
Perturbations in the Replication Program Contribute to Genomic Instability in Cancer

Abstract
:1. Introduction
2. Replication Stress
2.1. Mechanisms of Replication Stress
2.2. Resolution of Replication Stress
2.3. Replication Stress Leads to Genomic Instability, Common Fragile Sites Breakage, and Cancer
2.4. The DDR in Cancer Progression and Therapy
3. Replication Timing
3.1. Background
3.2. Effects of Replication Timing on Mutation Rates and Structural Variations
3.3. Replication Timing Changes and Cancer
3.4. Causes of Replication Timing Changes in Cancer
3.5. Effects of Changes in Replication Timing in Cancer
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| DNA | Deoxyribonucleic acid |
| S phase | Synthesis phase |
| ssDNA | Single stranded DNA |
| DSB | Double strand breaks |
| RPA | Replication protein A |
| ATR | Ataxia telangiectasia and Rad3-related protein |
| ROS | Reactive oxygen species |
| dNTP | Deoxynucleotide triphosphate |
| RNA | Ribonucleic acid |
| RNase H | Ribonuclease H |
| DDR | DNA damage response |
| Chk1 | Checkpoint kinase 1 |
| MRN | Mre11-Rad50-Nbs1 |
| Chk2 | Checkpoint kinase 2 |
| H2AX | H2A histone family member X |
| DDT | DNA damage tolerance |
| LOH | Loss of heterozygosity |
| CFS | Common fragile site |
| ERFS | Early replication fragile site |
| CNV | Copy number variations |
| PARP1 | Poly(ADP-ribose) polymerase 1 |
| GOF p53 | Gain of function p53 |
| CTR | Constant timing regions |
| TTR | Timing transition regions |
| MEF | Mouse embryonic fibroblasts |
| BrdU | Bromodeoxyuridine |
| SNP | Single nucleotide polymorphism |
| NHEJ | Non-homologous end-joining |
| RUNX1 | Runt-related transcription factor 1 |
| p53 | Tumor protein P53 |
| RB1 | Retinoblastoma 1 |
| Bcl2 | B-cell lymphoma 2 |
| Rif1 | Replication timing regulatory factor 1 |
| MCM | Minichromosome maintenance |
| ALL | Acute lymphoblastic leukemia |
References
- Tomasetti, C.; Li, L.; Vogelstein, B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 2017, 355, 1330–1334. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Vogelstein, B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, T.A. Evolving views of DNA replication (in)fidelity. Cold Spring Harb. Symp. Quant. Biol. 2009, 74, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Li, G.-M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Mutter-Rottmayer, E.; Zlatanou, A.; Vaziri, C.; Yang, Y. Mechanisms of post-replication DNA repair. Genes 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Farkash-Amar, S.; Simon, I. Genome-wide analysis of the replication program in mammals. Chromosome Res. 2010, 18, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Ryba, T.; Battaglia, D.; Chang, B.H.; Shirley, J.W.; Buckley, Q.; Pope, B.D.; Devidas, M.; Druker, B.J.; Gilbert, D.M. Abnormal developmental control of replication-timing domains in pediatric acute lymphoblastic leukemia. Genome Res. 2012, 22, 1833–1844. [Google Scholar] [CrossRef] [PubMed]
- Fritz, A.; Sinha, S.; Marella, N.; Berezney, R. Alterations in replication timing of cancer-related genes in malignant human breast cancer cells. J. Cell. Biochem. 2013, 114, 1074–1083. [Google Scholar] [CrossRef] [PubMed]
- Alvino, G.M.; Collingwood, D.; Murphy, J.M.; Delrow, J.; Brewer, B.J.; Raghuraman, M.K. Replication in Hydroxyurea: It is a Matter of Time. Mol. Cell. Biol. 2007, 27, 6396–6406. [Google Scholar] [CrossRef] [PubMed]
- Koren, A.; Soifer, I.; Barkai, N. MRC1-dependent scaling of the budding yeast DNA replication timing program. Genome Res. 2010, 20, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Heintz, N.H.; Dailey, L.; Held, P.; Heintz, N. Eukaryotic replication origins as promoters of bidirectional DNA synthesis. Trends Genet. TIG 1992, 8, 376–381. [Google Scholar] [CrossRef]
- Burhans, W.C.; Weinberger, M. DNA replication stress, genome instability and aging. Nucleic Acids Res. 2007, 35, 7545–7556. [Google Scholar] [CrossRef] [PubMed]
- Van, C.; Yan, S.; Michael, W.M.; Waga, S.; Cimprich, K.A. Continued primer synthesis at stalled replication forks contributes to checkpoint activation. J. Cell Biol. 2010, 189, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Byun, T.S.; Pacek, M.; Yee, M.; Walter, J.C.; Cimprich, K.A. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005, 19, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 2010, 11, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.-Q.; Zhang, K.; Wu, X.-C.; Mieczkowski, P.A.; Petes, T.D. Global analysis of genomic instability caused by DNA replication stress in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2016, 113, E8114–E8121. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A.; Lindsey-Boltz, L.A.; Ünsal-Kaçmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, S.M. DNA replication: Driving past four-stranded snags. Nature 2013, 497, 449–450. [Google Scholar] [CrossRef] [PubMed]
