DNA2—An Important Player in DNA Damage Response or Just Another DNA Maintenance Protein?



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
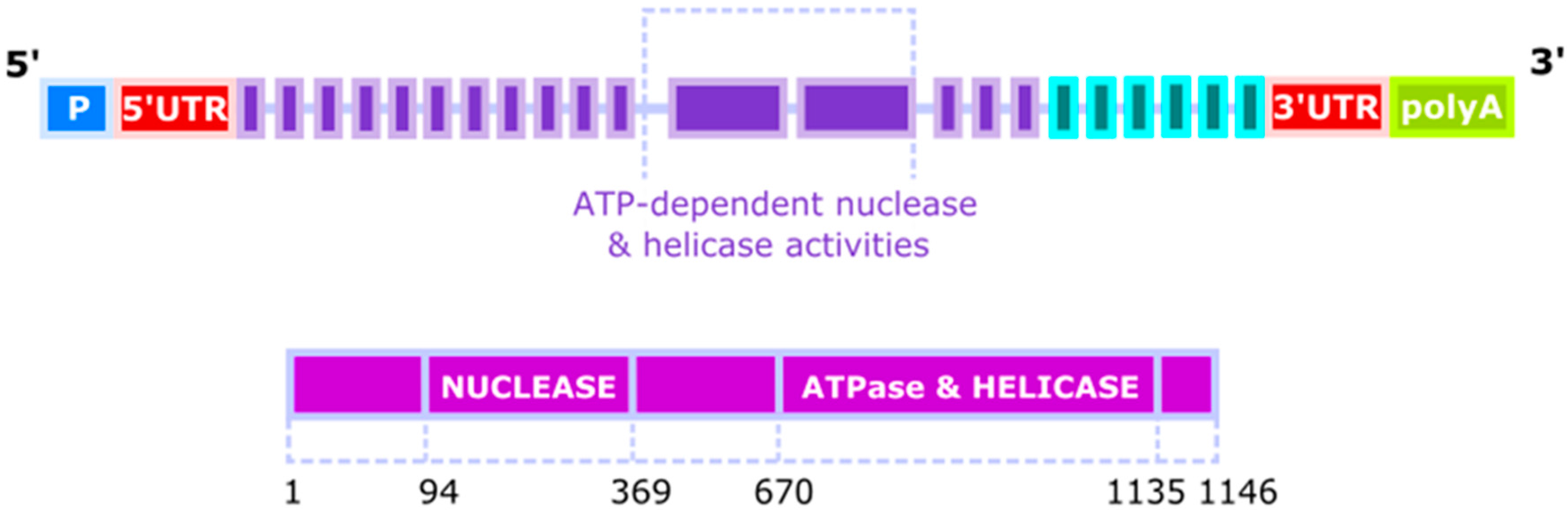
1. Gene, Protein and Activity
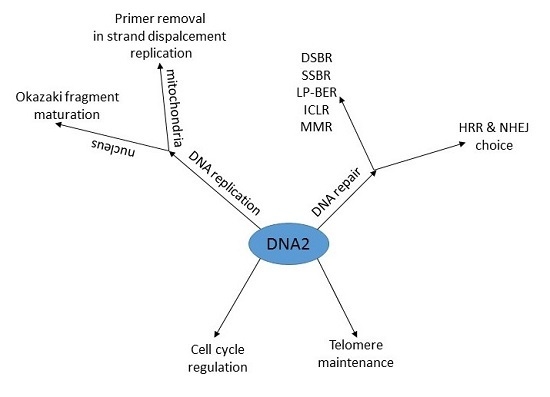
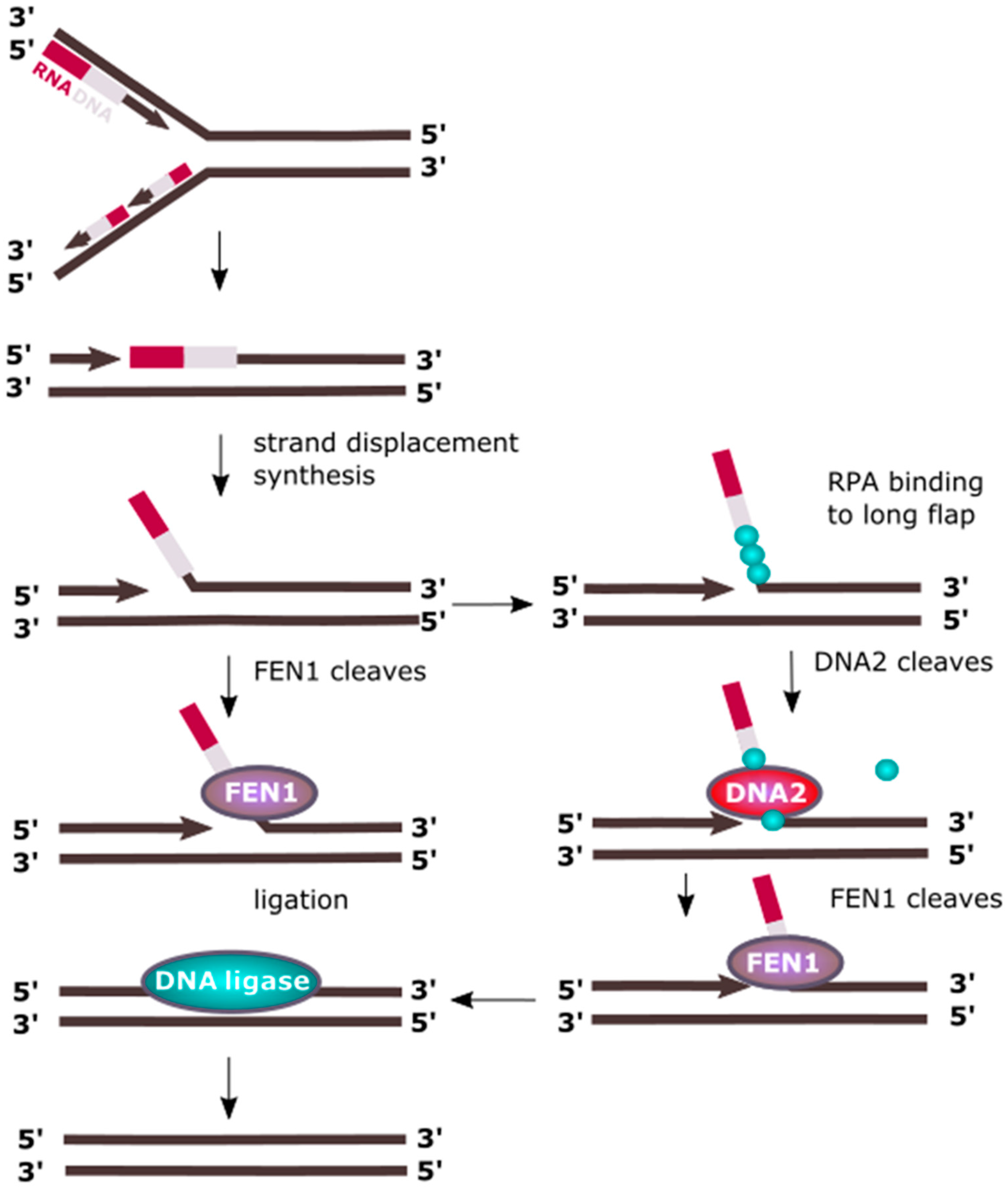
2. DNA Replication
2.1. The Nucleus
2.2. Mitochondria
3. DNA Damage Response
3.1. Post-Translational Modifications
3.2. Cell Cycle Regulation and Telomere Maintenance
3.3. DNA Repair
4. Cancer
5. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ATR | ATR serine/threonine kinase also known as ataxia telangiectasia and Rad3-related protein |
| Cdk1 | Cyclin-dependent kinase 1 |
| DNA2 | DNA replication helicase/nuclease 2 |
| DNA-PKCS | Catalytic subunit of DNA-dependent protein kinase |
| RPA | Replication protein A |
| FEN1 | Flap endonuclease 1 |
| WRN | Werner syndrome ATP-dependent helicase |
| BLM | Bloom syndrome protein |
| Pol γ, δ | DNA polymerase γ, δ |
| MRN | Mre11-Rad50-Nbs1 complex |
| NAM7 | Nuclear accommodation of mitochondria 7 |
| LP-BER | Long-Patch base excision repair |
| PIF1 | Petite integration frequency 1 |
| And-1 | Acidic nucleoplasmic DNA-binding protein 1 |
| RECQ1 | ATP-dependent DNA helicase Q1 |
| TOP1, 2 | DNA topoisomerase I, II |
| ss, ds | Single-stranded, double-stranded |
| MGME1 | Mitochondrial genome maintenance exonuclease 1 |
| mtSSB | Mitochondrial single stranded DNA-binding protein |
| SUV3 | Suppressor of var1 3-like |
| DSB | DNA double-strand break |
| G4 | Guanine quartet |
| U2OS | Osteosarcoma cell line |
| HeLa | Ovarian cancer cell line |
| DSBR | DNA double-strand break repair |
| MMR | Mismatch repair |
| ICL | DNA interstrand cross-link |
| NHEJ | Non-homologous end joining |
| HRR | Homologous recombination repair |
| Sgs1 | Slow growth suppressor 1 |
| ExoI | Exonuclease I |
| SRCAP | Snf2-related CREB-binding protein activator protein |
| CtIP | C-terminal binding interacting protein |
| SMARCAD1 | SWI/SNF-related matrix associated actin-dependent regulator of chromatin subfamily A containing DEAD/H box1 |
| Swi/Snf | Switch/sucrose non-fermentable |
| BRCA1, 2 | Breast cancer 1, 2 |
| RAD51 | Recombinase in homologous recombination |
| HELB | DNA helicase B |
| RMI1-2 | RecQ mediated genome instability 1–2 |
| Fun30 | Function unknown now 30 |
| γH2AX | Phosphorylated variant of the H2A histone |
| FA | Fanconi anemia |
| SP-, LP-BER | Short patch, long patch base excision repair |
| EXOG | 5′ exo/endonuclease |
| SSBR | DNA single-strand break repair |
| H-RAS | Harvey rat sarcoma viral oncogene homolog |
| K-RAS | Kirsten rat sarcoma viral oncogene homolog |
| PARP1 | Poly(ADP-ribose) polymerase 1 |
| TP53 | Tumor protein p53 |
References
- Budd, M.E.; Campbell, J.L. A yeast replicative helicase, DNA2 helicase, interacts with yeast FEN-1 nuclease in carrying out its essential function. Mol. Cell. Biol. 1997, 17, 2136–2142. [Google Scholar] [CrossRef] [PubMed]
- Dumas, L.B.; Lussky, J.P.; McFarland, E.J.; Shampay, J. New temperature-sensitive mutants of Saccharomyces cerevisiae affecting DNA replication. Mol. Gen. Genet. 1982, 187, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Budd, M.E.; Tong, A.H.; Polaczek, P.; Peng, X.; Boone, C.; Campbell, J.L. A network of multi-tasking proteins at the DNA replication fork preserves genome stability. PLoS Genet. 2005, 1, e61. [Google Scholar] [CrossRef] [PubMed]
- Eki, T.; Okumura, K.; Shiratori, A.; Abe, M.; Nogami, M.; Taguchi, H.; Shibata, T.; Murakami, Y.; Hanaoka, F. Assignment of the closest human homologue (DNA2L:KIAA0083) of the yeast DNA2 helicase gene to chromosome band 10q21.3-q22.1. Genomics 1996, 37, 408–410. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, H.D.; Ryu, G.H.; Kim, D.H.; Hurwitz, J.; Seo, Y.S. Isolation of human DNA2 endonuclease and characterization of its enzymatic properties. Nucleic Acids Res. 2006, 34, 1854–1864. [Google Scholar] [CrossRef] [PubMed]
