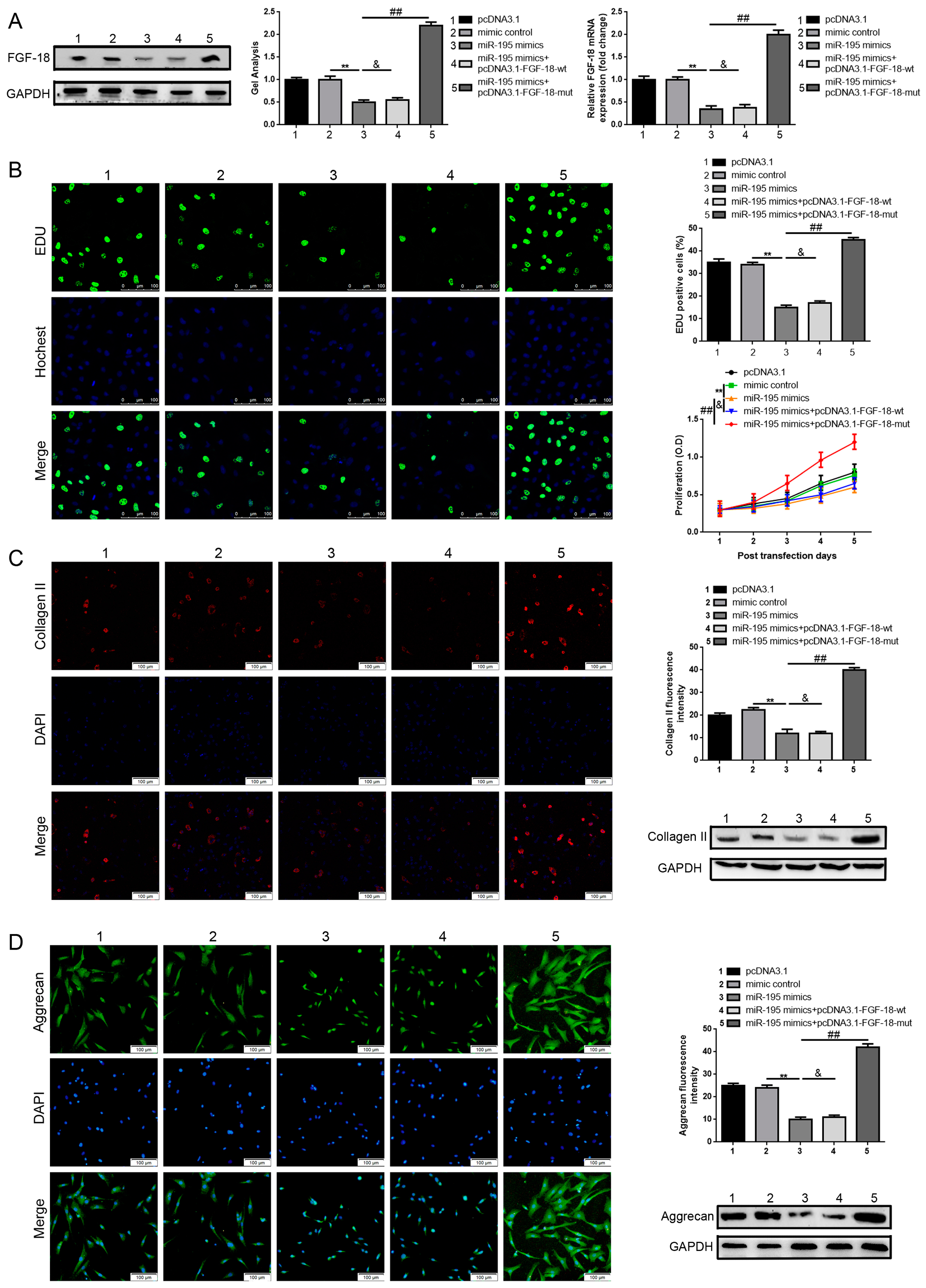
2.4. FGF-18 Rescues the Inhibitive Effect of miR-195 on Chondrocyte Proliferation and Col2a1/Aggrecan Expression
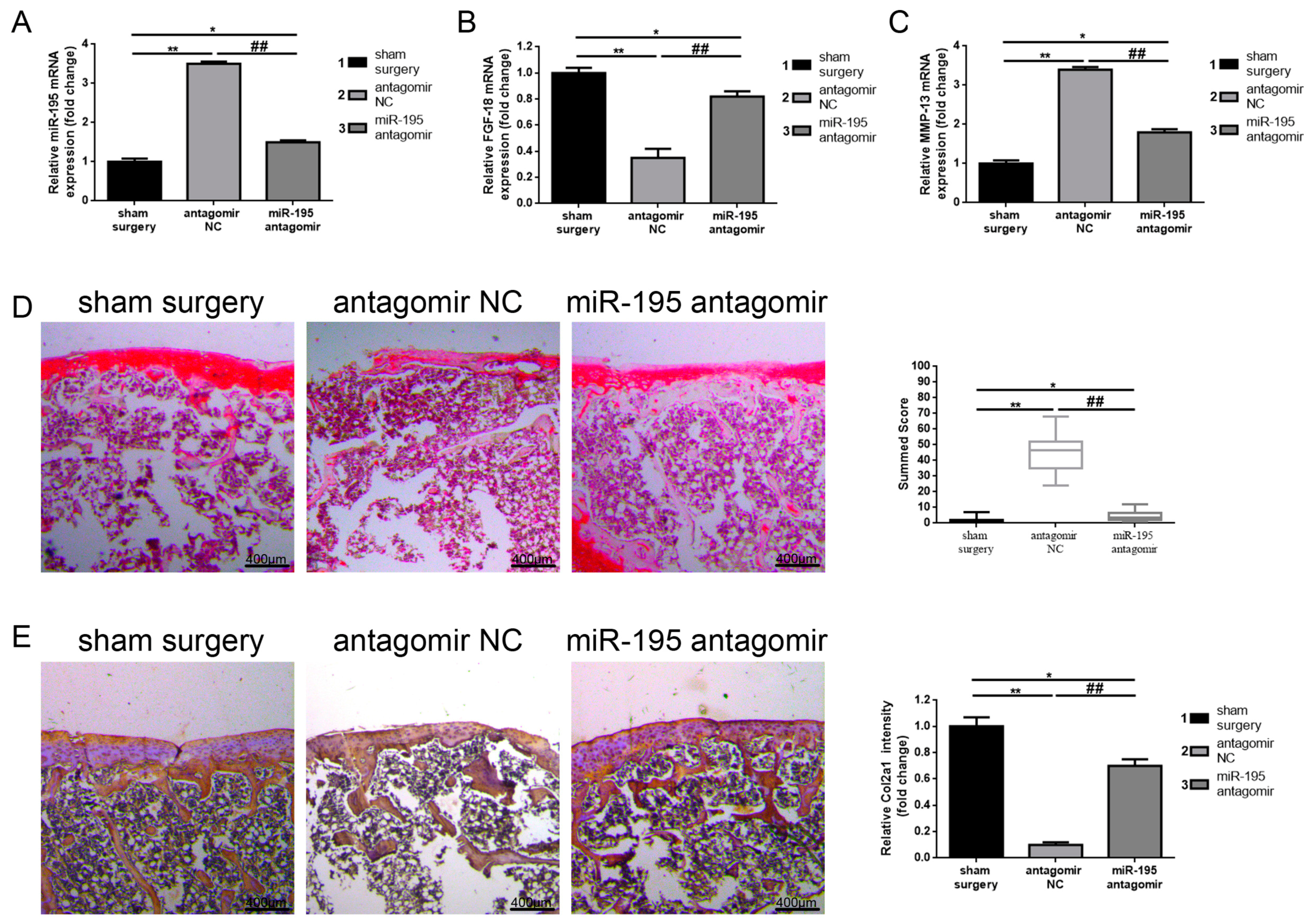
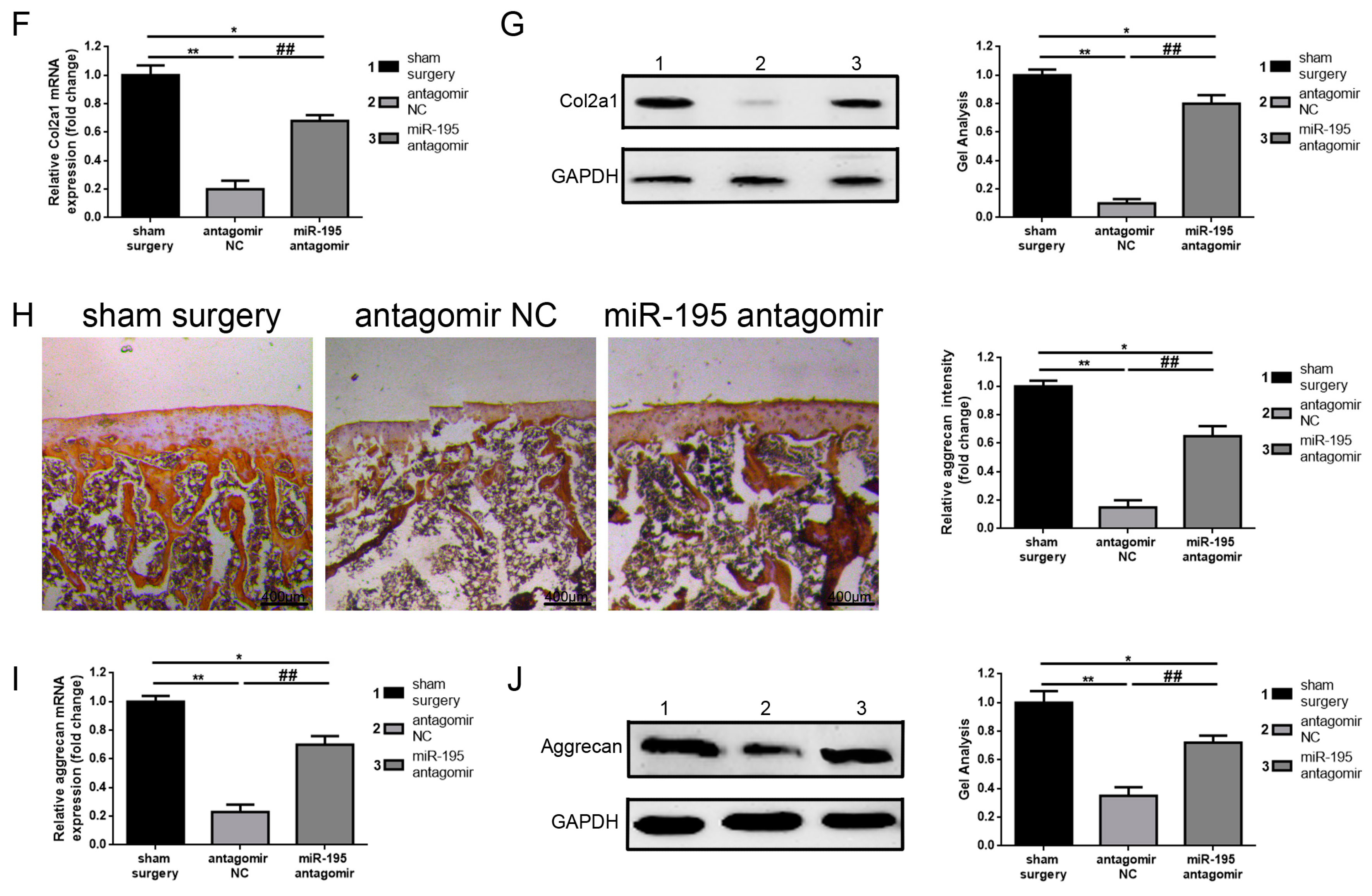
2.5. Downregulation of miR-195 Protects Chronic Cartilage Lesions In Vivo
3. Discussion
4. Materials and Methods
4.1. Patients and Tissue Samples
4.2. Cell Isolation and Culture
4.3. Plasmids Construction and Cell Transfection
4.4. Cell Proliferation Assays
4.5. Reverse Transcription and Quantitative Real-Time PCR
| Name | Forward Primer (5′–3′) | Reverse Primer (5′–3′) |
|---|---|---|
| FGF-18 | CGAGGACGGGGACAAGTATG | CAGCTCAGTCTGTCCCTTGG |
| SOX9 | TTCCTCCTCCCGGCATGAGTG | CAACTTTGCCAGCTTGCACG |
| Col2a1 | CCCCTGCAGTACATGCGG | CTCGACGTCATGCTGTCTCAAG |
| Aggrecan | TAAACCCGGTGTGAGAACCG | CCTGGGTGACAATCCAGTCC |
| MMP-13 | CAGTTGACAGGCTCCGAGAA | CGTGTGCCAGAAGACCAGAA |
| GAPDH | GGAATCCACTGGCGTCTTCA | GGTTCACGCCCATCACAAAC |
4.6. Western Blot Analysis
4.7. Immunofluorescence Analysis
4.8. Dual Luciferase Reporter Assay
4.9. Establishment of Chronic Cartilage Lesion Models in Rat
4.10. Samples Collection and Immunohistochemistry
4.11. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brophy, R.H.; Rodeo, S.A.; Barnes, R.P.; Powell, J.W.; Warren, R.F. Knee articular cartilage injuries in the National Football League: Epidemiology and treatment approach by team physicians. J. Knee Surg. 2009, 22, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Menetrey, J.; Unno-Veith, F.; Madry, H.; Van Breuseghem, I. Epidemiology and imaging of the subchondral bone in articular cartilage repair. Knee Surg. Sports Traumatol. Arthrosc. 2010, 18, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Cleland, K.A.; James, M.J.; Neumann, M.A.; Gibson, R.A.; Cleland, L.G. Differences in fatty acid composition of immature and mature articular cartilage in humans and sheep. Lipids 1995, 30, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Sandell, L.J.; Sugai, J.V.; Trippel, S.B. Expression of collagens I, II, X, and XI and aggrecan mRNAs by bovine growth plate chondrocytes in situ. J. Orthop. Res. 1994, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Biant, L.C.; Bentley, G.; Vijayan, S.; Skinner, J.A.; Carrington, R.W. Long-term results of autologous chondrocyte implantation in the knee for chronic chondral and osteochondral defects. Am. J. Sports Med. 2014, 42, 2178–2183. [Google Scholar] [CrossRef] [PubMed]
- Cole, B.J.; DeBerardino, T.; Brewster, R.; Farr, J.; Levine, D.W.; Nissen, C.; Roaf, P.; Zaslav, K. Outcomes of autologous chondrocyte implantation in study of the treatment of articular repair (STAR) patients with osteochondritis dissecans. Am. J. Sports Med. 2012, 40, 2015–2022. [Google Scholar] [CrossRef] [PubMed]
