
C1q/TNF-Related Protein-9 Ameliorates Ox-LDL-Induced Endothelial Dysfunction via PGC-1α/AMPK-Mediated Antioxidant Enzyme Induction

Abstract
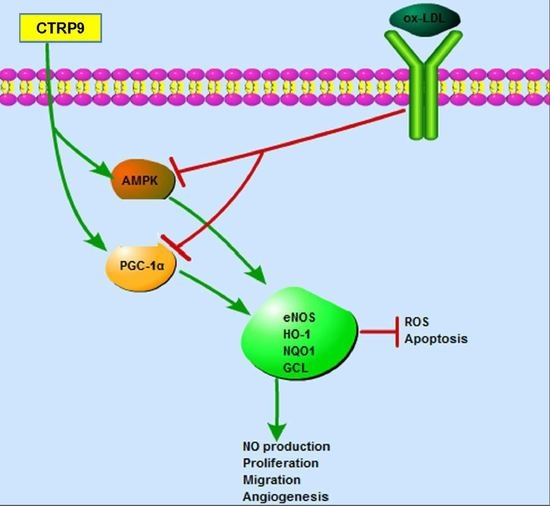
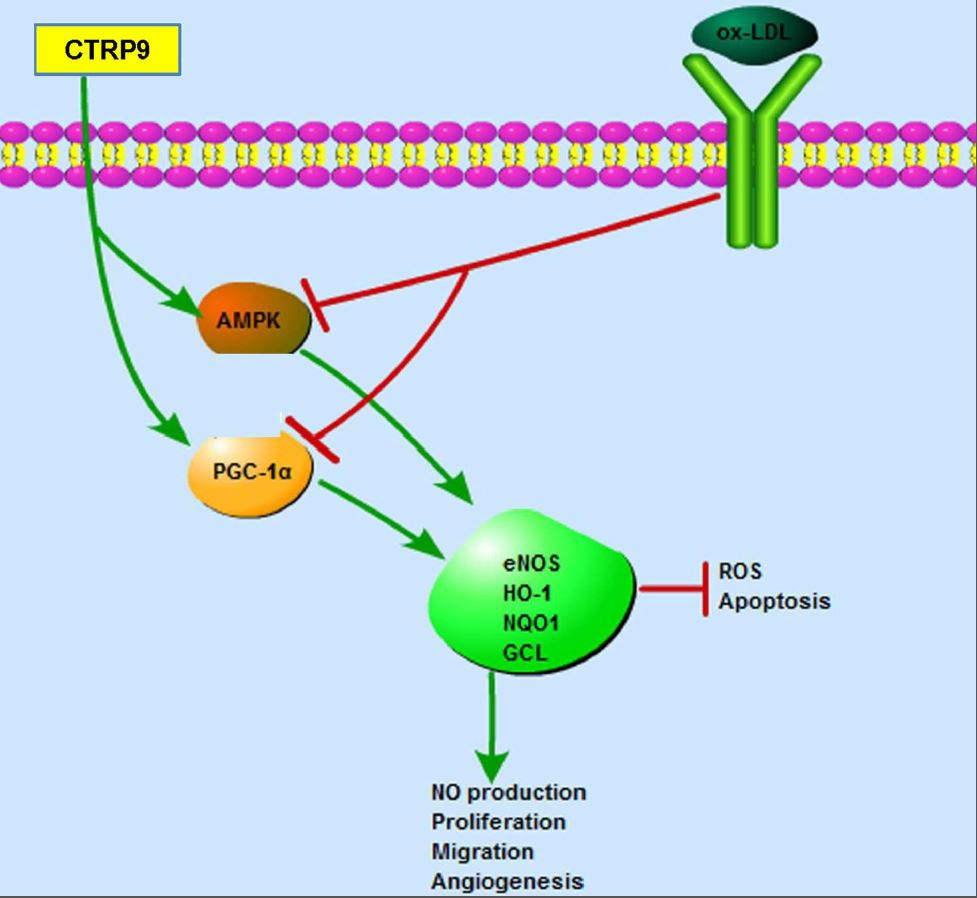
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. C1q/TNF-Related Protein 9 (CTRP9) Reversed the Proliferation, Migration, Angiogenesis, and Apoptosis in Human Umbilical Vein Endothelial Cells (HUVECs) Response to Oxidized Low-Density Lipoprotein (Ox-LDL)
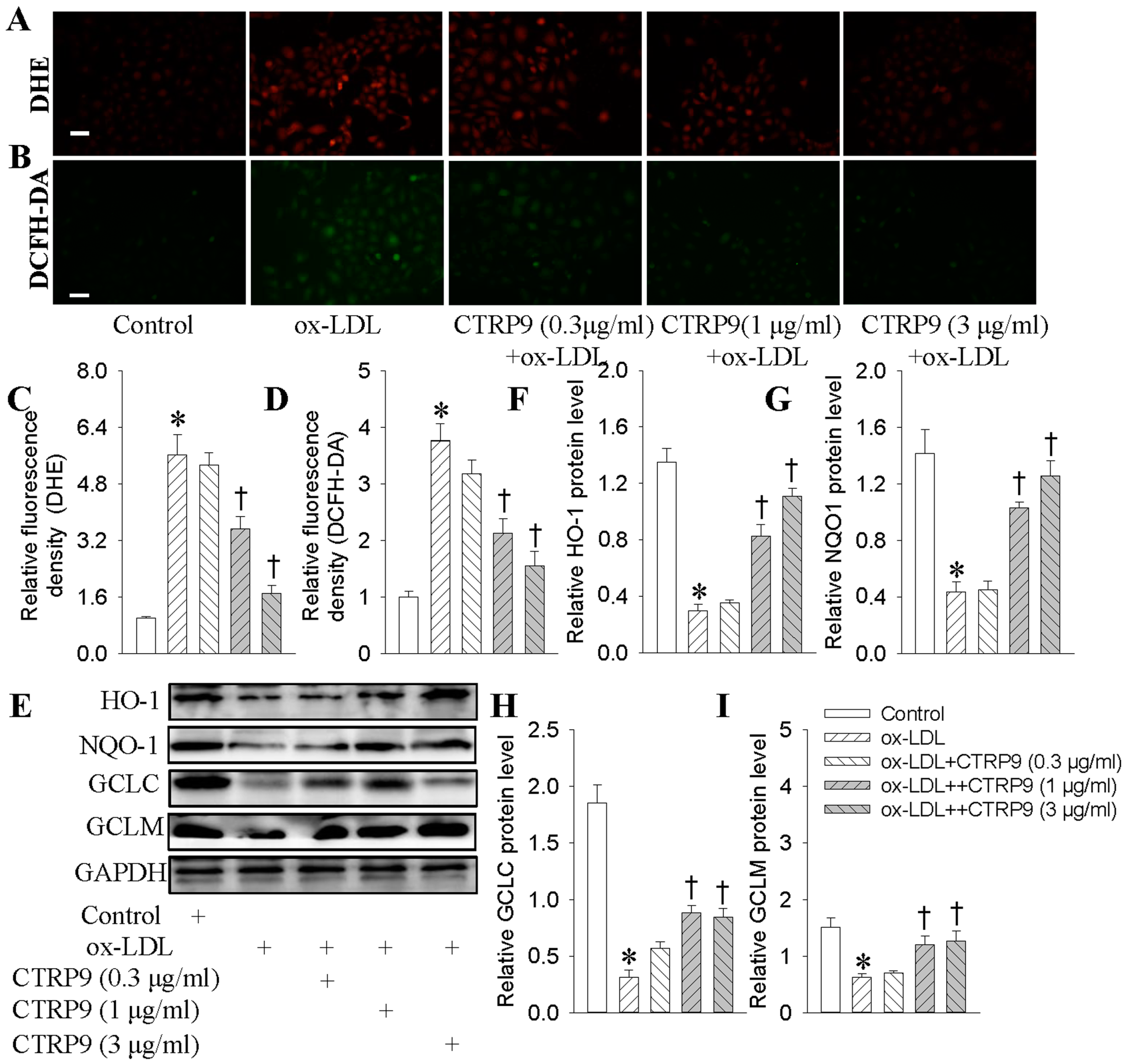
2.2. CTRP9 Alleviated Oxidative Stress in ox-LDL-Treated HUVECs
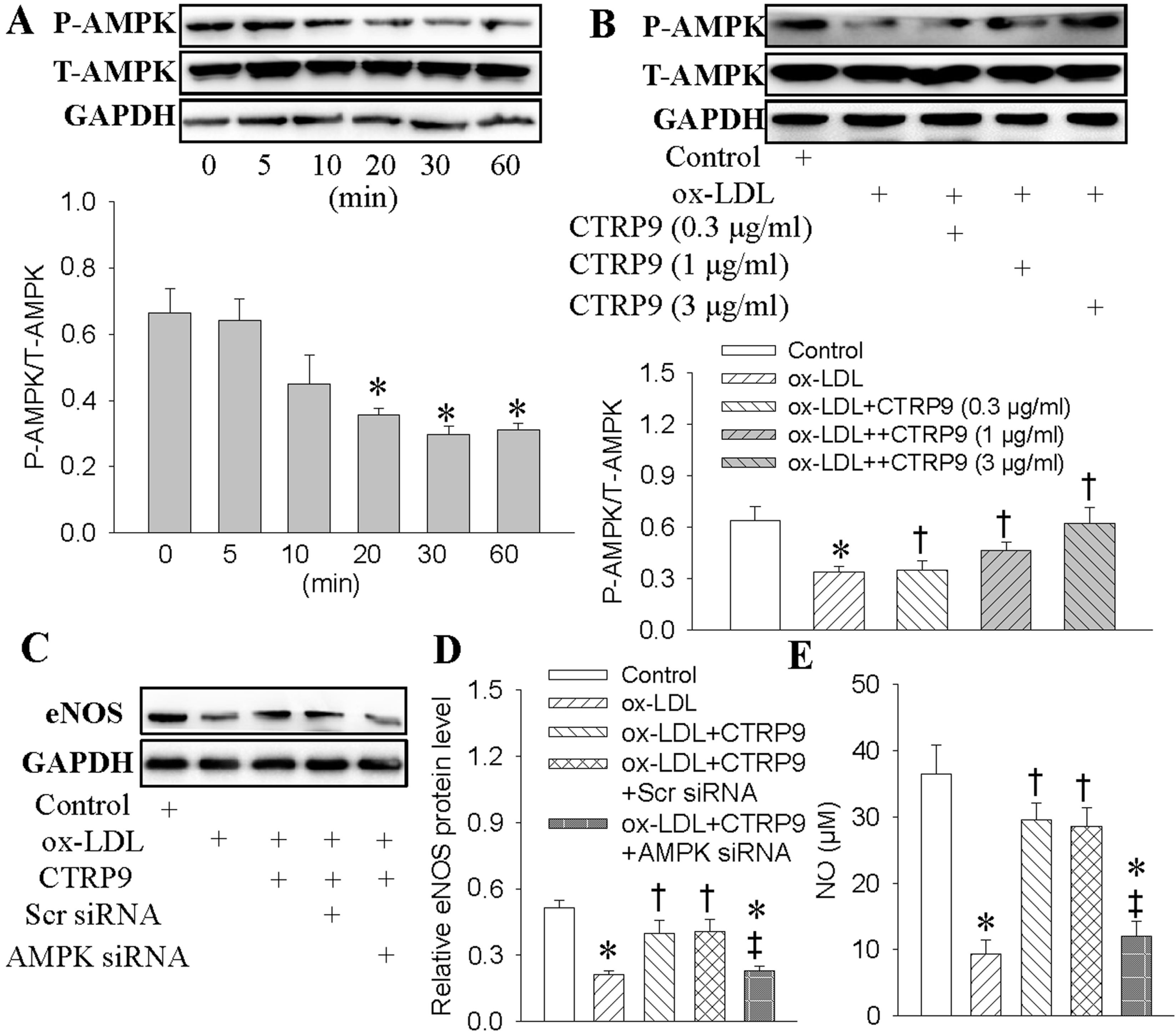
2.3. Effect of CTRP9 on Nitric Oxide (NO) Generation and Endothelial Nitric Oxide Synthase (eNOS) Levels
2.4. Activation of Adenosine Monophosphate-Activated Protein Kinase (AMPK) Mediated the Protective Effects of CTRP9
2.5. Proliferator-Activated Receptor γ Co-Activator 1α (PGC1-α) Was Involved in the Protective Effects of CTRP9
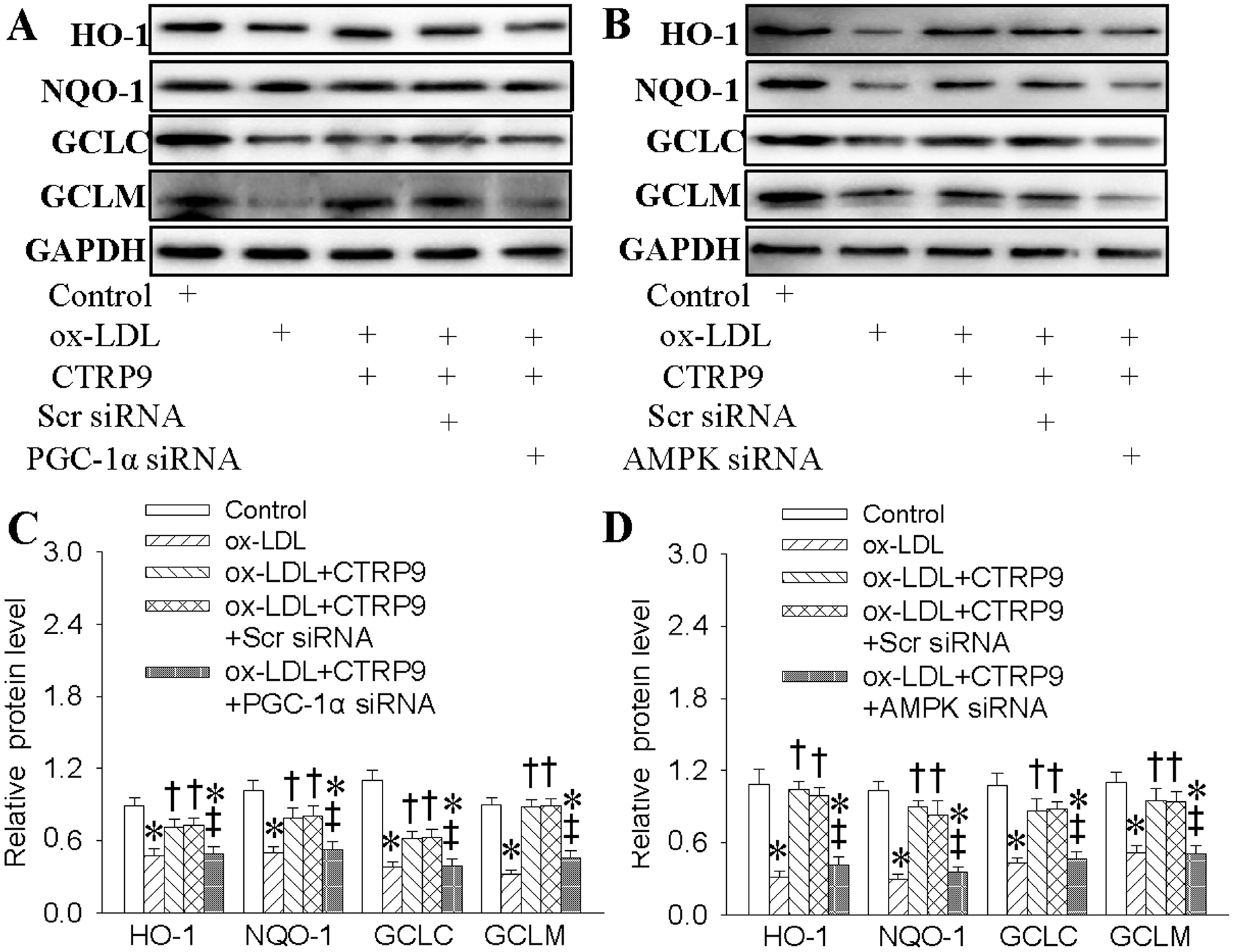
2.6. Both AMPK and PGC1-α Participated in CTRP9-Upregulated Antioxidant Enzymes Inductions
3. Discussion
4. Materials and Methods
4.1. Reagents and Chemicals
4.2. Cell Culture
4.3. Cytotoxicity of CTRP9
4.4. Cell Proliferation and Cell Cycle Assay
4.5. Migration Assay and Tube Formation Assay
4.6. Apoptosis Detection by Terminal Transferase-Mediated dUTP Nick End-Labeling (TUNEL) Staining
4.7. Nitric Oxide (NO) Generation Measurement
4.8. Intracellular Reactive Oxygen Species (ROS) Measurement
4.9. Immunofluorescence Assay
4.10. siRNA Transfections
4.11. Western Blot
4.12. Statistical Analysis
Supplementary Materials
Acknowledgment
Author Contributions
Conflicts of Interest
References
- Di Pietro, N.; Formoso, G.; Pandolfi, A. Physiology and pathophysiology of oxLDL uptake by vascular wall cells in atherosclerosis. Vasc. Pharmacol. 2016, 84, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Cochain, C.; Zernecke, A. Macrophages in vascular inflammation and atherosclerosis. Pflug. Arch. Eur. J. Phys. 2017, 469, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A.; Garcia-Cardena, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y. Transcriptional regulation of endothelial dysfunction in atherosclerosis: An epigenetic perspective. J. Biomed. Res. 2014, 28, 47–52. [Google Scholar] [PubMed]
