Prevention of Gastric Cancer: Eradication of Helicobacter Pylori and Beyond


Abstract
:
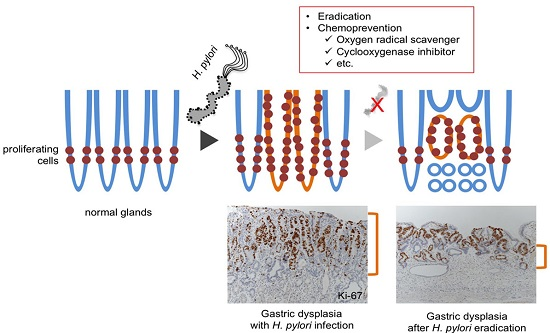{kind=link}
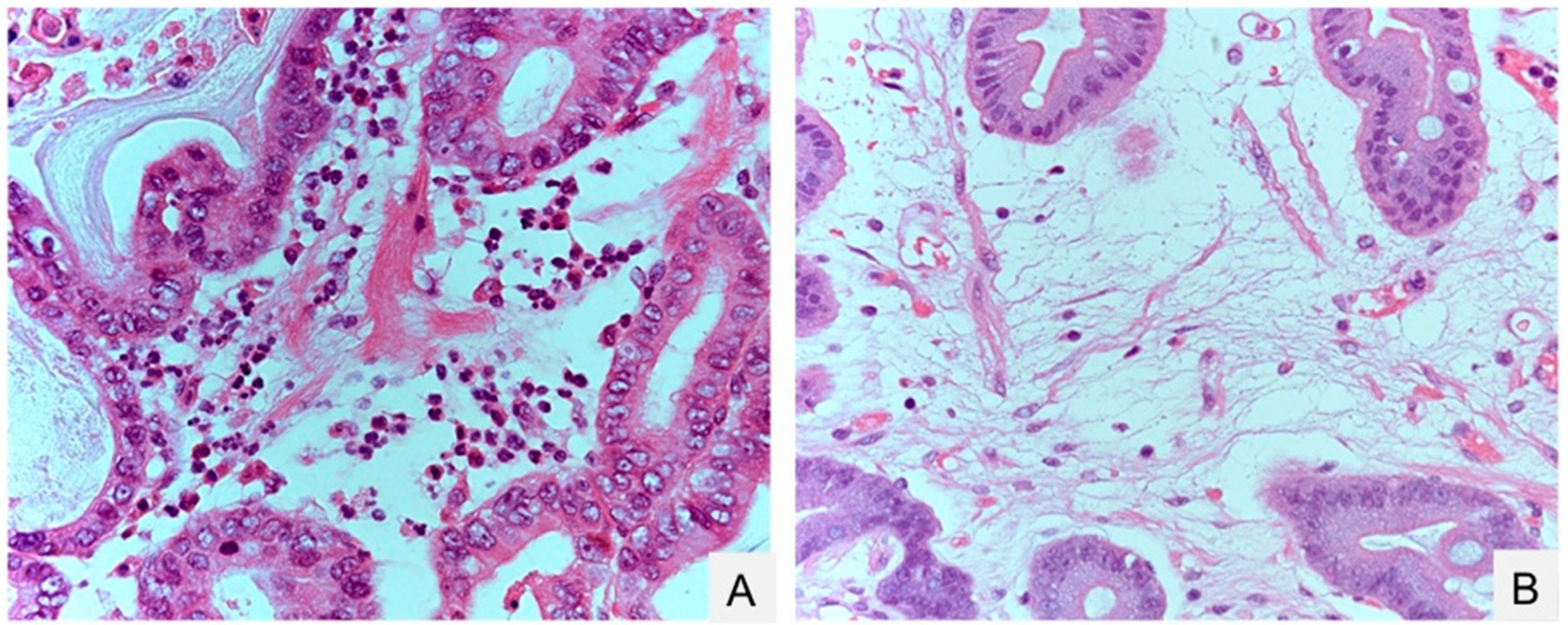{kind=link}
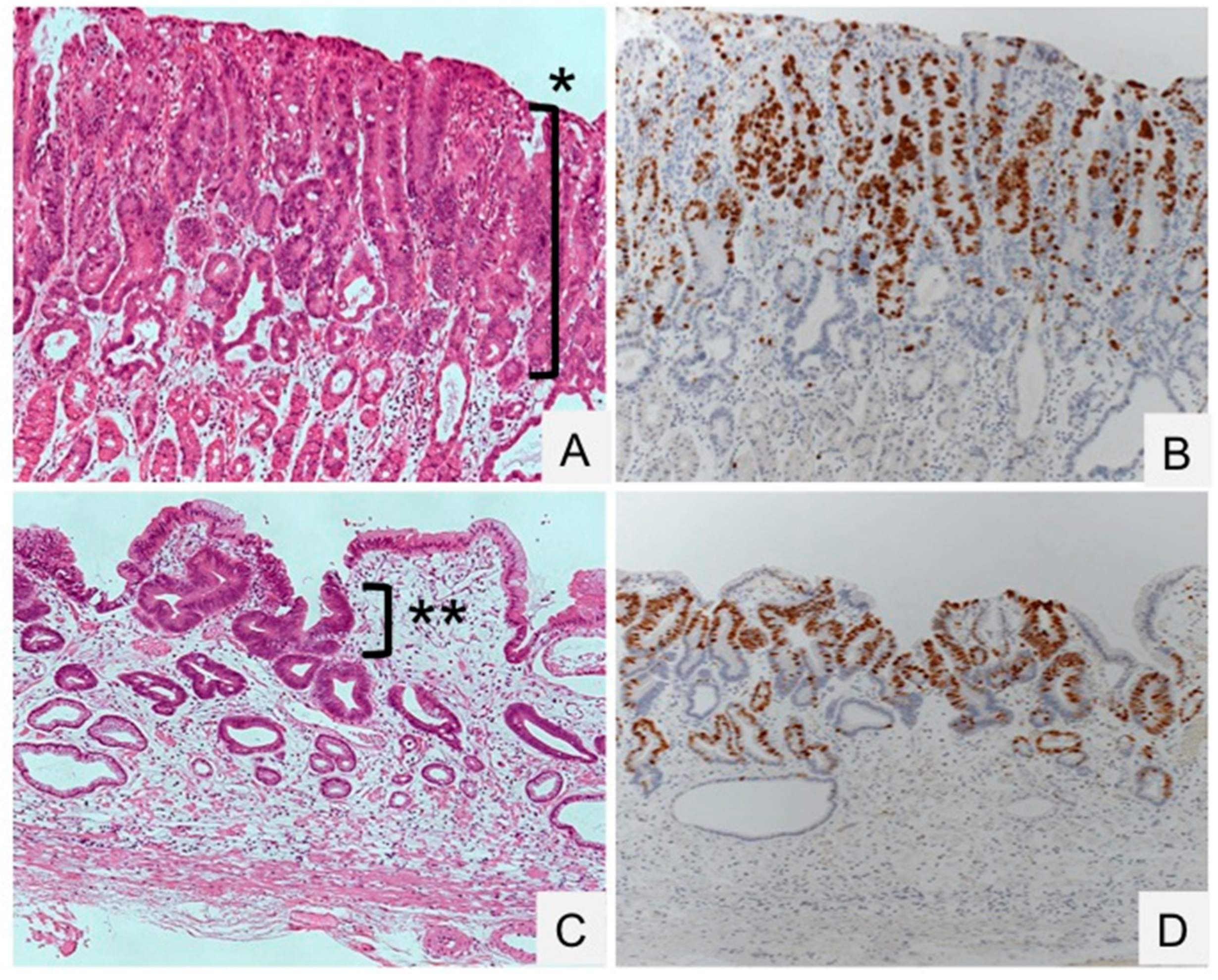{kind=link}
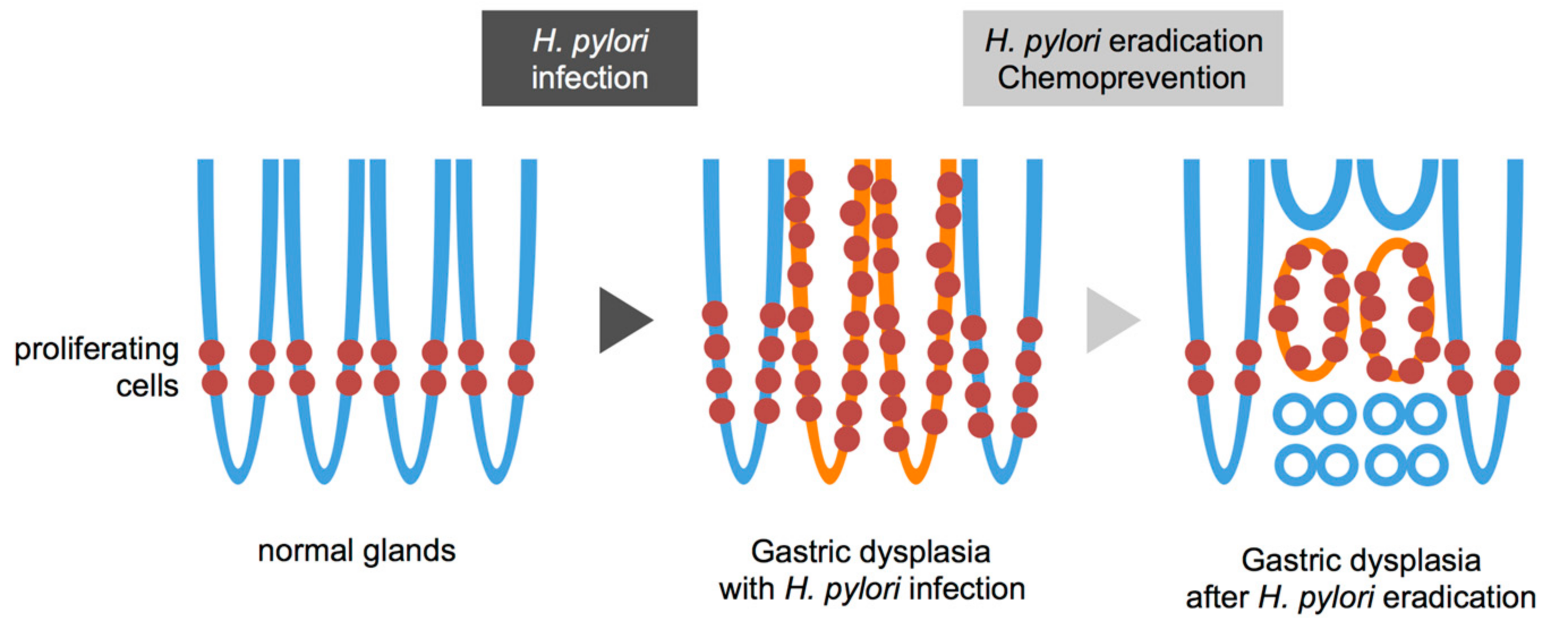{kind=link}
1. Introduction
2. Role of H. pylori Infection and Modifying Factors in Chronic Active Gastritis, Intestinal Metaplasia, and Gastric Carcinogenesis
2.1. Epidemiological Aspects
2.2. Geographical Difference of H. pylori
2.3. Animal Models
2.3.1. Mouse Models
2.3.2. Mongolian Gerbil Model
2.4. Pathological Changes Caused by H. pylori Infection
2.5. Host and Environmental Factors
2.6. Dietary Factors
2.6.1. Salt
2.6.2. Green Tea
2.6.3. Mastic Gum
2.6.4. Ginseng
2.6.5. Spices
3. Effects of Eradication of H. pylori on Gastric Inflammation, Intestinal Metaplasia, and Carcinogenesis
3.1. Humans
3.2. Animals
4. Chemoprevention of Gastric Carcinogenesis
4.1. Oxygen Radical Scavengers
4.2. COX-2 Inhibitors
5. Conclusions
Acknowledgments
Author contributions
Conflicts of interest
References
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global Cancer incidence and mortality rates and trends—An update. Cancer Epidemiol. Biomarkers Prev. 2016, 25, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, T.; Toyoda, T.; Mizoshita, T.; Tatematsu, M. Helicobacter pylori infection and gastric carcinogenesis in rodent models. Semin. Immunopathol. 2013, 35, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Asaka, M.; Mabe, K. Strategies for eliminating death from gastric cancer in Japan. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2014, 90, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Saka, A.; Yagi, K.; Nimura, S. Endoscopic and histological features of gastric cancers after successful Helicobacter pylori eradication therapy. Gastric Cancer 2016, 19, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Warren, J.R.; Marshall, B. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet 1983, 1, 1273–1275. [Google Scholar] [PubMed]
- Marshall, B.J.; Warren, J.R. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1984, 1, 1311–1315. [Google Scholar] [CrossRef]