- Masuda-Sasa, T.; Imamura, O.; Campbell, J.L. Biochemical analysis of human DNA2. Nucleic Acids Res. 2006, 34, 1865–1875. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Zhou, M.; Guo, Z.; Lu, H.; Qian, L.; Dai, H.; Qiu, J.; Yakubovskaya, E.; Bogenhagen, D.F.; Demple, B.; et al. Human DNA2 is a mitochondrial nuclease/helicase for efficient processing of DNA replication and repair intermediates. Mol. Cell 2008, 32, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Duxin, J.P.; Dao, B.; Martinsson, P.; Rajala, N.; Guittat, L.; Campbell, J.L.; Spelbrink, J.N.; Stewart, S.A. Human DNA2 is a nuclear and mitochondrial DNA maintenance protein. Mol. Cell. Biol. 2009, 29, 4274–4282. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, L.; Polaczek, P.; Pokharel, S.; Campbell, J.L.; Bambara, R.A. DNA2 exhibits a unique strand end-dependent helicase function. J. Biol. Chem. 2010, 285, 38861–38868. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.L.; Purohit, V.; Brandt, P.D.; Bambara, R.A. Lagging strand replication proteins in genome stability and DNA repair. Chem. Rev. 2006, 106, 453–473. [Google Scholar] [CrossRef] [PubMed]
- Copeland, W.C.; Longley, M.J. DNA2 resolves expanding flap in mitochondrial base excision repair. Mol. Cell 2008, 32, 457–458. [Google Scholar] [CrossRef] [PubMed]
- Garg, P.; Stith, C.M.; Sabouri, N.; Johansson, E.; Burgers, P.M. Idling by DNA polymerase delta maintains a ligatable nick during lagging-strand DNA replication. Genes Dev. 2004, 18, 2764–2773. [Google Scholar] [CrossRef] [PubMed]
- Ayyagari, R.; Gomes, X.V.; Gordenin, D.A.; Burgers, P.M. Okazaki fragment maturation in yeast. I. Distribution of functions between FEN1 AND DNA2. J. Biol. Chem. 2003, 278, 1618–1625. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Shen, B. Okazaki fragment maturation: Nucleases take centre stage. J. Mol. Cell. Biol. 2011, 3, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Burgers, P.M. Lagging strand maturation factor DNA2 is a component of the replication checkpoint initiation machinery. Genes Dev. 2013, 27, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, L.; Gloor, J.W.; Bambara, R.A. Reconstitution of eukaryotic lagging strand DNA replication. Methods 2010, 51, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Pike, J.E.; Burgers, P.M.; Campbell, J.L.; Bambara, R.A. Pif1 helicase lengthens some Okazaki fragment flaps necessitating DNA2 nuclease/helicase action in the two-nuclease processing pathway. J. Biol. Chem. 2009, 284, 25170–25180. [Google Scholar] [CrossRef] [PubMed]
- Pike, J.E.; Henry, R.A.; Burgers, P.M.; Campbell, J.L.; Bambara, R.A. An alternative pathway for Okazaki fragment processing: Resolution of fold-back flaps by Pif1 helicase. J. Biol. Chem. 2010, 285, 41712–41723. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Lee, M.; Kang, H.J.; Kim, D.H.; Kang, Y.H.; Bae, S.H.; Seo, Y.S. The N-terminal 45-kDa domain of DNA2 endonuclease/helicase targets the enzyme to secondary structure DNA. J. Biol. Chem. 2013, 288, 9468–9481. [Google Scholar] [CrossRef] [PubMed]
- Duxin, J.P.; Moore, H.R.; Sidorova, J.; Karanja, K.; Honaker, Y.; Dao, B.; Piwnica-Worms, H.; Campbell, J.L.; Monnat, R.J., Jr.; Stewart, S.A. Okazaki fragment processing-independent role for human DNA2 enzyme during DNA replication. J. Biol. Chem. 2012, 287, 21980–21991. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.M.; Lambert, S. Replication stress-induced genome instability: The dark side of replication maintenance by homologous recombination. J. Mol. Biol. 2013, 425, 4733–4744. [Google Scholar] [CrossRef] [PubMed]