- McElhinny, S.A.N.; Kumar, D.; Clark, A.B.; Watt, D.L.; Watts, B.E.; Lundström, E.-B.; Johansson, E.; Chabes, A.; Kunkel, T.A. Genome instability due to ribonucleotide incorporation into DNA. Nat. Chem. Biol. 2010, 6, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Beck, H.; Nähse-Kumpf, V.; Larsen, M.S.Y.; O’Hanlon, K.A.; Patzke, S.; Holmberg, C.; Mejlvang, J.; Groth, A.; Nielsen, O.; Syljuåsen, R.G.; et al. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol. Cell. Biol. 2012, 32, 4226–4236. [Google Scholar] [CrossRef] [PubMed]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Mejlvang, J.; Feng, Y.; Alabert, C.; Neelsen, K.J.; Jasencakova, Z.; Zhao, X.; Lees, M.; Sandelin, A.; Pasero, P.; Lopes, M.; et al. New histone supply regulates replication fork speed and PCNA unloading. J. Cell Biol. 2014, 204, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Groth, A.; Corpet, A.; Cook, A.J.L.; Roche, D.; Bartek, J.; Lukas, J.; Almouzni, G. Regulation of replication fork progression through histone supply and demand. Science 2007, 318, 1928–1931. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Altmeyer, M.; Rask, M.-B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Nakato, R.; Katou, Y.; Shirahige, K.; Araki, H. Origin Association of Sld3, Sld7, and Cdc45 Proteins Is a Key Step for Determination of Origin-Firing Timing. Curr. Biol. 2011, 21, 2055–2063. [Google Scholar] [CrossRef] [PubMed]
- Macheret, M.; Halazonetis, T.D. DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. 2015, 10, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Olavarrieta, L.; Hernández, P.; Krimer, D.B.; Schvartzman, J.B. DNA knotting caused by head-on collision of transcription and replication. J. Mol. Biol. 2002, 322, 1–6. [Google Scholar] [CrossRef]
- Aguilera, A.; García-Muse, T. R loops: From transcription byproducts to threats to genome stability. Mol. Cell 2012, 46, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Tuduri, S.; Crabbé, L.; Conti, C.; Tourrière, H.; Holtgreve-Grez, H.; Jauch, A.; Pantesco, V.; De Vos, J.; Thomas, A.; Theillet, C.; et al. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 2009, 11, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, R.; Capra, T.; Gonzalez-Huici, V.; Fachinetti, D.; Cocito, A.; Natoli, G.; Katou, Y.; Mori, H.; Kurokawa, K.; Shirahige, K.; et al. Genome-organizing factors Top2 and Hmo1 prevent chromosome fragility at sites of S phase transcription. Cell 2009, 138, 870–884. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Shatalin, K.; Epshtein, V.; Gottesman, M.E.; Nudler, E. Linking RNA polymerase backtracking to genome instability in E. coli. Cell 2011, 146, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Hills, S.A.; Diffley, J.F.X. DNA replication and oncogene-induced replicative stress. Curr. Biol. 2014, 24, R435–R444. [Google Scholar] [CrossRef] [PubMed]
- Stiff, T.; Alagoz, M.; Alcantara, D.; Outwin, E.; Brunner, H.G.; Bongers, E.M.H.F.; O’Driscoll, M.; Jeggo, P.A. Deficiency in origin licensing proteins impairs cilia formation: Implications for the aetiology of Meier-Gorlin syndrome. PLoS Genet. 2013, 9, e1003360. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.L.; Orr-Weaver, T.L. Replication fork instability and the consequences of fork collisions from rereplication. Genes Dev. 2016, 30, 2241–2252. [Google Scholar] [CrossRef] [PubMed]
- Mantiero, D.; Mackenzie, A.; Donaldson, A.; Zegerman, P. Limiting replication initiation factors execute the temporal programme of origin firing in budding yeast. EMBO J. 2011, 30, 4805–4814. [Google Scholar] [CrossRef] [PubMed]
- Helmrich, A.; Ballarino, M.; Nudler, E.; Tora, L. Transcription-replication encounters, consequences and genomic instability. Nat. Struct. Mol. Biol. 2013, 20, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Diffley, J.F.X. Deregulated G1-cyclin expression induces genomic instability by preventing efficient pre-RC formation. Genes Dev. 2002, 16, 2639–2649. [Google Scholar] [CrossRef] [PubMed]
- Lengronne, A.; Schwob, E. The yeast CDK inhibitor Sic1 prevents genomic instability by promoting replication origin licensing in late G1. Mol. Cell 2002, 9, 1067–1078. [Google Scholar] [CrossRef]
- Kunnev, D.; Rusiniak, M.E.; Kudla, A.; Freeland, A.; Cady, G.K.; Pruitt, S.C. DNA damage response and tumorigenesis in Mcm2-deficient mice. Oncogene 2010, 29, 3630–3638. [Google Scholar] [CrossRef] [PubMed]
- Shreeram, S.; Sparks, A.; Lane, D.P.; Blow, J.J. Cell type-specific responses of human cells to inhibition of replication licensing. Oncogene 2002, 21, 6624–6632. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Q.; Jackson, D.A.; Blow, J.J. Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev. 2007, 21, 3331–3341. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, A.; Schwob, E.; Méndez, J. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc. Natl. Acad. Sci. USA 2008, 105, 8956–8961. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, D.; Blow, J.J. Dormant origins, the licensing checkpoint, and the response to replicative stresses. Cold Spring Harb. Perspect. Biol. 2012, 4, a012955. [Google Scholar] [CrossRef] [PubMed]