- Gimbrone, M.A.; Garcia-Cardena, G. Vascular endothelium, hemodynamics, and the pathobiology of atherosclerosis. Cardiovasc. Pathol. 2013, 22, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, Q.; Wang, Z.; Wei, D. Shear stress in atherosclerotic plaque determination. DNA Cell Biol. 2014, 33, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Chen, C.; Yang, D.; Liao, Q.; Luo, H.; Wang, X.; Zhou, F.; Yang, X.; Yang, J.; Zeng, C.; et al. Mesenchymal stem cells-derived extracellular vesicles, via miR-210, improve infarcted cardiac function by promotion of angiogenesis. BBA Mol. Basis Dis. 2017. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Feng, H.; Sun, W.; Liu, K.; Lu, J.J.; Chen, X. Tert-butyl hydroperoxide (t-BHP) induced apoptosis and necroptosis in endothelial cells: Roles of NOX4 and mitochondrion. Redox Biol. 2017, 11, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Storniolo, C.E.; Rosello-Catafau, J.; Pinto, X.; Mitjavila, M.T.; Moreno, J.J. Polyphenol fraction of extra virgin olive oil protects against endothelial dysfunction induced by high glucose and free fatty acids through modulation of nitric oxide and endothelin-1. Redox Biol. 2014, 2, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Pandey, D.; Bhunia, A.; Oh, Y.J.; Chang, F.; Bergman, Y.; Kim, J.H.; Serbo, J.; Boronina, T.N.; Cole, R.N.; van Eyk, J.; et al. OxLDL triggers retrograde translocation of arginase2 in aortic endothelial cells via ROCK and mitochondrial processing peptidase. Circ. Res. 2014, 115, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Lubrano, V.; Balzan, S. LOX-1 and ROS, inseparable factors in the process of endothelial damage. Free Radic. Res. 2014, 48, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Yuan, Y.; Yi, W.; Lau, W.B.; Wang, Y.; Wang, X.; Sun, Y.; Lopez, B.L.; Christopher, T.A.; Peterson, J.M.; et al. C1q/TNF-related proteins, a family of novel adipokines, induce vascular relaxation through the adiponectin receptor-1/AMPK/eNOS/nitric oxide signaling pathway. Arterioscl. Throm. Vasc. Biol. 2011, 31, 2616–2623. [Google Scholar] [CrossRef] [PubMed]
- Kambara, T.; Ohashi, K.; Shibata, R.; Ogura, Y.; Maruyama, S.; Enomoto, T.; Uemura, Y.; Shimizu, Y.; Yuasa, D.; Matsuo, K.; et al. CTRP9 protein protects against myocardial injury following ischemia-reperfusion through AMP-activated protein kinase (AMPK)-dependent mechanism. J. Biol. Chem. 2012, 287, 18965–18973. [Google Scholar] [CrossRef] [PubMed]