- Hu, P.J.; Li, Y.Y.; Zhou, M.H.; Chen, M.H.; Du, G.G.; Huang, B.J.; Mitchell, H.M.; Hazell, S.L. Helicobacter pylori associated with a high prevalence of duodenal ulcer disease and a low prevalence of gastric cancer in a developing nation. Gut 1995, 36, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Craanen, M.E.; Dekker, W.; Blok, P.; Ferwerda, J.; Tytgat, G.N. Intestinal metaplasia and Helicobacter pylori: An endoscopic bioptic study of the gastric antrum. Gut 1992, 33, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Parsonnet, J.; Friedman, G.D.; Vandersteen, D.P.; Chang, Y.; Vogelman, J.H.; Orentreich, N.; Sibley, R.K. Helicobacter pylori infection and the risk of gastric carcinoma. N. Engl. J. Med. 1991, 325, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Nomura, A.; Stemmermann, G.N.; Chyou, P.H.; Kato, I.; Perez-Perez, G.I.; Blaser, M.J. Helicobacter pylori infection and gastric carcinoma among Japanese Americans in Hawaii. N. Engl. J. Med. 1991, 325, 1132–1136. [Google Scholar] [CrossRef] [PubMed]
- Forman, D.; Newell, D.G.; Fullerton, F.; Yarnell, J.W.; Stacey, A.R.; Wald, N.; Sitas, F. Association between infection with Helicobacter pylori and risk of gastric cancer: Evidence from a prospective investigation. BMJ 1991, 302, 1302–1305. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.Y.; Lew, G.M.; Klein, P.D.; Evans, D.G.; Evans, D.J., Jr.; Saeed, Z.A.; Malaty, H.M. Effect of treatment of Helicobacter pylori infection on the long-term recurrence of gastric or duodenal ulcer. A randomized, controlled study. Ann. Intern. Med. 1992, 116, 705–708. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, E.J.; Uyterlinde, A.M.; Pena, A.S.; Roosendaal, R.; Pals, G.; Nelis, G.F.; Festen, H.P.; Meuwissen, S.G. Long-term sequelae of Helicobacter pylori gastritis. Lancet 1995, 345, 1525–1528. [Google Scholar] [CrossRef]
- Asaka, M.; Kato, M.; Kudo, M.; Katagiri, M.; Nishikawa, K.; Koshiyama, H.; Takeda, H.; Yoshida, J.; Graham, D.Y. Atrophic changes of gastric mucosa are caused by Helicobacter pylori infection rather than aging: Studies in asymptomatic Japanese adults. Helicobacter 1996, 1, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.Q.; Sridhar, S.; Chen, Y.; Hunt, R.H. Meta-analysis of the relationship between Helicobacter pylori seropositivity and gastric cancer. Gastroenterology 1998, 114, 1169–1179. [Google Scholar] [CrossRef]
- Parsonnet, J.; Hansen, S.; Rodriguez, L.; Gelb, A.B.; Warnke, R.A.; Jellum, E.; Orentreich, N.; Vogelman, J.H.; Friedman, G.D. Helicobacter pylori infection and gastric lymphoma. N. Engl. J. Med. 1994, 330, 1267–1271. [Google Scholar] [CrossRef] [PubMed]