- Shima, N.; Alcaraz, A.; Liachko, I.; Buske, T.R.; Andrews, C.A.; Munroe, R.J.; Hartford, S.A.; Tye, B.K.; Schimenti, J.C. A viable allele of Mcm4 causes chromosome instability and mammary adenocarcinomas in mice. Nat. Genet. 2007, 39, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Rohban, S.; Campaner, S. Myc induced replicative stress response: How to cope with it and exploit it. Biochim. Biophys. Acta 2015, 1849, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Neiman, P.E.; Kimmel, R.; Icreverzi, A.; Elsaesser, K.; Bowers, S.-J.; Burnside, J.; Delrow, J. Genomic instability during Myc-induced lymphomagenesis in the bursa of Fabricius. Oncogene 2006, 25, 6325–6335. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.; Asawachaicharn, N.; Galloway, D.A.; Grandori, C. c-Myc accelerates S-phase and requires WRN to avoid replication stress. PLoS ONE 2009, 4, e5951. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.V.; Dominguez-Sola, D.; Wang, L.C.; Hyrien, O.; Gautier, J. Cdc45 is a critical effector of myc-dependent DNA replication stress. Cell Rep. 2013, 3, 1629–1639. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Sola, D.; Ying, C.Y.; Grandori, C.; Ruggiero, L.; Chen, B.; Li, M.; Galloway, D.A.; Gu, W.; Gautier, J.; Dalla-Favera, R. Non-transcriptional control of DNA replication by c-Myc. Nature 2007, 448, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Maya-Mendoza, A.; Ostrakova, J.; Kosar, M.; Hall, A.; Duskova, P.; Mistrik, M.; Merchut-Maya, J.M.; Hodny, Z.; Bartkova, J.; Christensen, C.; et al. Myc and Ras oncogenes engage different energy metabolism programs and evoke distinct patterns of oxidative and DNA replication stress. Mol. Oncol. 2015, 9, 601–616. [Google Scholar] [CrossRef] [PubMed]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Lindström, M.S.; Wiman, K.G. Myc and E2F1 induce p53 through p14ARF-independent mechanisms in human fibroblasts. Oncogene 2003, 22, 4993–5005. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.-V.F.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Davidson, I.F.; Li, A.; Blow, J.J. Deregulated replication licensing causes DNA fragmentation consistent with head-to-tail fork collision. Mol. Cell 2006, 24, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre’, M.; Nuciforo, P.G.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Miron, K.; Golan-Lev, T.; Dvir, R.; Ben-David, E.; Kerem, B. Oncogenes create a unique landscape of fragile sites. Nat. Commun. 2015, 6, 7094. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Horejsí, Z.; Koed, K.; Krämer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Matsumura, I.; Ezoe, S.; Satoh, Y.; Sakamaki, T.; Albanese, C.; Machii, T.; Pestell, R.G.; Kanakura, Y. E2F1 and c-Myc potentiate apoptosis through inhibition of NF-κB activity that facilitates MnSOD-mediated ROS elimination. Mol. Cell 2002, 9, 1017–1029. [Google Scholar] [CrossRef]
- Costantino, L.; Sotiriou, S.K.; Rantala, J.K.; Magin, S.; Mladenov, E.; Helleday, T.; Haber, J.E.; Iliakis, G.; Kallioniemi, O.P.; Halazonetis, T.D. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 2014, 343, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Ekholm-Reed, S.; Méndez, J.; Tedesco, D.; Zetterberg, A.; Stillman, B.; Reed, S.I. Deregulation of cyclin E in human cells interferes with prereplication complex assembly. J. Cell Biol. 2004, 165, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Mortusewicz, O.; Afzal, I.; Lorvellec, M.; García, P.; Helleday, T.; Petermann, E. Increased replication initiation and conflicts with transcription underlie Cyclin E-induced replication stress. Oncogene 2013, 32, 3744–3753. [Google Scholar] [CrossRef] [PubMed]
- Ray, D.; Terao, Y.; Fuhrken, P.G.; Ma, Z.-Q.; DeMayo, F.J.; Christov, K.; Heerema, N.A.; Franks, R.; Tsai, S.Y.; Papoutsakis, E.T.; et al. Deregulated CDC25A expression promotes mammary tumorigenesis with genomic instability. Cancer Res. 2007, 67, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Dungrawala, H.; Rose, K.L.; Bhat, K.P.; Mohni, K.N.; Glick, G.G.; Couch, F.B.; Cortez, D. The replication checkpoint prevents two types of fork collapse without regulating replisome stability. Mol. Cell 2015, 59, 998–1010. [Google Scholar] [CrossRef] [PubMed]
- Couch, F.B.; Bansbach, C.E.; Driscoll, R.; Luzwick, J.W.; Glick, G.G.; Bétous, R.; Carroll, C.M.; Jung, S.Y.; Qin, J.; Cimprich, K.A.; et al. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 2013, 27, 1610–1623. [Google Scholar] [CrossRef] [PubMed]
- Blow, J.J.; Ge, X.Q.; Jackson, D.A. How dormant origins promote complete genome replication. Trends Biochem. Sci. 2011, 36, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, T.; Luebben, S.W.; Yamaguchi, S.; Ilves, I.; Matise, I.; Buske, T.; Botchan, M.R.; Shima, N. Stalled fork rescue via dormant replication origins in unchallenged S phase promotes proper chromosome segregation and tumor suppression. Mol. Cell 2011, 41, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Washington, M.T. Translesion synthesis: Insights into the selection and switching of DNA polymerases. Genes 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Q.; Blow, J.J. Chk1 inhibits replication factory activation but allows dormant origin firing in existing factories. J. Cell Biol. 2010, 191, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. Interplay of replication checkpoints and repair proteins at stalled replication forks. DNA Repair 2007, 6, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Murga, M.; Gutierrez-Martinez, P.; Soria, R.; Fernandez-Capetillo, O. ATR signaling can drive cells into senescence in the absence of DNA breaks. Genes Dev. 2008, 22, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, C.; Lukas, J. Checking on DNA damage in S phase. Nat. Rev. Mol. Cell Biol. 2004, 5, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Lomonosov, M.; Anand, S.; Sangrithi, M.; Davies, R.; Venkitaraman, A.R. Stabilization of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Genes Dev. 2003, 17, 3017–3022. [Google Scholar] [CrossRef] [PubMed]