- Uemura, Y.; Shibata, R.; Ohashi, K.; Enomoto, T.; Kambara, T.; Yamamoto, T.; Ogura, Y.; Yuasa, D.; Joki, Y.; Matsuo, K.; et al. Adipose-derived factor CTRP9 attenuates vascular smooth muscle cell proliferation and neointimal formation. FASEB J. 2013, 27, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.W.; Krawczyk, S.A.; Kitidis-Mitrokostas, C.; Ge, G.; Spooner, E.; Hug, C.; Gimeno, R.; Lodish, H.F. Identification and characterization of CTRP9, a novel secreted glycoprotein, from adipose tissue that reduces serum glucose in mice and forms heterotrimers with adiponectin. FASEB J. 2009, 23, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, P.; Li, T.; Liu, Y.; Zhu, Q.; Chen, T.; Liu, T.; Huang, C.; Zhang, J.; Zhang, Y.; et al. CTRP9 enhances carotid plaque stability by reducing pro-inflammatory cytokines in macrophages. Biochem. Biophys. Res. Commun. 2015, 458, 890–895. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Lee, M.J.; Kang, Y.M.; Lee, Y.L.; Seol, S.M.; Yoon, H.K.; Kang, S.W.; Lee, W.J.; Park, J.Y. C1q/TNF-related protein-9 inhibits cytokine-induced vascular inflammation and leukocyte adhesiveness via AMP-activated protein kinase activation in endothelial cells. Mol. Cell. Endocrinol. 2016, 419, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Vita, J.A.; Keaney, J.F. Endothelial function: A barometer for cardiovascular risk? Circulation 2002, 106, 640–642. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.J.; Hou, B.; Wang, X.; Zhu, X.X.; Li, K.X.; Qiu, L.Y. Endothelial dysfunction and cardiometabolic diseases: Role of long non-coding RNAs. Life Sci. 2016, 167, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. Angiogenesis in life, disease and medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Yan, M.; Mehta, J.L.; Hu, C. Angiogenesis is a link between atherosclerosis and tumorigenesis: Role of LOX-1. Cardiovasc. Drug Ther. 2011, 25, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Xie, W.; Xia, N.; He, Q.; Sun, T. Silencing of Transient Receptor Potential Channel 4 Alleviates oxLDL-induced Angiogenesis in Human Coronary Artery Endothelial Cells by Inhibition of VEGF and NF-κB. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2016, 22, 930–936. [Google Scholar] [CrossRef]