- The Eurogast Study Group. An international association between Helicobacter pylori infection and gastric cancer. Lancet 1993, 341, 1359–1363. [Google Scholar]
- Uemura, N.; Okamoto, S.; Yamamoto, S.; Matsumura, N.; Yamaguchi, S.; Yamakido, M.; Taniyama, K.; Sasaki, N.; Schlemper, R.J. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 2001, 345, 784–789. [Google Scholar] [CrossRef] [PubMed]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Infection with Helicobacter pylori. In Schistosomes, Liver Flukes and Helibacter Pylori; World Health Organization/International Agency for Research on Cancer: Lyon, France, 1994; pp. 177–241. [Google Scholar]
- Backert, S.; Selbach, M. Tyrosine-phosphorylated bacterial effector proteins: The enemies within. Trends Microbiol. 2005, 13, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, M. Anthropological and clinical implications for the structural diversity of the Helicobacter pylori CagA oncoprotein. Cancer Sci. 2011, 102, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Naito, M.; Yamazaki, T.; Tsutsumi, R.; Higashi, H.; Onoe, K.; Yamazaki, S.; Azuma, T.; Hatakeyama, M. Influence of EPIYA-repeat polymorphism on the phosphorylation-dependent biological activity of Helicobacter pylori CagA. Gastroenterology 2006, 130, 1181–1190. [Google Scholar] [CrossRef] [PubMed]
- Krakowka, S.; Morgan, D.R.; Kraft, W.G.; Leunk, R.D. Establishment of gastric Campylobacter pylori infection in the neonatal gnotobiotic piglet. Infect. Immun. 1987, 55, 2789–2796. [Google Scholar] [PubMed]
- Radin, M.J.; Eaton, K.A.; Krakowka, S.; Morgan, D.R.; Lee, A.; Otto, G.; Fox, J. Helicobacter pylori gastric infection in gnotobiotic beagle dogs. Infect. Immun. 1990, 58, 2606–2612. [Google Scholar] [PubMed]
- Karita, M.; Kouchiyama, T.; Okita, K.; Nakazawa, T. New small animal model for human gastric Helicobacter pylori infection: Success in both nude and euthymic mice. Am. J. Gastroenterol. 1991, 86, 1596–1603. [Google Scholar] [PubMed]
- Karita, M.; Li, Q.; Cantero, D.; Okita, K. Establishment of a small animal model for human Helicobacter pylori infection using germ-free mouse. Am. J. Gastroenterol. 1994, 89, 208–213. [Google Scholar] [PubMed]