- Mazouzi, A.; Velimezi, G.; Loizou, J.I. DNA replication stress: Causes, resolution and disease. Exp. Cell Res. 2014, 329, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Zou, L. DNA damage sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef] [PubMed]
- Ammazzalorso, F.; Pirzio, L.M.; Bignami, M.; Franchitto, A.; Pichierri, P. ATR and ATM differently regulate WRN to prevent DSBs at stalled replication forks and promote replication fork recovery. EMBO J. 2010, 29, 3156–3169. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Kannouche, P.L.; Lehmann, A.R. Ubiquitination of PCNA and the polymerase switch in human cells. Cell Cycle Georget. Tex. 2004, 3, 1011–1013. [Google Scholar] [CrossRef]
- Sale, J.E. Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012708. [Google Scholar] [CrossRef] [PubMed]
- Waters, L.S.; Minesinger, B.K.; Wiltrout, M.E.; D’Souza, S.; Woodruff, R.V.; Walker, G.C. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol. Mol. Biol. Rev. 2009, 73, 134–154. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D. Ubiquitin family modifications and template switching. FEBS Lett. 2011, 585, 2810–2817. [Google Scholar] [CrossRef] [PubMed]
- Arlt, M.F.; Mulle, J.G.; Schaibley, V.M.; Ragland, R.L.; Durkin, S.G.; Warren, S.T.; Glover, T.W. Replication stress induces genome-wide copy number changes in human cells that resemble polymorphic and pathogenic variants. Am. J. Hum. Genet. 2009, 84, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Arlt, M.F.; Wilson, T.E.; Glover, T.W. Replication stress and mechanisms of CNV formation. Curr. Opin. Genet. Dev. 2012, 22, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, T.; Magdalou, I.; Barascu, A.; Técher, H.; Debatisse, M.; Lopez, B.S. Spontaneous slow replication fork progression elicits mitosis alterations in homologous recombination-deficient mammalian cells. Proc. Natl. Acad. Sci. USA 2014, 111, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.; van der Burg, M.; Szuhai, K.; Kops, G.J.P.L.; Medema, R.H. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 2011, 333, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Neelsen, K.J.; Zanini, I.M.Y.; Herrador, R.; Lopes, M. Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. J. Cell Biol. 2013, 200, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.-C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Ozeri-Galai, E.; Tur-Sinai, M.; Bester, A.C.; Kerem, B. Interplay between genetic and epigenetic factors governs common fragile site instability in cancer. Cell. Mol. Life Sci. 2014, 71, 4495–4506. [Google Scholar] [CrossRef] [PubMed]
- Admire, A.; Shanks, L.; Danzl, N.; Wang, M.; Weier, U.; Stevens, W.; Hunt, E.; Weinert, T. Cycles of chromosome instability are associated with a fragile site and are increased by defects in DNA replication and checkpoint controls in yeast. Genes Dev. 2006, 20, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Arlt, M.F.; Durkin, S.G.; Ragland, R.L.; Glover, T.W. Common fragile sites as targets for chromosome rearrangements. DNA Repair 2006, 5, 1126–1135. [Google Scholar] [CrossRef] [PubMed]
- Barlow, J.H.; Faryabi, R.B.; Callén, E.; Wong, N.; Malhowski, A.; Chen, H.T.; Gutierrez-Cruz, G.; Sun, H.-W.; McKinnon, P.; Wright, G.; et al. Identification of early replicating fragile sites that contribute to genome instability. Cell 2013, 152, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Mortusewicz, O.; Herr, P.; Helleday, T. Early replication fragile sites: Where replication-transcription collisions cause genetic instability. EMBO J. 2013, 32, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Le Beau, M.M.; Rassool, F.V.; Neilly, M.E.; Espinosa, R.; Glover, T.W.; Smith, D.I.; McKeithan, T.W. Replication of a common fragile site, FRA3B, occurs late in S phase and is delayed further upon induction: Implications for the mechanism of fragile site induction. Hum. Mol. Genet. 1998, 7, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.G.; Vassiliou, L.-V.F.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Tsantoulis, P.K.; Kotsinas, A.; Sfikakis, P.P.; Evangelou, K.; Sideridou, M.; Levy, B.; Mo, L.; Kittas, C.; Wu, X.-R.; Papavassiliou, A.G.; et al. Oncogene-induced replication stress preferentially targets common fragile sites in preneoplastic lesions. A genome-wide study. Oncogene 2008, 27, 3256–3264. [Google Scholar] [CrossRef] [PubMed]
- Burrow, A.A.; Williams, L.E.; Pierce, L.C.T.; Wang, Y.-H. Over half of breakpoints in gene pairs involved in cancer-specific recurrent translocations are mapped to human chromosomal fragile sites. BMC Genom. 2009, 10, 59. [Google Scholar] [CrossRef] [PubMed]
- Courtois-Cox, S.; Jones, S.L.; Cichowski, K. Many roads lead to oncogene-induced senescence. Oncogene 2008, 27, 2801–2809. [Google Scholar] [CrossRef] [PubMed]
- Sarkisian, C.J.; Keister, B.A.; Stairs, D.B.; Boxer, R.B.; Moody, S.E.; Chodosh, L.A. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat. Cell Biol. 2007, 9, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Berti, M.; Vindigni, A. Replication stress: Getting back on track. Nat. Struct. Mol. Biol. 2016, 23, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.-J.; Peng, G. Cellular responses to replication stress: Implications in cancer biology and therapy. DNA Repair 2017, 49, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Dai, Q.; Park, D.; Deng, X. Targeting DNA replication stress for cancer therapy. Genes 2016, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Klusmann, I.; Rodewald, S.; Müller, L.; Friedrich, M.; Wienken, M.; Li, Y.; Schulz-Heddergott, R.; Dobbelstein, M. p53 Activity results in DNA replication fork processivity. Cell Rep. 2016, 17, 1845–1857. [Google Scholar] [CrossRef] [PubMed]