- Wang, L.; Li, G.; Chen, Q.; Ke, D. Octanoylated ghrelin attenuates angiogenesis induced by oxLDL in human coronary artery endothelial cells via the GHSR1a-mediated NF-κB pathway. Metabolism 2015, 64, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Rikitake, Y.; Kawashima, S.; Yamashita, T.; Ueyama, T.; Ishido, S.; Hotta, H.; Hirata, K.; Yokoyama, M. Lysophosphatidylcholine inhibits endothelial cell migration and proliferation via inhibition of the extracellular signal-regulated kinase pathway. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yi, W.; Yuan, Y.; Lau, W.B.; Yi, D.; Wang, X.; Wang, Y.; Su, H.; Wang, X.; Gao, E.; et al. C1q/tumor necrosis factor-related protein-9, a novel adipocyte-derived cytokine, attenuates adverse remodeling in the ischemic mouse heart via protein kinase A activation. Circulation 2013, 128, S113–S120. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Geng, X.; Wang, H.; Cheng, G.; Xu, S. CTRP9 Ameliorates Pulmonary Arterial Hypertension Through Attenuating Inflammation and Improving Endothelial Cell Survival and Function. J. Cardiovasc. Pharm. 2016, 67, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Cheng, L.; Yang, Y.; Fan, C.; Zhao, D.; Qin, Z.; Feng, X.; Zhao, L.; Ma, J.; Wang, X.; et al. C1q/TNF-Related Protein 9 Protects Diabetic Rat Heart against Ischemia Reperfusion Injury: Role of Endoplasmic Reticulum Stress. Oxid. Med. Cell. Longev. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Fu, G.; Dai, T.; Huang, H. Migration of endothelial progenitor cells mediated by stromal cell-derived factor-1α/CXCR4 via PI3K/Akt/eNOS signal transduction pathway. J. Cardiovasc. Pharm. 2007, 50, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Song, C.L.; Liu, B.; Shi, Y.F.; Liu, N.; Yan, Y.Y.; Zhang, J.C.; Xue, X.; Wang, J.P.; Zhao, Z.; Liu, J.G.; et al. MicroRNA-130a alleviates human coronary artery endothelial cell injury and inflammatory responses by targeting PTEN via activating PI3K/Akt/eNOS signaling pathway. Oncotarget 2016, 7, 71922–71936. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.K.; Shin, W.S.; Lee, W.Y.; Clark, S.L. Oxidized low-density lipoprotein decreases the expression of endothelial nitric oxide synthase. J. Biol. Chem. 1995, 270, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Si, L.Y. Protective effects of AMP-activated protein kinase in the cardiovascular system. J. Cell. Mol. Med. 2010, 14, 2604–2613. [Google Scholar] [CrossRef] [PubMed]