- Neelsen, K.J.; Lopes, M. Replication fork reversal in eukaryotes: From dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 2015, 16, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Petermann, E.; Helleday, T. Pathways of mammalian replication fork restart. Nat. Rev. Mol. Cell Biol. 2010, 11, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Petermann, E. Replication fork dynamics and the DNA damage response. Biochem. J. 2012, 443, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Ashley, A.K.; Hromas, R.; Nickoloff, J.A. More forks on the road to replication stress recovery. J. Mol. Cell. Biol. 2011, 3, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.L.; Hegde, P.M.; Bellot, L.J.; Mandal, S.M.; Hazra, T.K.; Li, G.M.; Boldogh, I.; Tomkinson, A.E.; Mitra, S. Prereplicative repair of oxidized bases in the human genome is mediated by NEIL1 DNA glycosylase together with replication proteins. Proc. Natl. Acad. Sci. USA 2013, 110, E3090–E3099. [Google Scholar] [CrossRef] [PubMed]
- Lundin, C.; Erixon, K.; Arnaudeau, C.; Schultz, N.; Jenssen, D.; Meuth, M.; Helleday, T. Different roles for nonhomologous end joining and homologous recombination following replication arrest in mammalian cells. Mol. Cell. Biol. 2002, 22, 5869–5878. [Google Scholar] [CrossRef] [PubMed]
- Michel, B.; Boubakri, H.; Baharoglu, Z.; LeMasson, M.; Lestini, R. Recombination proteins and rescue of arrested replication forks. DNA Repair 2007, 6, 967–980. [Google Scholar] [CrossRef] [PubMed]
- Michel, B.; Flores, M.J.; Viguera, E.; Grompone, G.; Seigneur, M.; Bidnenko, V. Rescue of arrested replication forks by homologous recombination. Proc. Natl. Acad. Sci. USA 2001, 98, 8181–8188. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.G.; Cortes, U.; Patnaik, S.; Jasin, M.; Wang, Z.Q. Ablation of PARP-1 does not interfere with the repair of DNA double-strand breaks, but compromises the reactivation of stalled replication forks. Oncogene 2004, 23, 3872–3882. [Google Scholar] [CrossRef] [PubMed]
- Zellweger, R.; Dalcher, D.; Mutreja, K.; Berti, M.; Schmid, J.A.; Herrador, R.; Vindigni, A.; Lopes, M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 2015, 208, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Berti, M.; Ray Chaudhuri, A.; Thangavel, S.; Gomathinayagam, S.; Kenig, S.; Vujanovic, M.; Odreman, F.; Glatter, T.; Graziano, S.; Mendoza-Maldonado, R.; et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol. 2013, 20, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Thangavel, S.; Berti, M.; Levikova, M.; Pinto, C.; Gomathinayagam, S.; Vujanovic, M.; Zellweger, R.; Moore, H.; Lee, E.H.; Hendrickson, E.A.; et al. DNA2 drives processing and restart of reversed replication forks in human cells. J. Cell Biol. 2015, 208, 545–562. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Toczylowski, T.; Yan, H. Identification of the Xenopus DNA2 protein as a major nuclease for the 5′→3′ strand-specific processing of DNA ends. Nucleic Acids Res. 2008, 36, 6091–6100. [Google Scholar] [CrossRef] [PubMed]
- Sturzenegger, A.; Burdova, K.; Kanagaraj, R.; Levikova, M.; Pinto, C.; Cejka, P.; Janscak, P. DNA2 cooperates with the WRN and BLM RecQ helicases to mediate long-range DNA end resection in human cells. J. Biol. Chem. 2014, 289, 27314–27326. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Dai, H.; Zhang, W.; Hsieh, H.J.; Pan, M.R.; Park, Y.Y.; Tsai, R.Y.; Bedrosian, I.; Lee, J.S.; Ira, G.; et al. Human nuclease/helicase DNA2 alleviates replication stress by promoting DNA end resection. Cancer Res. 2012, 72, 2802–2813. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Liu, Y. Borrowing nuclear DNA helicases to protect mitochondrial DNA. Int. J. Mol. Sci. 2015, 16, 10870–10887. [Google Scholar] [CrossRef] [PubMed]
- McKinney, E.A.; Oliveira, M.T. Replicating animal mitochondrial DNA. Genet. Mol. Biol. 2013, 36, 308–315. [Google Scholar]