- Rhind, N.; Gilbert, D.M. DNA replication timing. Cold Spring Harb. Perspect. Biol. 2013, 5, a010132. [Google Scholar] [CrossRef] [PubMed]
- Hiratani, I.; Ryba, T.; Itoh, M.; Yokochi, T.; Schwaiger, M.; Chang, C.-W.; Lyou, Y.; Townes, T.M.; Schübeler, D.; Gilbert, D.M. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 2008, 6, e245. [Google Scholar] [CrossRef] [PubMed]
- Yehuda, Y.; Blumenfeld, B.; Lehmann, D.; Simon, I. Genome-wide determination of mammalian replication timing by DNA content measurement. J. Vis. Exp. JoVE 2017. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Mulia, J.C.; Buckley, Q.; Sasaki, T.; Zimmerman, J.; Didier, R.A.; Nazor, K.; Loring, J.F.; Lian, Z.; Weissman, S.; Robins, A.J.; et al. Dynamic changes in replication timing and gene expression during lineage specification of human pluripotent stem cells. Genome Res. 2015, 25, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Hiratani, I.; Ryba, T.; Itoh, M.; Rathjen, J.; Kulik, M.; Papp, B.; Fussner, E.; Bazett-Jones, D.P.; Plath, K.; Dalton, S.; et al. Genome-wide dynamics of replication timing revealed by in vitro models of mouse embryogenesis. Genome Res. 2010, 20, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Dileep, V.; Rivera-Mulia, J.C.; Sima, J.; Gilbert, D.M. Large-Scale Chromatin Structure-Function Relationships during the cell cycle and development: Insights from replication timing. Cold Spring Harb. Symp. Quant. Biol. 2015, 80, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Hiratani, I.; Takebayashi, S.; Lu, J.; Gilbert, D.M. Replication timing and transcriptional control: Beyond cause and effect—Part II. Curr. Opin. Genet. Dev. 2009, 19, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Ryba, T.; Battaglia, D.; Pope, B.D.; Hiratani, I.; Gilbert, D.M. Genome-scale analysis of replication timing: From bench to bioinformatics. Nat. Protoc. 2011, 6, 870–895. [Google Scholar] [CrossRef] [PubMed]
- Sima, J.; Gilbert, D.M. Complex correlations: Replication timing and mutational landscapes during cancer and genome evolution. Curr. Opin. Genet. Dev. 2014, 25, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Stamatoyannopoulos, J.A.; Adzhubei, I.; Thurman, R.E.; Kryukov, G.V.; Mirkin, S.M.; Sunyaev, S.R. Human mutation rate associated with DNA replication timing. Nat. Genet. 2009, 41, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-L.; Rappailles, A.; Duquenne, L.; Huvet, M.; Guilbaud, G.; Farinelli, L.; Audit, B.; d’Aubenton-Carafa, Y.; Arneodo, A.; Hyrien, O.; et al. Impact of replication timing on non-CpG and CpG substitution rates in mammalian genomes. Genome Res. 2010, 20, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Koren, A.; Polak, P.; Nemesh, J.; Michaelson, J.J.; Sebat, J.; Sunyaev, S.R.; McCarroll, S.A. Differential relationship of DNA replication timing to different forms of human mutation and variation. Am. J. Hum. Genet. 2012, 91, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Fujiyama, A.; Ichiba, Y.; Hattori, M.; Yada, T.; Sakaki, Y.; Ikemura, T. Chromosome-wide assessment of replication timing for human chromosomes 11q and 21q: Disease-related genes in timing-switch regions. Hum. Mol. Genet. 2002, 11, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Pink, C.J.; Hurst, L.D. Timing of replication is a determinant of neutral substitution rates but does not explain slow Y chromosome evolution in rodents. Mol. Biol. Evol. 2010, 27, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.C.; Pink, C.J.; Hurst, L.D. Late-replicating domains have higher divergence and diversity in Drosophila melanogaster. Mol. Biol. Evol. 2012, 29, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.H.; Li, W.-H. DNA replication timing and selection shape the landscape of nucleotide variation in cancer genomes. Nat. Commun. 2012, 3, 1004. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, A.; Furuta, M.; Totoki, Y.; Tsunoda, T.; Kato, M.; Shiraishi, Y.; Tanaka, H.; Taniguchi, H.; Kawakami, Y.; Ueno, M.; et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet. 2016, 48, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Lehner, B. Differential DNA mismatch repair underlies mutation rate variation across the human genome. Nature 2015, 521, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; De, S.; Michor, F. DNA replication timing and higher-order nuclear organization determine single-nucleotide substitution patterns in cancer genomes. Nat. Commun. 2013, 4, 1502. [Google Scholar] [CrossRef] [PubMed]