- Dasar, U.; Gursoy, S.; Akkaya, M.; Algin, O.; Isik, C.; Bozkurt, M. Microfracture technique versus carbon fibre rod implantation for treatment of knee articular cartilage lesions. J. Orthop. Surg. 2016, 24, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, N.; Yamane, S.; Nishida, K.; Masuko, T.; Funakoshi, T.; Kamishima, T.; Minami, A. Transplantation of tissue-engineered cartilage for the treatment of osteochondritis dissecans in the elbow: Outcomes over a four-year follow-up in two patients. J. Shoulder Elbow Surg. 2010, 19, e1–e6. [Google Scholar] [CrossRef] [PubMed]
- Mithoefer, K.; Gill, T.J.; Cole, B.J.; Williams, R.J.; Mandelbaum, B.R. Clinical Outcome and Return to Competition after Microfracture in the Athlete’s Knee: An Evidence-Based Systematic Review. Cartilage 2010, 1, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Mithoefer, K.; Venugopal, V.; Manaqibwala, M. Incidence, Degree, and Clinical Effect of Subchondral Bone Overgrowth after Microfracture in the Knee. Am. J. Sports Med. 2016, 44, 2057–2063. [Google Scholar] [CrossRef] [PubMed]
- Polat, G.; Ersen, A.; Erdil, M.E.; Kizilkurt, T.; Kilicoglu, O.; Asik, M. Long-term results of microfracture in the treatment of talus osteochondral lesions. Knee Surg. Sports Traumatol. Arthrosc. 2016, 24, 1299–1303. [Google Scholar] [CrossRef] [PubMed]
- Takazawa, K.; Adachi, N.; Deie, M.; Kamei, G.; Uchio, Y.; Iwasa, J.; Kumahashi, N.; Tadenuma, T.; Kuwata, S.; Yasuda, K.; et al. Evaluation of magnetic resonance imaging and clinical outcome after tissue-engineered cartilage implantation: Prospective 6-year follow-up study. J. Orthop. Sci. 2012, 17, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.A.; Reisler, T.; Bader, D.L. Expansion of chondrocytes for tissue engineering in alginate beads enhances chondrocytic phenotype compared to conventional monolayer techniques. Acta Orthop. Scand. 2003, 74, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Oldershaw, R.A. Cell sources for the regeneration of articular cartilage: The past, the horizon and the future. Int. J. Exp. Pathol. 2012, 93, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Tubo, R.; Binette, F. Culture and identification of autologous human articular chondrocytes for implantation. Methods Mol. Med. 1999, 18, 205–215. [Google Scholar] [PubMed]