- Wanrooij, S.; Goffart, S.; Pohjoismaki, J.L.; Yasukawa, T.; Spelbrink, J.N. Expression of catalytic mutants of the mtDNA helicase Twinkle and polymerase POLG causes distinct replication stalling phenotypes. Nucleic Acids Res. 2007, 35, 3238–3251. [Google Scholar] [CrossRef] [PubMed]
- Uchiumi, T.; Kang, D. Mitochondrial nucleic acid binding proteins associated with diseases. Front. Biosci. 2017, 22, 168–179. [Google Scholar] [CrossRef]
- Milenkovic, D.; Matic, S.; Kuhl, I.; Ruzzenente, B.; Freyer, C.; Jemt, E.; Park, C.B.; Falkenberg, M.; Larsson, N.G. TWINKLE is an essential mitochondrial helicase required for synthesis of nascent D-loop strands and complete mtDNA replication. Hum. Mol. Genet. 2013, 22, 1983–1993. [Google Scholar] [CrossRef] [PubMed]
- Goffart, S.; Cooper, H.M.; Tyynismaa, H.; Wanrooij, S.; Suomalainen, A.; Spelbrink, J.N. Twinkle mutations associated with autosomal dominant progressive external ophthalmoplegia lead to impaired helicase function and in vivo mtDNA replication stalling. Hum. Mol. Genet. 2009, 18, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Aanen, D.K.; Spelbrink, J.N.; Beekman, M. What cost mitochondria? The maintenance of functional mitochondrial DNA within and across generations. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130438. [Google Scholar] [CrossRef] [PubMed]
- Akhmedov, A.T.; Marin-Garcia, J. Mitochondrial DNA maintenance: An appraisal. Mol. Cell. Biochem. 2015, 409, 283–305. [Google Scholar] [CrossRef] [PubMed]
- Copeland, W.C.; Longley, M.J. Mitochondrial genome maintenance in health and disease. DNA Repair 2014, 19, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Kaguni, L.S.; Oliveira, M.T. Structure, function and evolution of the animal mitochondrial replicative DNA helicase. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Cerritelli, S.M.; Crouch, R.J. Ribonuclease H: The enzymes in eukaryotes. FEBS J. 2009, 276, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Uhler, J.P.; Falkenberg, M. Primer removal during mammalian mitochondrial DNA replication. DNA Repair 2015, 34, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Kornblum, C.; Nicholls, T.J.; Haack, T.B.; Scholer, S.; Peeva, V.; Danhauser, K.; Hallmann, K.; Zsurka, G.; Rorbach, J.; Iuso, A.; et al. Loss-of-function mutations in MGME1 impair mtDNA replication and cause multisystemic mitochondrial disease. Nat. Genet. 2013, 45, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Szczesny, R.J.; Hejnowicz, M.S.; Steczkiewicz, K.; Muszewska, A.; Borowski, L.S.; Ginalski, K.; Dziembowski, A. Identification of a novel human mitochondrial endo-/exonuclease Ddk1/c20orf72 necessary for maintenance of proper 7S DNA levels. Nucleic Acids Res. 2013, 41, 3144–3161. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, L.; Stewart, J.; Polaczek, P.; Campbell, J.L.; Bambara, R.A. Acetylation of DNA2 endonuclease/helicase and flap endonuclease 1 by p300 promotes DNA stability by creating long flap intermediates. J. Biol. Chem. 2010, 285, 4398–4404. [Google Scholar] [CrossRef] [PubMed]
- Hasan, S.; Stucki, M.; Hassa, P.O.; Imhof, R.; Gehrig, P.; Hunziker, P.; Hubscher, U.; Hottiger, M.O. Regulation of human flap endonuclease-1 activity by acetylation through the transcriptional coactivator p300. Mol. Cell 2001, 7, 1221–1231. [Google Scholar] [CrossRef]
- Friedrich-Heineken, E.; Henneke, G.; Ferrari, E.; Hubscher, U. The acetylatable lysines of human Fen1 are important for endo- and exonuclease activities. J. Mol. Biol. 2003, 328, 73–84. [Google Scholar] [CrossRef]
- Villa, M.; Cassani, C.; Gobbini, E.; Bonetti, D.; Longhese, M.P. Coupling end resection with the checkpoint response at DNA double-strand breaks. Cell. Mol. Life Sci. 2016, 73, 3655–3663. [Google Scholar] [CrossRef] [PubMed]
- Trovesi, C.; Manfrini, N.; Falcettoni, M.; Longhese, M.P. Regulation of the DNA damage response by cyclin-dependent kinases. J. Mol. Biol. 2013, 425, 4756–4766. [Google Scholar] [CrossRef] [PubMed]