- Jia, P.; Jin, H.; Meador, C.B.; Xia, J.; Ohashi, K.; Liu, L.; Pirazzoli, V.; Dahlman, K.B.; Politi, K.; Michor, F.; et al. Next-generation sequencing of paired tyrosine kinase inhibitor-sensitive and -resistant EGFR mutant lung cancer cell lines identifies spectrum of DNA changes associated with drug resistance. Genome Res. 2013, 23, 1434–1445. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Kucab, J.E.; Morganella, S.; Glodzik, D.; Alexandrov, L.B.; Arlt, V.M.; Weninger, A.; Hollstein, M.; Stratton, M.R.; Phillips, D.H. The genome as a record of environmental exposure. Mutagenesis 2015, 30, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Lang, G.I.; Murray, A.W. Mutation rates across budding yeast chromosome VI are correlated with replication timing. Genome Biol. Evol. 2011, 3, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Agier, N.; Fischer, G. The mutational profile of the yeast genome is shaped by replication. Mol. Biol. Evol. 2012, 29, 905–913. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Michor, F. DNA replication timing and long-range DNA interactions predict mutational landscapes of cancer genomes. Nat. Biotechnol. 2011, 29, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Ikemura, T.; Sugimura, H. Amplicons on human chromosome 11q are located in the early/late-switch regions of replication timing. Genomics 2004, 84, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Li, H.; Hu, M.; Sasaki, T.; Baccei, A.; Gilbert, D.M.; Liu, J.S.; Collins, J.J.; Lerou, P.H. The distribution of genomic variations in human iPSCs is related to replication-timing reorganization during reprogramming. Cell Rep. 2014, 7, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Cardoso-Moreira, M.M.; Long, M. Mutational bias shaping fly copy number variation: Implications for genome evolution. Trends Genet. TIG 2010, 26, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Cardoso-Moreira, M.; Emerson, J.J.; Clark, A.G.; Long, M. Drosophila duplication hotspots are associated with late-replicating regions of the genome. PLoS Genet. 2011, 7, e1002340. [Google Scholar] [CrossRef] [PubMed]
- Drier, Y.; Lawrence, M.S.; Carter, S.L.; Stewart, C.; Gabriel, S.B.; Lander, E.S.; Meyerson, M.; Beroukhim, R.; Getz, G. Somatic rearrangements across cancer reveal classes of samples with distinct patterns of DNA breakage and rearrangement-induced hypermutability. Genome Res. 2013, 23, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Janoueix-Lerosey, I.; Hupé, P.; Maciorowski, Z.; La Rosa, P.; Schleiermacher, G.; Pierron, G.; Liva, S.; Barillot, E.; Delattre, O. Preferential occurrence of chromosome breakpoints within early replicating regions in neuroblastoma. Cell Cycle Georget. Tex. 2005, 4, 1842–1846. [Google Scholar] [CrossRef] [PubMed]
- Shugay, M.; Ortiz de Mendíbil, I.; Vizmanos, J.L.; Novo, F.J. Genomic hallmarks of genes involved in chromosomal translocations in hematological cancer. PLoS Comput. Biol. 2012, 8, e1002797. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, E.; Farkash-Amar, S.; Polten, A.; Yakhini, Z.; Tanay, A.; Simon, I. Comparative analysis of DNA replication timing reveals conserved large-scale chromosomal architecture. PLoS Genet. 2010, 6, e1001011. [Google Scholar] [CrossRef] [PubMed]
- Letessier, A.; Millot, G.A.; Koundrioukoff, S.; Lachagès, A.-M.; Vogt, N.; Hansen, R.S.; Malfoy, B.; Brison, O.; Debatisse, M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 2011, 470, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Palakodeti, A.; Han, Y.; Jiang, Y.; Le Beau, M.M. The role of late/slow replication of the FRA16D in common fragile site induction. Genes Chromosomes Cancer 2004, 39, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Darling, J.; Zhang, J.S.; Huang, H.; Liu, W.; Smith, D.I. Allele-specific late replication and fragility of the most active common fragile site, FRA3B. Hum. Mol. Genet. 1999, 8, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.S.; De, S. Loss of heterozygosity preferentially occurs in early replicating regions in cancer genomes. Nucleic Acids Res. 2013, 41, 7615–7624. [Google Scholar] [CrossRef] [PubMed]
- Morganella, S.; Alexandrov, L.B.; Glodzik, D.; Zou, X.; Davies, H.; Staaf, J.; Sieuwerts, A.M.; Brinkman, A.B.; Martin, S.; Ramakrishna, M.; et al. The topography of mutational processes in breast cancer genomes. Nat. Commun. 2016, 7, 11383. [Google Scholar] [CrossRef] [PubMed]
- Hellman, A.; Rahat, A.; Scherer, S.W.; Darvasi, A.; Tsui, L.C.; Kerem, B. Replication delay along FRA7H, a common fragile site on human chromosome 7, leads to chromosomal instability. Mol. Cell. Biol. 2000, 20, 4420–4427. [Google Scholar] [CrossRef] [PubMed]
- Hellman, A.; Zlotorynski, E.; Scherer, S.W.; Cheung, J.; Vincent, J.B.; Smith, D.I.; Trakhtenbrot, L.; Kerem, B. A role for common fragile site induction in amplification of human oncogenes. Cancer Cell 2002, 1, 89–97. [Google Scholar] [CrossRef]
- Pelliccia, F.; Bosco, N.; Curatolo, A.; Rocchi, A. Replication timing of two human common fragile sites: FRA1H and FRA2G. Cytogenet. Genome Res. 2008, 121, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Engreitz, J.M.; Agarwala, V.; Mirny, L.A. Three-dimensional genome architecture influences partner selection for chromosomal translocations in human disease. PLoS ONE 2012, 7, e44196. [Google Scholar] [CrossRef] [PubMed]
- Pope, B.D.; Ryba, T.; Dileep, V.; Yue, F.; Wu, W.; Denas, O.; Vera, D.L.; Wang, Y.; Hansen, R.S.; Canfield, T.K.; et al. Topologically associating domains are stable units of replication-timing regulation. Nature 2014, 515, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Farkash-Amar, S.; David, Y.; Polten, A.; Hezroni, H.; Eldar, Y.C.; Meshorer, E.; Yakhini, Z.; Simon, I. Systematic determination of replication activity type highlights interconnections between replication, chromatin structure and nuclear localization. PLoS ONE 2012, 7, e48986. [Google Scholar] [CrossRef] [PubMed]