- Zheng, M.H.; King, E.; Kirilak, Y.; Huang, L.; Papadimitriou, J.M.; Wood, D.J.; Xu, J. Molecular characterisation of chondrocytes in autologous chondrocyte implantation. Int. J. Mol. Med. 2004, 13, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Hong, E.; Reddi, A.H. MicroRNAs in chondrogenesis, articular cartilage, and osteoarthritis: Implications for tissue engineering. Tissue Eng. Part B Rev. 2012, 18, 445–453. [Google Scholar] [CrossRef] [PubMed]
- McAlinden, A.; Varghese, N.; Wirthlin, L.; Chang, L.W. Differentially expressed microRNAs in chondrocytes from distinct regions of developing human cartilage. PLoS ONE 2013, 8, e75012. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Liu, H.; Zhou, Y. Roles of microRNAs in prenatal chondrogenesis, postnatal chondrogenesis and cartilage-related diseases. J. Cell. Mol. Med. 2013, 17, 1515–1524. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Qu, J.; Xu, F.; Guo, Y.; Wang, Y.; Yu, H.; Qian, B. MiR-195 suppresses non-small cell lung cancer by targeting CHEK1. Oncotarget 2015, 6, 9445–9456. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, X.; Zou, C.; Kung, H.F.; Lin, M.C.; Dress, A.; Wardle, F.; Jiang, B.H.; Lai, L. miR-195 inhibits tumor growth and angiogenesis through modulating IRS1 in breast cancer. Biomed. Pharmacother. 2016, 80, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Song, W.; Xu, X.; Zhao, X.; Yang, L. IGF2BP2 promotes colorectal cancer cell proliferation and survival through interfering with RAF-1 degradation by miR-195. FEBS Lett. 2016, 590, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tao, T.; Liu, C.; Guan, H.; Huang, Y.; Xu, B.; Chen, M. Downregulation of miR-195 promotes prostate cancer progression by targeting HMGA1. Oncol. Rep. 2016, 36, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Chen, F.; Zhao, J.; Li, B.; Liang, Y.; Pan, W.; Zhang, S.; Wang, X.; Zheng, D. Long non-coding RNA PVT1 promotes osteosarcoma development by acting as a molecular sponge to regulate miR-195. Oncotarget 2016, 7, 82620–82633. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.L.; Rong, X.X.; Wen, L.T.; Zhu, G.X.; Qian, M.Q. miR-195 inhibits the proliferation and migration of chondrocytes by targeting GIT1. Mol. Med. Rep. 2017, 15, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Zhao, A.Q.; Zhao, Z.Q.; Liu, W.L.; Jian, D.M. MicroRNA-195 induced apoptosis in hypoxic chondrocytes by targeting hypoxia-inducible factor 1 α. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 545–551. [Google Scholar] [PubMed]