- Rojas, E.; Rodriguez-Molina, D.; Bolli, P.; Israili, Z. H.; Faria, J.; Fidilio, E.; Bermudez, V.; Velasco, M. The role of adiponectin in endothelial dysfunction and hypertension. Curr. Hypertens. Rep. 2014, 16, 463. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Lakshmanan, A.P.; Hwang, M.J.; Kubba, H.; Mushannen, A.; Triggle, C.R.; Ding, H. Metformin improves endothelial function in aortic tissue and microvascular endothelial cells subjected to diabetic hyperglycaemic conditions. Biochem. Pharmacol. 2015, 98, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wang, S.; Li, B.; Sun, A.; Zou, Y.; Ge, J. A protective role of ciglitazone in ox-LDL-induced rat microvascular endothelial cells via modulating PPARγ-dependent AMPK/eNOS pathway. J. Cell. Mol. Med. 2015, 19, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Huang, C.; Li, J.; Li, T.; Guo, H.; Liu, T.; Li, N.; Zhu, Q.; Guo, Y. Globular CTRP9 inhibits oxLDL-induced inflammatory response in RAW 264.7 macrophages via AMPK activation. Mol. Cell. Biochem. 2016, 417, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Patten, I.S.; Arany, Z. PGC-1 coactivators in the cardiovascular system. Trends Endocrin. Met. 2012, 23, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Chengye, Z.; Daixing, Z.; Qiang, Z.; Shusheng, L. PGC-1-related coactivator (PRC) negatively regulates endothelial adhesion of monocytes via inhibition of NF kappaB activity. Biochem. Biophys. Res. Commun. 2013, 439, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Park, K.G.; Yoo, E.K.; Kim, Y.H.; Kim, Y.N.; Kim, H.S.; Kim, H.T.; Park, J.Y.; Lee, K.U.; Jang, W.G.; et al. Effects of PGC-1α on TNF-α-induced MCP-1 and VCAM-1 expression and NF-kappaB activation in human aortic smooth muscle and endothelial cells. Antioxid. Redox Sign. 2007, 9, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.; Yang, X.; Li, W.; Bian, F.; Wu, D.; Chi, J.; Xu, G.; Zhang, Y.; Jin, S. Salidroside stimulates mitochondrial biogenesis and protects against H2O2-induced endothelial dysfunction. Oxid. Med. Cell. Longev. 2014, 2014, 904834. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Li, B.; Chen, X.; Su, J.; Wang, H.; Yu, S.; Zheng, Q. CTRP9 induces mitochondrial biogenesis and protects high glucose-induced endothelial oxidative damage via AdipoR1-SIRT1-PGC-1α activation. Biochem. Biophys. Res. Commun. 2016, 477, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Kellow, N.J.; Savige, G.S. Dietary advanced glycation end-product restriction for the attenuation of insulin resistance, oxidative stress and endothelial dysfunction: A systematic review. Eur. J. Clin. Nutr. 2013, 67, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, H.; Li, X.; Jia, B.; Yang, Y.; Zhou, P.; Li, P.; Chen, J. Miltirone protects human EA.hy926 endothelial cells from oxidized low-density lipoprotein-derived oxidative stress via a heme oxygenase-1 and MAPK/Nrf2 dependent pathway. Phytomedicine 2016, 23, 1806–1813. [Google Scholar] [CrossRef] [PubMed]