- Ira, G.; Pellicioli, A.; Balijja, A.; Wang, X.; Fiorani, S.; Carotenuto, W.; Liberi, G.; Bressan, D.; Wan, L.; Hollingsworth, N.M.; et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 2004, 431, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Aylon, Y.; Liefshitz, B.; Kupiec, M. The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO J. 2004, 23, 4868–4875. [Google Scholar] [CrossRef] [PubMed]
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.; Lukas, J.; Jackson, S.P. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Niu, H.; Chung, W.H.; Zhu, Z.; Papusha, A.; Shim, E.Y.; Lee, S.E.; Sung, P.; Ira, G. Cell cycle regulation of DNA double-strand break end resection by Cdk1-dependent DNA2 phosphorylation. Nat. Struct. Mol. Biol. 2011, 18, 1015–1019. [Google Scholar] [CrossRef] [PubMed]
- Choe, W.; Budd, M.; Imamura, O.; Hoopes, L.; Campbell, J.L. Dynamic localization of an Okazaki fragment processing protein suggests a novel role in telomere replication. Mol. Cell. Biol. 2002, 22, 4202–4217. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Sampathi, S.; Dai, H.; Liu, C.; Zhou, M.; Hu, J.; Huang, Q.; Campbell, J.; Shin-Ya, K.; Zheng, L.; et al. Mammalian DNA2 helicase/nuclease cleaves G-quadruplex DNA and is required for telomere integrity. EMBO J. 2013, 32, 1425–1439. [Google Scholar] [CrossRef] [PubMed]
- Meyne, J.; Ratliff, R.L.; Moyzis, R.K. Conservation of the human telomere sequence (TTAGGG)n among vertebrates. Proc. Natl. Acad. Sci. USA 1989, 86, 7049–7053. [Google Scholar] [CrossRef] [PubMed]
- Moyzis, R.K.; Buckingham, J.M.; Cram, L.S.; Dani, M.; Deaven, L.L.; Jones, M.D.; Meyne, J.; Ratliff, R.L.; Wu, J.R. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc. Natl. Acad. Sci. USA 1988, 85, 6622–6626. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.T. G-quartets 40 years later: From 5′-GMP to molecular biology and supramolecular chemistry. Angew. Chem. Int. Ed. Engl. 2004, 43, 668–698. [Google Scholar] [CrossRef] [PubMed]
- Masuda-Sasa, T.; Polaczek, P.; Peng, X.P.; Chen, L.; Campbell, J.L. Processing of G4 DNA by DNA2 helicase/nuclease and replication protein A (RPA) provides insights into the mechanism of DNA2/RPA substrate recognition. J. Biol. Chem. 2008, 283, 24359–24373. [Google Scholar] [CrossRef] [PubMed]
- Budd, M.E.; Campbell, J.L. The pattern of sensitivity of yeast DNA2 mutants to DNA damaging agents suggests a role in DSB and postreplication repair pathways. Mutat. Res. 2000, 459, 173–186. [Google Scholar] [CrossRef]
- Formosa, T.; Nittis, T. DNA2 mutants reveal interactions with DNA polymerase alpha and Ctf4, a Pol alpha accessory factor, and show that full DNA2 helicase activity is not essential for growth. Genetics 1999, 151, 1459–1470. [Google Scholar] [PubMed]
- Poot, M.; Haaf, T. Mechanisms of Origin, Phenotypic Effects and Diagnostic Implications of Complex Chromosome Rearrangements. Mol. Syndromol. 2015, 6, 110–134. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.J.; Chen, B.P.; Chen, D.J. DNA-PK: A dynamic enzyme in a versatile DSB repair pathway. DNA Repair 2014, 17, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Hammel, M.; Yu, Y.; Radhakrishnan, S.K.; Chokshi, C.; Tsai, M.S.; Matsumoto, Y.; Kuzdovich, M.; Remesh, S.G.; Fang, S.; Tomkinson, A.E.; et al. An Intrinsically Disordered APLF Links Ku, DNA-PKcs, and XRCC4-DNA Ligase IV in an Extended Flexible Non-homologous End Joining Complex. J. Biol. Chem. 2016, 291, 26987–27006. [Google Scholar] [CrossRef] [PubMed]