- Seidl, C.I.; Martinez-Sanchez, A.; Murphy, C.L. Derepression of MicroRNA-138 Contributes to Loss of the Human Articular Chondrocyte Phenotype. Arthritis Rheumatol. 2016, 68, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Hong, E.; Reddi, A.H. Dedifferentiation and redifferentiation of articular chondrocytes from surface and middle zones: Changes in microRNAs-221/-222, -140, and -143/145 expression. Tissue Eng. Part A 2013, 19, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Brewer, J.R.; Mazot, P.; Soriano, P. Genetic insights into the mechanisms of FGF signaling. Genes Dev. 2016, 30, 751–771. [Google Scholar] [CrossRef] [PubMed]
- Teven, C.M.; Farina, E.M.; Rivas, J.; Reid, R.R. Fibroblast growth factor (FGF) signaling in development and skeletal diseases. Genes Dis. 2014, 1, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Haque, T.; Nakada, S.; Hamdy, R.C. A review of FGF18: Its expression; signaling pathways and possible functions during embryogenesis and post-natal development. Histol. Histopathol. 2007, 22, 97–105. [Google Scholar] [PubMed]
- Liu, Z.; Lavine, K.J.; Hung, I.H.; Ornitz, D.M. FGF18 is required for early chondrocyte proliferation, hypertrophy and vascular invasion of the growth plate. Dev. Boil. 2007, 302, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Bradley, E.W.; Carpio, L.R.; Newton, A.C.; Westendorf, J.J. Deletion of the PH-domain and Leucine-rich Repeat Protein Phosphatase 1 (Phlpp1) Increases Fibroblast Growth Factor (FGF) 18 Expression and Promotes Chondrocyte Proliferation. J. Boil. Chem. 2015, 290, 16272–16280. [Google Scholar] [CrossRef] [PubMed]
- Davidson, D.; Blanc, A.; Filion, D.; Wang, H.; Plut, P.; Pfeffer, G.; Buschmann, M.D.; Henderson, J.E. Fibroblast growth factor (FGF) 18 signals through FGF receptor 3 to promote chondrogenesis. J. Boil. Chem. 2005, 280, 20509–20515. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Saito, T.; Chang, S.H.; Kobayashi, H.; Ladel, C.H.; Guehring, H.; Chung, U.I.; Kawaguchi, H. Identification of fibroblast growth factor-18 as a molecule to protect adult articular cartilage by gene expression profiling. J. Boil. Chem. 2014, 289, 10192–10200. [Google Scholar] [CrossRef] [PubMed]
- Power, J.; Hernandez, P.; Guehring, H.; Getgood, A.; Henson, F. Intra-articular injection of rhFGF-18 improves the healing in microfracture treated chondral defects in an ovine model. J. Orthop. Res. 2014, 32, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, D.H.; Ou, S.J.; Quinn, T.M. P38 mitogen-activated protein kinase promotes dedifferentiation of primary articular chondrocytes in monolayer culture. J. Cell. Mol. Med. 2013, 17, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, D.H.; Solar-Cafaggi, S.; Quinn, T.M. Functionalization of dynamic culture surfaces with a cartilage extracellular matrix extract enhances chondrocyte phenotype against dedifferentiation. Acta Biomater. 2012, 8, 3333–3341. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, H.; Nishizawa, S.; Asawa, Y.; Fujihara, Y.; Ogasawara, T.; Yamaoka, K.; Nagata, S.; Takato, T.; Hoshi, K. Involvement of fibroblast growth factor 18 in dedifferentiation of cultured human chondrocytes. Cell Prolif. 2010, 43, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Valverde-Franco, G.; Binette, J.S.; Li, W.; Wang, H.; Chai, S.; Laflamme, F.; Tran-Khanh, N.; Quenneville, E.; Meijers, T.; Poole, A.R.; et al. Defects in articular cartilage metabolism and early arthritis in fibroblast growth factor receptor 3 deficient mice. Hum. Mol. Genet. 2006, 15, 1783–1792. [Google Scholar] [CrossRef] [PubMed]
- Kaul, G.; Cucchiarini, M.; Arntzen, D.; Zurakowski, D.; Menger, M.D.; Kohn, D.; Trippel, S.B.; Madry, H. Local stimulation of articular cartilage repair by transplantation of encapsulated chondrocytes overexpressing human fibroblast growth factor 2 (FGF-2) in vivo. J. Gene Med. 2006, 8, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Knecht, S.; Vanwanseele, B.; Stussi, E. A review on the mechanical quality of articular cartilage—Implications for the diagnosis of osteoarthritis. Clin. Biomech. 2006, 21, 999–1012. [Google Scholar] [CrossRef] [PubMed]
- Sandell, L.J.; Aigner, T. Articular cartilage and changes in arthritis. An introduction: Cell biology of osteoarthritis. Arthritis Res. 2001, 3, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Madry, H.; Orth, P.; Cucchiarini, M. Gene Therapy for Cartilage Repair. Cartilage 2011, 2, 201–225. [Google Scholar] [CrossRef] [PubMed]
- Trippel, S.; Cucchiarini, M.; Madry, H.; Shi, S.; Wang, C. Gene therapy for articular cartilage repair. Proc. Inst. Mech. Eng. H 2007, 221, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B.; Marcu, K.B. Epigenomic and microRNA-mediated regulation in cartilage development; homeostasis; and osteoarthritis. Trends Mol. Med. 2012, 18, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Sanchez, A.; Dudek, K.A.; Murphy, C.L. Regulation of human chondrocyte function through direct inhibition of cartilage master regulator SOX9 by microRNA-145 (miRNA-145). J. Boil. Chem. 2012, 287, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Amer, M.; Elhefnawi, M.; El-Ahwany, E.; Awad, A.F.; Gawad, N.A.; Zada, S.; Tawab, F.M. Hsa-miR-195 targets PCMT1 in hepatocellular carcinoma that increases tumor life span. Tumour Boil. 2014, 35, 11301–11309. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Zhao, H.; Tang, J.; Wu, H. Serum miR-195 is a diagnostic and prognostic marker for osteosarcoma. J. Surg. Res. 2015, 194, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Cecene, G.; Ak, S.; Eskiler, G.G.; Demirdogen, E.; Erturk, E.; Gokgoz, S.; Polatkan, V.; Egeli, U.; Tunca, B.; Tezcan, G.; et al. Circulating miR-195 as a Therapeutic Biomarker in Turkish Breast Cancer Patients. Asian Pac. J. Cancer Prev. 2016, 17, 4241–4246. [Google Scholar] [PubMed]