- Battelli, M.G.; Polito, L.; Bolognesi, A. Xanthine oxidoreductase in atherosclerosis pathogenesis: Not only oxidative stress. Atherosclerosis 2014, 237, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, W.J.; Frei, B. Quercetin inhibits LPS-induced adhesion molecule expression and oxidant production in human aortic endothelial cells by p38-mediated Nrf2 activation and antioxidant enzyme induction. Redox Biol. 2016, 9, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Maamoun, H.; Zachariah, M.; McVey, J.H.; Green, F.R.; Agouni, A. Heme oxygenase (HO)-1 induction prevents Endoplasmic Reticulum stress-mediated endothelial cell death and impaired angiogenic capacity. Biochem. Pharmacol. 2017, 127, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Pu, C.; Zhou, P.; Wang, P.; Liang, D.; Wang, Q.; Hu, Y.; Li, B.; Hao, X. Cinnamaldehyde prevents endothelial dysfunction induced by high glucose by activating Nrf2. Cell. Physiol. Biochem. 2015, 36, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, W.J.; Choi, J.; Frei, B. Quercetin affects glutathione levels and redox ratio in human aortic endothelial cells not through oxidation but formation and cellular export of quercetin-glutathione conjugates and upregulation of glutamate-cysteine ligase. Redox Biol. 2016, 9, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Yuan, Y.; Wang, X.M.; Lau, W.B.; Wang, Y.; Wang, X.; Gao, E.; Koch, W.J.; Ma, X.L. Inhibition of CTRP9, a novel and cardiac-abundantly expressed cell survival molecule, by TNF α-initiated oxidative signaling contributes to exacerbated cardiac injury in diabetic mice. Basic Res. Cardiol. 2013, 108, 315. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Yang, Y.; Yuan, Z.; Gao, Y.; Zhou, J.; Chen, Q.; Xu, Y. Myocardin-related transcription factor A mediates OxLDL-induced endothelial injury. Circ. Res. 2011, 108, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Yao, J.; Liu, J.Y.; Li, X.M.; Wang, X.Q.; Li, Y.J.; Tao, Z.F.; Song, Y.C.; Chen, Q.; Jiang, Q. lncRNA-MIAT regulates microvascular dysfunction by functioning as a competing endogenous RNA. Circ. Res. 2015, 116, 1143–1156. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.Y.; Shi, C.X.; Gao, R.; Sun, H.J.; Xiong, X.Q.; Ding, L.; Chen, Q.; Li, Y.H.; Wang, J.J.; Kang, Y.M.; et al. Irisin inhibits hepatic gluconeogenesis and increases glycogen synthesis via the PI3K/Akt pathway in type 2 diabetic mice and hepatocytes. Clin. Sci. 2015, 129, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.J.; Liu, T.Y.; Zhang, F.; Xiong, X.Q.; Wang, J.J.; Chen, Q.; Li, Y.H.; Kang, Y.M.; Zhou, Y.B.; Han, Y.; et al. Salusin-β contributes to vascular remodeling associated with hypertension via promoting vascular smooth muscle cell proliferation and vascular fibrosis. BBA Mol. Basis Dis. 2015, 1852, 1709–1718. [Google Scholar] [CrossRef] [PubMed]
- Shuang, E.; Kijima, R.; Honma, T.; Yamamoto, K.; Hatakeyama, Y.; Kitano, Y.; Kimura, T.; Nakagawa, K.; Miyazawa, T.; Tsuduki, T. 1-Deoxynojirimycin attenuates high glucose-accelerated senescence in human umbilical vein endothelial cells. Exp. Gerontol. 2014, 55, 63–69. [Google Scholar]
- Slator, C.; Molphy, Z.; McKee, V.; Kellett, A. Triggering autophagic cell death with a di-manganese(II) developmental therapeutic. Redox Biol. 2017, 12, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Shan, K.; Jiang, Q.; Wang, X.Q.; Wang, Y.N.; Yang, H.; Yao, M.D.; Liu, C.; Li, X.M.; Yao, J.; Liu, B.; et al. Role of long non-coding RNA-RNCR3 in atherosclerosis-related vascular dysfunction. Cell Death Dis. 2016, 7, e2248. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.N.; Shan, K.; Yao, M.D.; Yao, J.; Wang, J.J.; Li, X.; Liu, B.; Zhang, Y.Y.; Ji, Y.; Jiang, Q.; et al. Long Noncoding RNA-GAS5: A Novel Regulator of Hypertension-Induced Vascular Remodeling. Hypertension 2016, 68, 736–748. [Google Scholar] [CrossRef] [PubMed]
- Nagamine, A.; Hasegawa, H.; Hashimoto, N.; Yamada-Inagawa, T.; Hirose, M.; Kobara, Y.; Tadokoro, H.; Kobayashi, Y.; Takano, H. The effects of DPP-4 inhibitor on hypoxia-induced apoptosis in human umbilical vein endothelial cells. J. Pharmacol. Sci. 2017, 133, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Zeng, K.W.; Yu, Q.; Liao, L.X.; Song, F.J.; Lv, H.N.; Jiang, Y.; Tu, P.F. Anti-Neuroinflammatory Effect of MC13, a Novel Coumarin Compound From Condiment Murraya, Through Inhibiting Lipopolysaccharide-Induced TRAF6-TAK1-NF-κB, P38/ERK MAPKS and Jak2-Stat1/Stat3 Pathways. J. Cell. Biochem. 2015, 116, 1286–1299. [Google Scholar] [CrossRef] [PubMed]