- Ryba, T.; Hiratani, I.; Lu, J.; Itoh, M.; Kulik, M.; Zhang, J.; Schulz, T.C.; Robins, A.J.; Dalton, S.; Gilbert, D.M. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res. 2010, 20, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Guilbaud, G.; Rappailles, A.; Baker, A.; Chen, C.-L.; Arneodo, A.; Goldar, A.; d’Aubenton-Carafa, Y.; Thermes, C.; Audit, B.; Hyrien, O. Evidence for sequential and increasing activation of replication origins along replication timing gradients in the human genome. PLoS Comput. Biol. 2011, 7, e1002322. [Google Scholar] [CrossRef] [PubMed]
- Donley, N.; Thayer, M.J. DNA replication timing, genome stability and cancer: Late and/or delayed DNA replication timing is associated with increased genomic instability. Semin. Cancer Biol. 2013, 23, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Kenigsberg, E.; Yehuda, Y.; Marjavaara, L.; Keszthelyi, A.; Chabes, A.; Tanay, A.; Simon, I. The mutation spectrum in genomic late replication domains shapes mammalian GC content. Nucleic Acids Res. 2016, 44, 4222–4232. [Google Scholar] [CrossRef] [PubMed]
- Murga, M.; Jaco, I.; Fan, Y.; Soria, R.; Martinez-Pastor, B.; Cuadrado, M.; Yang, S.-M.; Blasco, M.A.; Skoultchi, A.I.; Fernandez-Capetillo, O. Global chromatin compaction limits the strength of the DNA damage response. J. Cell Biol. 2007, 178, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.; Plug, A.; Thayer, M. Delayed replication timing leads to delayed mitotic chromosome condensation and chromosomal instability of chromosome translocations. Proc. Natl. Acad. Sci. USA 2001, 98, 13300–13305. [Google Scholar] [CrossRef] [PubMed]
- Reish, O.; Orlovski, A.; Mashevitz, M.; Sher, C.; Libman, V.; Rosenblat, M.; Avivi, L. Modified allelic replication in lymphocytes of patients with neurofibromatosis type 1. Cancer Genet. Cytogenet. 2003, 143, 133–139. [Google Scholar] [CrossRef]
- Grinberg-Rashi, H.; Cytron, S.; Gelman-Kohan, Z.; Litmanovitch, T.; Avivi, L. Replication timing aberrations and aneuploidy in peripheral blood lymphocytes of breast cancer patients. Neoplasia N. Y. 2010, 12, 668–674. [Google Scholar] [CrossRef]
- Korenstein-Ilan, A.; Amiel, A.; Lalezari, S.; Lishner, M.; Avivi, L. Allele-specific replication associated with aneuploidy in blood cells of patients with hematologic malignancies. Cancer Genet. Cytogenet. 2002, 139, 97–103. [Google Scholar] [CrossRef]
- Dotan, Z.A.; Dotan, A.; Litmanovitch, T.; Ravia, Y.; Oniashvili, N.; Leibovitch, I.; Ramon, J.; Avivi, L. Modification in the inherent mode of allelic replication in lymphocytes of patients suffering from renal cell carcinoma: A novel genetic alteration associated with malignancy. Genes Chromosomes Cancer 2000, 27, 270–277. [Google Scholar] [CrossRef]
- Sun, Y.; Wyatt, R.T.; Bigley, A.; Krontiris, T.G. Expression and replication timing patterns of wildtype and translocated BCL2 genes. Genomics 2001, 73, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Gispan, A.; Carmi, M.; Barkai, N. Model-based analysis of DNA replication profiles: Predicting replication fork velocity and initiation rate by profiling free-cycling cells. Genome Res. 2017, 27, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Lau, E.; Tsuji, T.; Guo, L.; Lu, S.-H.; Jiang, W. The role of pre-replicative complex (pre-RC) components in oncogenesis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2007, 21, 3786–3794. [Google Scholar] [CrossRef] [PubMed]
- Ostrow, A.Z.; Kalhor, R.; Gan, Y.; Villwock, S.K.; Linke, C.; Barberis, M.; Chen, L.; Aparicio, O.M. Conserved forkhead dimerization motif controls DNA replication timing and spatial organization of chromosomes in S. cerevisiae. Proc. Natl. Acad. Sci. USA 2017, 114, E2411–E2419. [Google Scholar] [CrossRef] [PubMed]
- Mattarocci, S.; Shyian, M.; Lemmens, L.; Damay, P.; Altintas, D.M.; Shi, T.; Bartholomew, C.R.; Thomä, N.H.; Hardy, C.F.J.; Shore, D. Rif1 controls DNA replication timing in yeast through the PP1 phosphatase Glc7. Cell Rep. 2014, 7, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Cornacchia, D.; Dileep, V.; Quivy, J.-P.; Foti, R.; Tili, F.; Santarella-Mellwig, R.; Antony, C.; Almouzni, G.; Gilbert, D.M.; Buonomo, S.B.C. Mouse Rif1 is a key regulator of the replication-timing programme in mammalian cells. EMBO J. 2012, 31, 3678–3690. [Google Scholar] [CrossRef] [PubMed]
- Foti, R.; Gnan, S.; Cornacchia, D.; Dileep, V.; Bulut-Karslioglu, A.; Diehl, S.; Buness, A.; Klein, F.A.; Huber, W.; Johnstone, E.; et al. Nuclear architecture organized by Rif1 underpins the replication-timing program. Mol. Cell 2016, 61, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Ishii, A.; Kanoh, Y.; Oda, M.; Nishito, Y.; Masai, H. Rif1 regulates the replication timing domains on the human genome. EMBO J. 2012, 31, 3667–3677. [Google Scholar] [CrossRef] [PubMed]
- Das, S.P.; Borrman, T.; Liu, V.W.T.; Yang, S.C.-H.; Bechhoefer, J.; Rhind, N. Replication timing is regulated by the number of MCMs loaded at origins. Genome Res. 2015, 25, 1886–1892. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Vidal, A.; Guitton-Sert, L.; Cadoret, J.-C.; Drac, M.; Schwob, E.; Baldacci, G.; Cazaux, C.; Hoffmann, J.-S. A role for DNA polymerase θ in the timing of DNA replication. Nat. Commun. 2014, 5, 4285. [Google Scholar] [CrossRef] [PubMed]