- Marchetti, M.; Arico, B.; Burroni, D.; Figura, N.; Rappuoli, R.; Ghiara, P. Development of a mouse model of Helicobacter pylori infection that mimics human disease. Science 1995, 267, 1655–1658. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Fox, J.G.; Otto, G.; Murphy, J. A small animal model of human Helicobacter pylori active chronic gastritis. Gastroenterology 1990, 99, 1315–1323. [Google Scholar] [CrossRef]
- Lee, A.; O’Rourke, J.; de Ungria, M.C.; Robertson, B.; Daskalopoulos, G.; Dixon, M.F. A standardized mouse model of Helicobacter pylori infection: Introducing the Sydney strain. Gastroenterology 1997, 112, 1386–1397. [Google Scholar] [CrossRef]
- Draper, J.L.; Hansen, L.M.; Bernick, D.L.; Abedrabbo, S.; Underwood, J.G.; Kong, N.; Huang, B.C.; Weis, A.M.; Weimer, B.C.; van Vliet, A.H.; et al. Fallacy of the unique genome: Sequence diversity within single Helicobacter pylori strains. MBio 2017, 8, e02321–e02337. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.J.; Danon, S.J.; Wilson, J.E.; O’Rourke, J.L.; Salama, N.R.; Falkow, S.; Mitchell, H.; Lee, A. Chronic Helicobacter pylori infection with Sydney strain 1 and a newly identified mouse-adapted strain (Sydney strain 2000) in C57BL/6 and BALB/c mice. Infect. Immun. 2004, 72, 4668–4679. [Google Scholar] [CrossRef] [PubMed]
- Suerbaum, S.; Josenhans, C. Helicobacter pylori evolution and phenotypic diversification in a changing host. Nat. Rev. Microbiol. 2007, 5, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Lina, T.T.; Alzahrani, S.; House, J.; Yamaoka, Y.; Sharpe, A.H.; Rampy, B.A.; Pinchuk, I.V.; Reyes, V.E. Helicobacter pylori cag pathogenicity island’s role in B7-H1 induction and immune evasion. PLoS ONE 2015, 10, e0121841. [Google Scholar] [CrossRef] [PubMed]
- Sugimura, T.; Fujimura, S. Tumour production in glandular stomach of rat by N-methyl-N′-nitro-N-nitrosoguanidine. Nature 1967, 216, 943–944. [Google Scholar] [CrossRef] [PubMed]
- Tatematsu, M.; Ogawa, K.; Hoshiya, T.; Shichino, Y.; Kato, T.; Imaida, K.; Ito, N. Induction of adenocarcinomas in the glandular stomach of BALB/c mice treated with N-methyl-N-nitrosourea. Jpn. J. Cancer Res. 1992, 83, 915–918. [Google Scholar] [CrossRef] [PubMed]
- Tatematsu, M.; Yamamoto, M.; Iwata, H.; Fukami, H.; Yuasa, H.; Tezuka, N.; Masui, T.; Nakanishi, H. Induction of glandular stomach cancers in C3H mice treated with N-methyl-N-nitrosourea in the drinking water. Jpn. J. Cancer Res. 1993, 84, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- Takasu, S.; Tsukamoto, T.; Cao, X.Y.; Toyoda, T.; Hirata, A.; Ban, H.; Yamamoto, M.; Sakai, H.; Yanai, T.; Masegi, T.; et al. Roles of cyclooxygenase-2 and microsomal prostaglandin E synthase-1 expression and β-catenin activation in gastric carcinogenesis in N-methyl-N-nitrosourea-treated K19-C2mE transgenic mice. Cancer Sci. 2008, 99, 2356–2364. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Yu, Y. Transgenic and gene knockout mice in gastric cancer research. Oncotarget 2017, 8, 3696–3710. [Google Scholar] [CrossRef] [PubMed]
- Oshima, H.; Oshima, M.; Inaba, K.; Taketo, M.M. Hyperplastic gastric tumors induced by activated macrophages in COX-2/mPGES-1 transgenic mice. EMBO J. 2004, 23, 1669–1678. [Google Scholar] [CrossRef] [PubMed]
- Oshima, H.; Matsunaga, A.; Fujimura, T.; Tsukamoto, T.; Taketo, M.M.; Oshima, M. Carcinogenesis in mouse stomach by simultaneous activation of the Wnt signaling and prostaglandin E2 pathway. Gastroenterology 2006, 131, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- El-Omar, E.M.; Carrington, M.; Chow, W.H.; McColl, K.E.; Bream, J.H.; Young, H.A.; Herrera, J.; Lissowska, J.; Yuan, C.C.; Rothman, N.; et al. The role of interleukin-1 polymorphisms in the pathogenesis of gastric cancer. Nature 2001, 412, 99. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.; Bhagat, G.; Cui, G.; Takaishi, S.; Kurt-Jones, E.A.; Rickman, B.; Betz, K.S.; Penz-Oesterreicher, M.; Bjorkdahl, O.; Fox, J.G.; et al. Overexpression of interleukin-1β induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 2008, 14, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.C.; Brand, S.J. Function and regulation of gastrin in transgenic mice: A review. Yale J. Biol. Med. 1992, 65, 705–713, discussion 737–740. [Google Scholar] [PubMed]
- Wang, T.C.; Dangler, C.A.; Chen, D.; Goldenring, J.R.; Koh, T.; Raychowdhury, R.; Coffey, R.J.; Ito, S.; Varro, A.; Dockray, G.J.; et al. Synergistic interaction between hypergastrinemia and Helicobacter infection in a mouse model of gastric cancer. Gastroenterology 2000, 118, 36–47. [Google Scholar] [CrossRef]
- Hirayama, F.; Takagi, S.; Yokoyama, Y.; Iwao, E.; Ikeda, Y. Establishment of gastric Helicobacter pylori infection in Mongolian gerbils. J. Gastroenterol. 1996, 31 (Suppl. S9), 24–28. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, K.; Shimizu, N.; Tsukamoto, T.; Inada, K.; Cao, X.; Ikehara, Y.; Kaminishi, M.; Sugiyama, A.; Tatematsu, M. Reversibility of Heterotopic proliferative glands in glandular stomach of Helicobacter pylori-infected mongolian gerbils on eradication. Jpn. J. Cancer Res. 2002, 93, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Honda, S.; Fujioka, T.; Tokieda, M.; Satoh, R.; Nishizono, A.; Nasu, M. Development of Helicobacter pylori-induced gastric carcinoma in Mongolian gerbils. Cancer Res. 1998, 58, 4255–4259. [Google Scholar] [PubMed]