- Kurland, D.B.; Gerzanich, V.; Karimy, J.K.; Woo, S.K.; Vennekens, R.; Freichel, M.; Nilius, B.; Bryan, J.; Simard, J.M. The Sur1-Trpm4 channel regulates NOS2 transcription in TLR4-activated microglia. J. Neuroinflamm. 2016, 13, 130. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.J.; Zhao, M.X.; Ren, X.S.; Liu, T.Y.; Chen, Q.; Li, Y.H.; Kang, Y.M.; Wang, J.J.; Zhu, G.Q. Salusin-β Promotes Vascular Smooth Muscle Cell Migration and Intimal Hyperplasia After Vascular Injury via ROS/NFκB/MMP-9 Pathway. Antioxid. Redox Sign. 2016, 24, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Ryu, J.C.; Kwon, Y.; Lee, S.; Bae, Y.S.; Yoon, J.H.; Ryu, J.H. Dual oxidase 2 in lung epithelia is essential for hyperoxia-induced acute lung injury in mice. Antioxid. Redox Sign. 2014, 21, 1803–1818. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yang, R.; Liu, H.; Ren, Z.; Wang, C.; Li, D.; Ma, X. Knockdown of peroxisome proliferator-activated receptor γ coactivator-1α increased apoptosis of human endometrial cancer HEC-1A cells. Onco Targets Ther. 2016, 9, 5329–5338. [Google Scholar]
- Huang, N.L.; Chiang, S.H.; Hsueh, C.H.; Liang, Y.J.; Chen, Y.J.; Lai, L.P. Metformin inhibits TNF-α-induced I κB kinase phosphorylation, I κB-α degradation and IL-6 production in endothelial cells through PI3K-dependent AMPK phosphorylation. Int. J. Cardiol. 2009, 134, 169–175. [Google Scholar] [CrossRef] [PubMed]






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, H.; Zhu, X.; Zhou, Y.; Cai, W.; Qiu, L. C1q/TNF-Related Protein-9 Ameliorates Ox-LDL-Induced Endothelial Dysfunction via PGC-1α/AMPK-Mediated Antioxidant Enzyme Induction. Int. J. Mol. Sci. 2017, 18, 1097. https://doi.org/10.3390/ijms18061097
Sun H, Zhu X, Zhou Y, Cai W, Qiu L. C1q/TNF-Related Protein-9 Ameliorates Ox-LDL-Induced Endothelial Dysfunction via PGC-1α/AMPK-Mediated Antioxidant Enzyme Induction. International Journal of Molecular Sciences. 2017; 18(6):1097. https://doi.org/10.3390/ijms18061097
Chicago/Turabian StyleSun, Haijian, Xuexue Zhu, Yuetao Zhou, Weiwei Cai, and Liying Qiu. 2017. "C1q/TNF-Related Protein-9 Ameliorates Ox-LDL-Induced Endothelial Dysfunction via PGC-1α/AMPK-Mediated Antioxidant Enzyme Induction" International Journal of Molecular Sciences 18, no. 6: 1097. https://doi.org/10.3390/ijms18061097
APA StyleSun, H., Zhu, X., Zhou, Y., Cai, W., & Qiu, L. (2017). C1q/TNF-Related Protein-9 Ameliorates Ox-LDL-Induced Endothelial Dysfunction via PGC-1α/AMPK-Mediated Antioxidant Enzyme Induction. International Journal of Molecular Sciences, 18(6), 1097. https://doi.org/10.3390/ijms18061097