- Liu, C.; Guan, H.; Wang, Y.; Chen, M.; Xu, B.; Zhang, L.; Lu, K.; Tao, T.; Zhang, X.; Huang, Y. miR-195 Inhibits EMT by Targeting FGF2 in Prostate Cancer Cells. PLoS ONE 2015, 10, e0144073. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Tang, J.; Li, H.; Zhang, H.; Lu, C.; Chen, H.; Li, W.; Xia, Y.; Tang, W. MiR-195 affects cell migration and cell proliferation by down-regulating DIEXF in Hirschsprung’s disease. BMC Gastroenterol. 2014, 14, 123. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Kim, H.W.; Matsu-ura, K.; Wang, Y.G.; Xu, M.; Ashraf, M. Abrogation of Age-Induced MicroRNA-195 Rejuvenates the Senescent Mesenchymal Stem Cells by Reactivating Telomerase. Stem Cells 2016, 34, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Zampetaki, A.; Attia, R.; Mayr, U.; Gomes, R.S.; Phinikaridou, A.; Yin, X.; Langley, S.R.; Willeit, P.; Lu, R.; Fanshawe, B.; et al. Role of miR-195 in aortic aneurysmal disease. Circ. Res. 2014, 115, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.I.; Silva, A.M.; Vasconcelos, D.M.; Almeida, C.R.; Caires, H.; Pinto, M.T.; Calin, G.A.; Santos, S.G.; Barbosa, M.A. miR-195 in human primary mesenchymal stromal/stem cells regulates proliferation, osteogenesis and paracrine effect on angiogenesis. Oncotarget 2016, 7, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Han, L.R.; Zhou, Y.X.; Li, Y. MiR-195 Suppresses Cervical Cancer Migration and Invasion Through Targeting Smad3. Int. J. Gynecol. Cancer 2016, 26, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, J.; Hu, B.; Wu, X.; Chen, Y.; Li, R.; Yuan, W. MiR-34a promotes Fas-mediated cartilage endplate chondrocyte apoptosis by targeting Bcl-2. Mol. Cell. Biochem. 2015, 406, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.J.; Zhuang, H.; Wang, G.X.; Li, Z.; Zhang, H.T.; Yu, T.Q.; Zhang, B.D. MiRNA-140 is a negative feedback regulator of MMP-13 in IL-1beta-stimulated human articular chondrocyte C28/I2 cells. Inflamm. Res. 2012, 61, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, W.; Fu, Q. miR-335 suppresses migration and invasion by targeting ROCK1 in osteosarcoma cells. Mol. Cell. Biochem. 2013, 384, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Barnard, J.C.; Williams, A.J.; Rabier, B.; Chassande, O.; Samarut, J.; Cheng, S.Y.; Bassett, J.H.; Williams, G.R. Thyroid hormones regulate fibroblast growth factor receptor signaling during chondrogenesis. Endocrinology 2005, 146, 5568–5580. [Google Scholar] [CrossRef] [PubMed]