- Watanabe, T.; Tada, M.; Nagai, H.; Sasaki, S.; Nakao, M. Helicobacter pylori infection induces gastric cancer in mongolian gerbils. Gastroenterology 1998, 115, 642–648. [Google Scholar] [CrossRef]
- Tatematsu, M.; Yamamoto, M.; Shimizu, N.; Yoshikawa, A.; Fukami, H.; Kaminishi, M.; Oohara, T.; Sugiyama, A.; Ikeno, T. Induction of glandular stomach cancers in Helicobacter pylori-sensitive Mongolian gerbils treated with N-methyl-N-nitrosourea and N-methyl-N′-nitro-N-nitrosoguanidine in drinking water. Jpn. J. Cancer Res. 1998, 89, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, A.; Maruta, F.; Ikeno, T.; Ishida, K.; Kawasaki, S.; Katsuyama, T.; Shimizu, N.; Tatematsu, M. Helicobacter pylori infection enhances N-methyl-N-nitrosourea-induced stomach carcinogenesis in the Mongolian gerbil. Cancer Res. 1998, 58, 2067–2069. [Google Scholar] [PubMed]
- Shimizu, N.; Inada, K.; Nakanishi, H.; Tsukamoto, T.; Ikehara, Y.; Kaminishi, M.; Kuramoto, S.; Sugiyama, A.; Katsuyama, T.; Tatematsu, M. Helicobacter pylori infection enhances glandular stomach carcinogenesis in Mongolian gerbils treated with chemical carcinogens. Carcinogenesis 1999, 20, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, N.; Inada, K.I.; Tsukamoto, T.; Nakanishi, H.; Ikehara, Y.; Yoshikawa, A.; Kaminishi, M.; Kuramoto, S.; Tatematsu, M. New animal model of glandular stomach carcinogenesis in Mongolian gerbils infected with Helicobacter pylori and treated with a chemical carcinogen. J. Gastroenterol. 1999, 34, 61–66. [Google Scholar] [PubMed]
- Tatematsu, M.; Tsukamoto, T.; Inada, K. Stem cells and gastric cancer-role of gastric and intestinal mixed intestinal metaplasia. Cancer Sci. 2003, 94, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Mizoshita, T.; Tsukamoto, T.; Takenaka, Y.; Cao, X.; Kato, S.; Kaminishi, M.; Tatematsu, M. Gastric and intestinal phenotypes and histogenesis of advanced glandular stomach cancers in carcinogen-treated, Helicobacter pylori-infected Mongolian gerbils. Cancer Sci. 2006, 97, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Tsuji, I.; Wakai, K.; Nagata, C.; Mizoue, T.; Tanaka, K.; Tsugane, S. Evaluation based on systematic review of epidemiological evidence among Japanese populations: Tobacco smoking and total cancer risk. Jpn. J. Clin. Oncol. 