- Szankasi, P.; Smith, G.R. A single-stranded DNA exonuclease from Schizosaccharomyces pombe. Biochemistry 1992, 31, 6769–6773. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Huang, J. DNA End Resection: Facts and Mechanisms. Genom. Proteom. Bioinform. 2016, 14, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Costelloe, T.; Louge, R.; Tomimatsu, N.; Mukherjee, B.; Martini, E.; Khadaroo, B.; Dubois, K.; Wiegant, W.W.; Thierry, A.; Burma, S.; et al. The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature 2012, 489, 581–584. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.K.; Hanada, K.; Kanaar, R.; Hickson, I.D. BLM has early and late functions in homologous recombination repair in mouse embryonic stem cells. Oncogene 2010, 29, 4705–4714. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.K.; Hickson, I.D. RecQ helicases: Multifunctional genome caretakers. Nat. Rev. Cancer 2009, 9, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Gravel, S.; Chapman, J.R.; Magill, C.; Jackson, S.P. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008, 22, 2767–2772. [Google Scholar] [CrossRef] [PubMed]
- Nimonkar, A.V.; Ozsoy, A.Z.; Genschel, J.; Modrich, P.; Kowalczykowski, S.C. Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc. Natl. Acad. Sci. USA 2008, 105, 16906–16911. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Doty, T.; Gibson, B.; Heyer, W.D. Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat. Struct. Mol. Biol. 2010, 17, 1260–1262. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.B.; Carreira, A.; Kowalczykowski, S.C. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 2010, 467, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Daley, J.M.; Niu, H.; Miller, A.S.; Sung, P. Biochemical mechanism of DSB end resection and its regulation. DNA Repair 2015, 32, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, R.A.; Lee, J.H.; Paull, T.T. Rad50 ATPase activity is regulated by DNA ends and requires coordination of both active sites. Nucleic Acids Res. 2017, 45, 5255–5268. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lee, J.H.; Jiang, W.; Crowe, J.L.; Zha, S.; Paull, T.T. Regulation of the DNA Damage Response by DNA-PKcs Inhibitory Phosphorylation of ATM. Mol. Cell 2017, 65, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.T.; Erdeniz, N.; Symington, L.S.; Liskay, R.M. EXO1-A multi-tasking eukaryotic nuclease. DNA Repair 2004, 3, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011, 25, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Davies, S.L.; Levitt, N.C.; Hickson, I.D. Potential role for the BLM helicase in recombinational repair via a conserved interaction with RAD51. J. Biol. Chem. 2001, 276, 19375–19381. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.S.; Daley, J.M.; Pham, N.T.; Niu, H.; Xue, X.; Ira, G.; Sung, P. A novel role of the DNA2 translocase function in DNA break resection. Genes Dev. 2017, 31, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Tkac, J.; Xu, G.; Adhikary, H.; Young, J.T.; Gallo, D.; Escribano-Diaz, C.; Krietsch, J.; Orthwein, A.; Munro, M.; Sol, W.; et al. HELB Is a Feedback Inhibitor of DNA End Resection. Mol. Cell 2016, 61, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yang, Y.; Singh, T.R.; Busygina, V.; Guo, R.; Wan, K.; Wang, W.; Sung, P.; Meetei, A.R.; Lei, M. Crystal structures of RMI1 and RMI2, two OB-fold regulatory subunits of the BLM complex. Structure 2010, 18, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Daley, J.M.; Chiba, T.; Xue, X.; Niu, H.; Sung, P. Multifaceted role of the Topo IIIalpha-RMI1-RMI2 complex and DNA2 in the BLM-dependent pathway of DNA break end resection. Nucleic Acids Res. 2014, 42, 11083–11091. [Google Scholar] [CrossRef] [PubMed]
- Blasiak, J. DNA-Damaging anticancer drugs—A Perspective for DNA repair-oriented therapy. Curr. Med. Chem. 2017. [Google Scholar] [CrossRef]
- Tammaro, M.; Liao, S.; Beeharry, N.; Yan, H. DNA double-strand breaks with 5′ adducts are efficiently channeled to the DNA2-mediated resection pathway. Nucleic Acids Res. 2016, 44, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Symington, L.S. Overcoming the chromatin barrier to end resection. Cell Res. 2013, 23, 317–319. [Google Scholar] [CrossRef] [PubMed]