- Ohbayashi, N.; Shibayama, M.; Kurotaki, Y.; Imanishi, M.; Fujimori, T.; Itoh, N.; Takada, S. FGF18 is required for normal cell proliferation and differentiation during osteogenesis and chondrogenesis. Genes Dev. 2002, 16, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Eldridge, S.; Nalesso, G.; Ismail, H.; Vicente-Greco, K.; Kabouridis, P.; Ramachandran, M.; Niemeier, A.; Herz, J.; Pitzalis, C.; Perretti, M.; et al. Agrin mediates chondrocyte homeostasis and requires both LRP4 and α-dystroglycan to enhance cartilage formation in vitro and in vivo. Ann. Rheum. Dis. 2016, 75, 1228–1235. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, I.; Tsuji, K.; Koopman, P.; Watanabe, H.; Yamada, Y.; Shinomiya, K.; Nifuji, A.; Noda, M. SOX9 enhances aggrecan gene promoter/enhancer activity and is up-regulated by retinoic acid in a cartilage-derived cell line, TC6. J. Boil. Chem. 2000, 275, 10738–10744. [Google Scholar] [CrossRef]
- Shi, S.; Wang, C.; Acton, A.J.; Eckert, G.J.; Trippel, S.B. Role of SOX9 in growth factor regulation of articular chondrocytes. J. Cell. Biochem. 2015, 116, 1391–1400. [Google Scholar] [CrossRef] [PubMed]
- Simental-Mendia, M.; Lara-Arias, J.; Alvarez-Lozano, E.; Said-Fernandez, S.; Soto-Dominguez, A.; Padilla-Rivas, G.R.; Martinez-Rodriguez, H.G. Cotransfected human chondrocytes: Over-expression of IGF-I and SOX9 enhances the synthesis of cartilage matrix components collagen-II and glycosaminoglycans. Braz. J. Med. Boil. Res. 2015, 48, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, H.; Oh, C.D.; Chen, D.; de Crombrugghe, B.; Kim, J.H. A Novel Regulatory Mechanism of Type II Collagen Expression via a SOX9-dependent Enhancer in Intron 6. J. Boil. Chem. 2017, 292, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Correa, D.; Somoza, R.A.; Lin, P.; Greenberg, S.; Rom, E.; Duesler, L.; Welter, J.F.; Yayon, A.; Caplan, A.I. Sequential exposure to fibroblast growth factors (FGF) 2, 9 and 18 enhances hMSC chondrogenic differentiation. Osteoarthr. Cartil. 2015, 23, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Elluru, R.G.; Thompson, F.; Reece, A. Fibroblast growth factor 18 gives growth and directional cues to airway cartilage. Laryngoscope 2009, 119, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Filip, A.; Bianchi, A.; Mainard, D.; Lacolley, P.; Magdalou, J.; Mercier, N. A simple two dimensional culture method to study the hypertrophic differentiation of rat articular chondrocytes. Biomed. Mater. Eng. 2015, 25, 87–102. [Google Scholar] [PubMed]