2005, 35, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Sasazuki, S.; Sasaki, S.; Tsugane, S. Cigarette smoking, alcohol consumption and subsequent gastric cancer risk by subsite and histologic type. Int. J. Cancer 2002, 101, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Nishino, Y.; Inoue, M.; Tsuji, I.; Wakai, K.; Nagata, C.; Mizoue, T.; Tanaka, K.; Tsugane, S. Tobacco smoking and gastric cancer risk: An evaluation based on a systematic review of epidemiologic evidence among the Japanese population. Jpn. J. Clin. Oncol. 2006, 36, 800–807. [Google Scholar] [CrossRef] [PubMed]
- Tamer, L.; Ates, N.A.; Ates, C.; Ercan, B.; Elipek, T.; Yildirim, H.; Camdeviren, H.; Atik, U.; Aydin, S. Glutathione S-transferase M1, T1 and P1 genetic polymorphisms, cigarette smoking and gastric cancer risk. Cell Biochem. Funct. 2005, 23, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Wan, H.; Lin, Y.; Xie, X.; Li, Z.; Tan, G. Androgen receptor may be responsible for gender disparity in gastric cancer. Med. Hypotheses 2013, 80, 672–674. [Google Scholar] [CrossRef] [PubMed]
- Jukic, Z.; Radulovic, P.; Stojkovic, R.; Mijic, A.; Grah, J.; Kruslin, B.; Ferencic, Z.; Fucic, A. Gender difference in distribution of estrogen and androgen receptors in intestinal-type gastric cancer. Anticancer Res. 2017, 37, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Tajima, K.; Tominaga, S. Dietary habits and gastro-intestinal cancers: A comparative case-control study of stomach and large intestinal cancers in Nagoya, Japan. Jpn. J. Cancer Res. 1985, 76, 705–716. [Google Scholar] [PubMed]
- Tsugane, S.; Tsuda, M.; Gey, F.; Watanabe, S. Cross-sectional study with multiple measurements of biological markers for assessing stomach cancer risks at the population level. Environ. Health Perspect. 1992, 98, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Joossens, J.V.; Hill, M.J.; Elliott, P.; Stamler, R.; Lesaffre, E.; Dyer, A.; Nichols, R.; Kesteloot, H. Dietary salt, nitrate and stomach cancer mortality in 24 countries. Int. J. Epidemiol. 