- Casas-Delucchi, C.S.; van Bemmel, J.G.; Haase, S.; Herce, H.D.; Nowak, D.; Meilinger, D.; Stear, J.H.; Leonhardt, H.; Cardoso, M.C. Histone hypoacetylation is required to maintain late replication timing of constitutive heterochromatin. Nucleic Acids Res. 2012, 40, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Goren, A.; Tabib, A.; Hecht, M.; Cedar, H. DNA replication timing of the human β-globin domain is controlled by histone modification at the origin. Genes Dev. 2008, 22, 1319–1324. [Google Scholar] [CrossRef] [PubMed]
- Farkash-Amar, S.; Lipson, D.; Polten, A.; Goren, A.; Helmstetter, C.; Yakhini, Z.; Simon, I. Global organization of replication time zones of the mouse genome. Genome Res. 2008, 18, 1562–1570. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Mulia, J.C.; Gilbert, D.M. Replication timing and transcriptional control: Beyond cause and effect-part III. Curr. Opin. Cell Biol. 2016, 40, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xu, F.; Hashimshony, T.; Keshet, I.; Cedar, H. Establishment of transcriptional competence in early and late S phase. Nature 2002, 420, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Polak, P.; Karlić, R.; Koren, A.; Thurman, R.; Sandstrom, R.; Lawrence, M.S.; Reynolds, A.; Rynes, E.; Vlahoviček, K.; Stamatoyannopoulos, J.A.; et al. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature 2015, 518, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Rivera-Mulia, J.C.; Vera, D.; Zimmerman, J.; Das, S.; Padget, M.; Nakamichi, N.; Chang, B.H.; Tyner, J.; Druker, B.J.; et al. Stability of patient-specific features of altered DNA replication timing in xenografts of primary human acute lymphoblastic leukemia. Exp. Hematol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Haradhvala, N.J.; Polak, P.; Stojanov, P.; Covington, K.R.; Shinbrot, E.; Hess, J.M.; Rheinbay, E.; Kim, J.; Maruvka, Y.E.; Braunstein, L.Z.; et al. Mutational strand asymmetries in cancer genomes reveal mechanisms of DNA damage and repair. Cell 2016, 164, 538–549. [Google Scholar] [CrossRef] [PubMed]



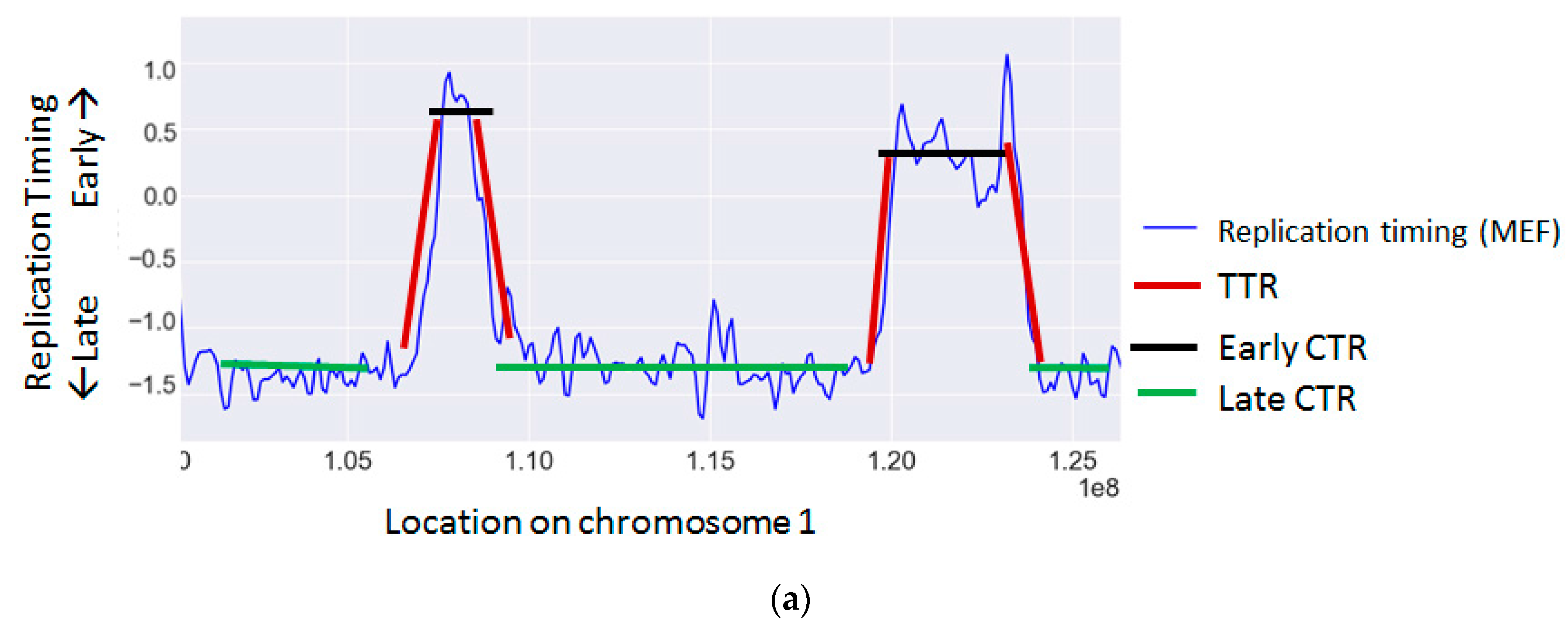{kind=link}
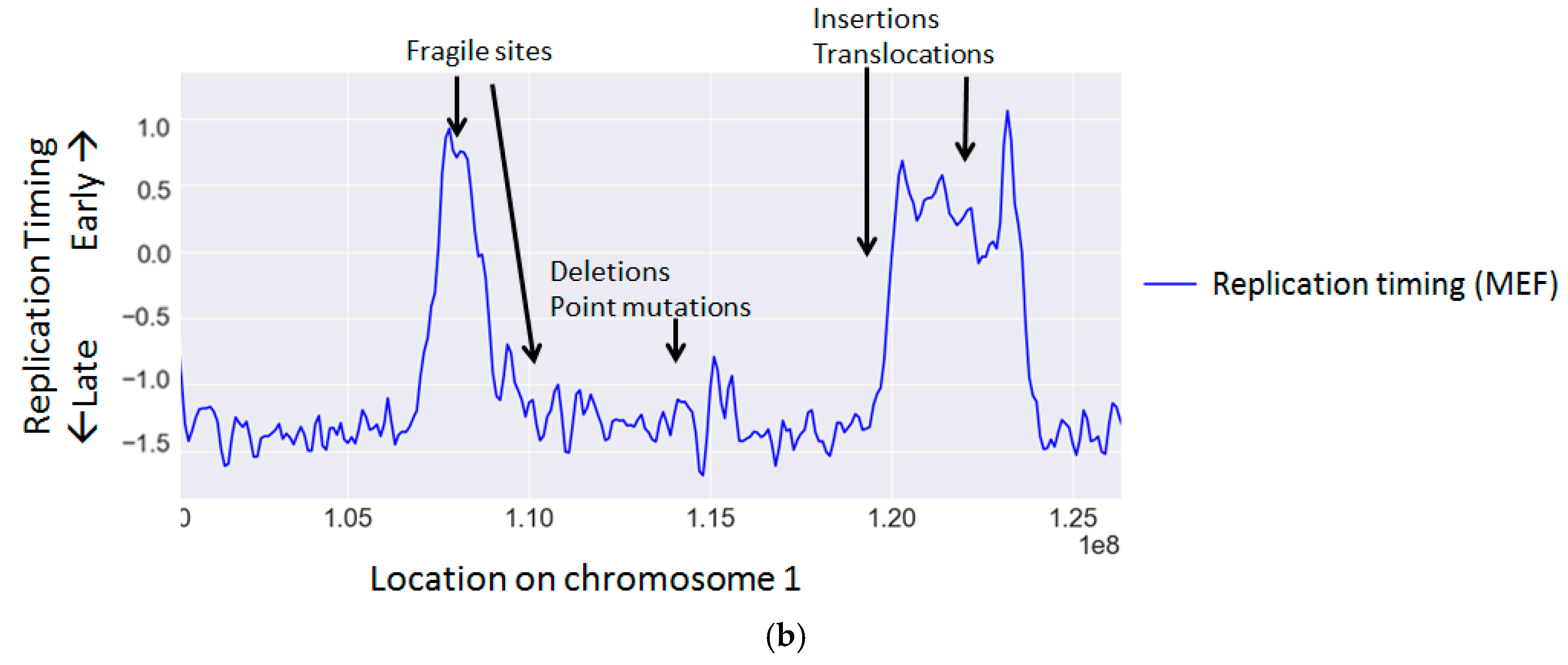{kind=link}
| Gene | Mechanisms | References |
|---|---|---|
| c-myc | Accelerated S phase, increased origin activity, ROS, transcriptional interference | [51,52,53,54,55,56,57,58] |
| Cdt1 | Re-replication | [59] |
| H-RasV12 | Oxidative stress, hyperreplication | [55,58,60,61] |
| E2F1 | Deregulated replication , Oxidative stress | [57,62,63] |
| MOS | Unknown | [58] |
| Cdc6 | Unknown | [58] |
| Cyclin E | Deregulated replication, deficient licensing, transcriptional interference, origin over usage, nucleotide depletion | [26,58,61,62,64,65,66] |
| Cdc25A | Deregulated replication | [62,67] |
| HPV-16 E6/E7 | Nucleotide depletion | [26] |
| Mutation type | Measurement | Higher in | References |
|---|---|---|---|
| Germline point mutations | Human SNP | Late and TTR | [118,119,120,121] |
| Mouse SNP | Late | [119] | |
| Mouse–rat divergence | Late | [119,122] | |
| Human–chimp divergence | Late | [118,119] | |
| Drosophila divergence | Late | [123] | |
| Somatic point mutations | Human cancer | Late | [124,125,126,127,128,129,130] |
| Yeast point mutations | Yeast URA3 gene | Late | [131,132] |
| Insertions | Human cancer | Early and TTR | [126,133,134] |
| Human iPSC | Early | [135] | |
| Fly | Late | [136,137] | |
| Translocations | Human cancer | Early (and late in [138]) | [126,133,138,139,140] |
| Mammalian divergence | TTR/early | [134,141] | |
| Deletions | Human cancer | Late | [126] |
| Human iPSC | Late | [135] | |
| Fly | Early | [136] | |
| Fragile sites | Cancer | Early or late | [96,99,142,143,144] |
| LOH | Cancer | Early | [145] |
| Replication Timing Change | Genes | Cancer Type | Reference |
|---|---|---|---|
| Global replication timing changes | Global changes | Bone marrow from ALL patients | [10] |
| Asynchronous replication | HER-2, AML1, RB1, c-myc, p53 | Peripheral blood lymphocyte of cancer patients, MCF10CA1a | [11,159,160,161,162] |
| Replication timing changes of cancer-related genes | p53, ATM, c-myc, RAD51, PTEN, translocated Bcl2 | MCF10CA1a, SU-DHL-6, Jurkat | [11,163] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blumenfeld, B.; Ben-Zimra, M.; Simon, I. Perturbations in the Replication Program Contribute to Genomic Instability in Cancer. Int. J. Mol. Sci. 2017, 18, 1138. https://doi.org/10.3390/ijms18061138
Blumenfeld B, Ben-Zimra M, Simon I. Perturbations in the Replication Program Contribute to Genomic Instability in Cancer. International Journal of Molecular Sciences. 2017; 18(6):1138. https://doi.org/10.3390/ijms18061138
Chicago/Turabian StyleBlumenfeld, Britny, Micha Ben-Zimra, and Itamar Simon. 2017. "Perturbations in the Replication Program Contribute to Genomic Instability in Cancer" International Journal of Molecular Sciences 18, no. 6: 1138. https://doi.org/10.3390/ijms18061138
APA StyleBlumenfeld, B., Ben-Zimra, M., & Simon, I. (2017). Perturbations in the Replication Program Contribute to Genomic Instability in Cancer. International Journal of Molecular Sciences, 18(6), 1138. https://doi.org/10.3390/ijms18061138