- Kuhne, M.; John, T.; El-Sayed, K.; Marzahn, U.; Aue, A.; Kohl, B.; Stoelzel, K.; Ertel, W.; Blottner, D.; Haisch, A.; et al. Characterization of auricular chondrocytes and auricular/articular chondrocyte co-cultures in terms of an application in articular cartilage repair. Int. J. Mol. Med. 2010, 25, 701–708. [Google Scholar] [PubMed]
- Wei, X.; Peng, G.; Zheng, S.; Wu, X. Differentiation of umbilical cord mesenchymal stem cells into steroidogenic cells in comparison to bone marrow mesenchymal stem cells. Cell Prolif. 2012, 45, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, T.; Zhang, Z.; Lu, M.; Zhao, W.; Zeng, X.; Zhang, W. Long non-coding RNA TUG1 promotes migration and invasion by acting as a ceRNA of miR-335-5p in osteosarcoma cells. Cancer Sci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Tian, K.; Zhong, W.; Zheng, X.; Zhang, J.; Liu, P.; Zhang, W.; Liu, H. Neuroleukin/Autocrine Motility Factor Receptor Pathway Promotes Proliferation of Articular Chondrocytes through Activation of AKT and SMAD2/3. Sci. Rep. 2015, 5, 15101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Cao, X.; Li, J.; Zhao, G. MiR-210 inhibits NF-κB signaling pathway by targeting DR6 in osteoarthritis. Sci. Rep. 2015, 5, 12775. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Xu, H. High expression of TRPM8 predicts poor prognosis in patients with osteosarcoma. Oncol. Lett. 2016, 12, 1373–1379. [Google Scholar] [CrossRef] [PubMed]








© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Yang, T.; Liu, Y.; Zhao, W.; Zhang, Z.; Lu, M.; Zhang, W. Decrease of miR-195 Promotes Chondrocytes Proliferation and Maintenance of Chondrogenic Phenotype via Targeting FGF-18 Pathway. Int. J. Mol. Sci. 2017, 18, 975. https://doi.org/10.3390/ijms18050975
Wang Y, Yang T, Liu Y, Zhao W, Zhang Z, Lu M, Zhang W. Decrease of miR-195 Promotes Chondrocytes Proliferation and Maintenance of Chondrogenic Phenotype via Targeting FGF-18 Pathway. International Journal of Molecular Sciences. 2017; 18(5):975. https://doi.org/10.3390/ijms18050975
Chicago/Turabian StyleWang, Yong, Tao Yang, Yadong Liu, Wei Zhao, Zhen Zhang, Ming Lu, and Weiguo Zhang. 2017. "Decrease of miR-195 Promotes Chondrocytes Proliferation and Maintenance of Chondrogenic Phenotype via Targeting FGF-18 Pathway" International Journal of Molecular Sciences 18, no. 5: 975. https://doi.org/10.3390/ijms18050975
APA StyleWang, Y., Yang, T., Liu, Y., Zhao, W., Zhang, Z., Lu, M., & Zhang, W. (2017). Decrease of miR-195 Promotes Chondrocytes Proliferation and Maintenance of Chondrogenic Phenotype via Targeting FGF-18 Pathway. International Journal of Molecular Sciences, 18(5), 975. https://doi.org/10.3390/ijms18050975