- Doiguchi, M.; Nakagawa, T.; Imamura, Y.; Yoneda, M.; Higashi, M.; Kubota, K.; Yamashita, S.; Asahara, H.; Iida, M.; Fujii, S.; et al. SMARCAD1 is an ATP-dependent stimulator of nucleosomal H2A acetylation via CBP, resulting in transcriptional regulation. Sci. Rep. 2016, 6, 20179. [Google Scholar] [CrossRef] [PubMed]
- Ferroudj, S.; Yildiz, G.; Bouras, M.; Iscan, E.; Ekin, U.; Ozturk, M. Role of Fanconi anemia/BRCA pathway genes in hepatocellular carcinoma chemoresistance. Hepatol. Res. 2016, 46, 1264–1274. [Google Scholar] [CrossRef] [PubMed]
- Martens-de Kemp, S.R.; Brink, A.; van der Meulen, I.H.; de Menezes, R.X.; Te Beest, D.E.; Leemans, C.R.; van Beusechem, V.W.; Braakhuis, B.J.; Brakenhoff, R.H. The FA/BRCA Pathway Identified as the Major Predictor of Cisplatin Response in Head and Neck Cancer by Functional Genomics. Mol. Cancer Ther. 2017, 16, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Bierie, B.; Li, A.G.; Pathania, S.; Toomire, K.; Dimitrov, S.D.; Liu, B.; Gelman, R.; Giobbie-Hurder, A.; Feunteun, J.; et al. BRCA1/FANCD2/BRG1-Driven DNA Repair Stabilizes the Differentiation State of Human Mammary Epithelial Cells. Mol. Cell 2016, 63, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Karanja, K.K.; Cox, S.W.; Duxin, J.P.; Stewart, S.A.; Campbell, J.L. DNA2 and EXO1 in replication-coupled, homology-directed repair and in the interplay between HDR and the FA/BRCA network. Cell Cycle 2012, 11, 3983–3996. [Google Scholar] [CrossRef] [PubMed]
- Karanja, K.K.; Lee, E.H.; Hendrickson, E.A.; Campbell, J.L. Preventing over-resection by DNA2 helicase/nuclease suppresses repair defects in Fanconi anemia cells. Cell Cycle 2014, 13, 1540–1550. [Google Scholar] [CrossRef] [PubMed]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The maintenance of mitochondrial DNA integrity—Critical analysis and update. Cold Spring Harb. Perspect. Biol. 2013, 5, a012641. [Google Scholar]
- Shokolenko, I.N.; Wilson, G.L.; Alexeyev, M.F. Aging: A mitochondrial DNA perspective, critical analysis and an update. World J. Exp. Med. 2014, 4, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Abbotts, R.; Wilson, D.M., III. Coordination of DNA single strand break repair. Free Radic. Biol. Med. 2017, 107, 228–244. [Google Scholar] [CrossRef] [PubMed]
- Pages, V. Single-strand gap repair involves both RecF and RecBCD pathways. Curr. Genet. 2016, 62, 519–521. [Google Scholar] [CrossRef] [PubMed]
- Tann, A.W.; Boldogh, I.; Meiss, G.; Qian, W.; Van Houten, B.; Mitra, S.; Szczesny, B. Apoptosis induced by persistent single-strand breaks in mitochondrial genome: Critical role of EXOG (5′-EXO/endonuclease) in their repair. J. Biol. Chem. 2011, 286, 31975–31983. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.R.; Yu, J.; Shanker, K.; Deshpande, N.; Varambally, R.; Ghosh, D.; Barrette, T.; Pandey, A.; Chinnaiyan, A.M. Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proc. Natl. Acad. Sci. USA 2004, 101, 9309–9314. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.R.; Yu, J.; Shanker, K.; Deshpande, N.; Varambally, R.; Ghosh, D.; Barrette, T.; Pandey, A.; Chinnaiyan, A.M. ONCOMINE: A cancer microarray database and integrated data-mining platform. Neoplasia 2004, 6, 1–6. [Google Scholar] [CrossRef]
- Campbell, P.M.; Groehler, A.L.; Lee, K.M.; Ouellette, M.M.; Khazak, V.; Der, C.J. K-Ras promotes growth transformation and invasion of immortalized human pancreatic cells by Raf and phosphatidylinositol 3-kinase signaling. Cancer Res. 2007, 67, 2098–2106. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Peng, X.; Daley, J.; Yang, L.; Shen, J.; Nguyen, N.; Bae, G.; Niu, H.; Peng, Y.; Hsieh, H.J.; et al. Inhibition of DNA2 nuclease as a therapeutic strategy targeting replication stress in cancer cells. Oncogenesis 2017, 6, e319. [Google Scholar] [CrossRef] [PubMed]
- Fang, B. Development of synthetic lethality anticancer therapeutics. J. Med. Chem. 2014, 57, 7859–7873. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O′Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Petermann, E.; Schultz, N.; Jemth, A.S.; Loseva, O.; Issaeva, N.; Johansson, F.; Fernandez, S.; McGlynn, P.; Helleday, T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009, 28, 2601–2615. [Google Scholar] [CrossRef] [PubMed]
- Wanrooij, P.H.; Burgers, P.M. Yet another job for DNA2: Checkpoint activation. DNA Repair 2015, 32, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Hamdy, F.C.; Helleday, T. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res. 2012, 72, 2814–2821. [Google Scholar] [CrossRef] [PubMed]
- Cunniff, C.; Bassetti, J.A.; Ellis, N.A. Bloom’s Syndrome: Clinical Spectrum, Molecular Pathogenesis, and Cancer Predisposition. Mol. Syndromol. 2017, 8, 4–23. [Google Scholar] [CrossRef] [PubMed]
- Sugasawa, K. Molecular mechanisms of DNA damage recognition for mammalian nucleotide excision repair. DNA Repair 2016, 44, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Ronchi, D.; Di Fonzo, A.; Lin, W.; Bordoni, A.; Liu, C.; Fassone, E.; Pagliarani, S.; Rizzuti, M.; Zheng, L.; Filosto, M.; et al. Mutations in DNA2 link progressive myopathy to mitochondrial DNA instability. Am. J. Hum. Genet. 2013, 92, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Strauss, C.; Kornowski, M.; Benvenisty, A.; Shahar, A.; Masury, H.; Ben-Porath, I.; Ravid, T.; Arbel-Eden, A.; Goldberg, M. The DNA2 nuclease/helicase is an estrogen-dependent gene mutated in breast and ovarian cancers. Oncotarget 2014, 5, 9396–9409. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhou, M.; Li, Z.; Li, H.; Polaczek, P.; Dai, H.; Wu, Q.; Liu, C.; Karanja, K.K.; Popuri, V.; et al. A Selective Small Molecule DNA2 Inhibitor for Sensitization of Human Cancer Cells to Chemotherapy. EBioMedicine 2016, 6, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Molchadsky, A.; Rotter, V. p53 and its mutants on the slippery road from stemness to carcinogenesis. Carcinogenesis 2017, 38, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Zhao, Y.; Xu, Y.; Zheng, M.; Feng, Z.; Hu, W. Mutant p53 in Cancer: Accumulation, Gain-of-Function, and Therapy. J. Mol. Biol. 2017, 429, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Lin, F.T.; Graves, J.D.; Lee, Y.J.; Lin, W.C. Mutant p53 perturbs DNA replication checkpoint control through TopBP1 and Treslin. Proc. Natl. Acad. Sci. USA 2017, 114, E3766–E3775. [Google Scholar] [CrossRef] [PubMed]
- Jia, N.; Liu, X.; Gao, H. A DNA2 Homolog Is Required for DNA Damage Repair, Cell Cycle Regulation, and Meristem Maintenance in Plants. Plant Physiol. 2016, 171, 318–333. [Google Scholar] [CrossRef] [PubMed]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pawłowska, E.; Szczepanska, J.; Blasiak, J. DNA2—An Important Player in DNA Damage Response or Just Another DNA Maintenance Protein? Int. J. Mol. Sci. 2017, 18, 1562. https://doi.org/10.3390/ijms18071562
Pawłowska E, Szczepanska J, Blasiak J. DNA2—An Important Player in DNA Damage Response or Just Another DNA Maintenance Protein? International Journal of Molecular Sciences. 2017; 18(7):1562. https://doi.org/10.3390/ijms18071562
Chicago/Turabian StylePawłowska, Elzbieta, Joanna Szczepanska, and Janusz Blasiak. 2017. "DNA2—An Important Player in DNA Damage Response or Just Another DNA Maintenance Protein?" International Journal of Molecular Sciences 18, no. 7: 1562. https://doi.org/10.3390/ijms18071562
APA StylePawłowska, E., Szczepanska, J., & Blasiak, J. (2017). DNA2—An Important Player in DNA Damage Response or Just Another DNA Maintenance Protein? International Journal of Molecular Sciences, 18(7), 1562. https://doi.org/10.3390/ijms18071562