1996, 25, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Kono, S.; Hirohata, T. Nutrition and stomach cancer. Cancer Causes Control 1996, 7, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Tatematsu, M.; Takahashi, M.; Fukushima, S.; Hananouchi, M.; Shirai, T. Effects in rats of sodium chloride on experimental gastric cancers induced by N-methyl-N-nitro-N-nitrosoguanidine or 4-nitroquinoline-1-oxide. J. Natl. Cancer Inst. 1975, 55, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Tsukamoto, T.; Mizoshita, T.; Tanaka, H.; Kumagai, T.; Ota, H.; Katsuyama, T.; Asaka, M.; Tatematsu, M. High salt diets dose-dependently promote gastric chemical carcinogenesis in Helicobacter pylori-infected Mongolian gerbils associated with a shift in mucin production from glandular to surface mucous cells. Int. J. Cancer 2006, 119, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, K.; Shimizu, N.; Inada, K.; Tsukamoto, T.; Inoue, M.; Kumagai, T.; Sugiyama, A.; Mizoshita, T.; Kaminishi, M.; Tatematsu, M. Synergistic Promoting effects of Helicobacter pylori infection and high-salt diet on gastric carcinogenesis in mongolian gerbils. Jpn. J. Cancer Res. 2002, 93, 1083–1089. [Google Scholar] [CrossRef] [PubMed]
- Kawakubo, M.; Ito, Y.; Okimura, Y.; Kobayashi, M.; Sakura, K.; Kasama, S.; Fukuda, M.N.; Fukuda, M.; Katsuyama, T.; Nakayama, J. Natural antibiotic function of a human gastric mucin against Helicobacter pylori infection. Science 2004, 305, 1003–1006. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Tajima, K.; Hirose, K.; Hamajima, N.; Takezaki, T.; Kuroishi, T.; Tominaga, S. Tea and coffee consumption and the risk of digestive tract cancers: Data from a comparative case-referent study in Japan. Cancer Causes Control 1998, 9, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Shibata, K.; Moriyama, M.; Fukushima, T.; Kaetsu, A.; Miyazaki, M.; Une, H. Green tea consumption and chronic atrophic gastritis: A cross-sectional study in a green tea production village. J. Epidemiol. 2000, 10, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Lee, M.J.; Chen, L.; Yang, G.Y. Polyphenols as inhibitors of carcinogenesis. Environ. Health Perspect. 1997, 105, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Du, W.; Yang, D. Inhibition of green tea polyphenol EGCG((−)-epigallocatechin-3-gallate) on the proliferation of gastric cancer cells by suppressing canonical Wnt/β-catenin signalling pathway. Int. J. Food Sci. Nutr. 2016, 67, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Ohno, T.; Ohtani, M.; Suto, H.; Ohta, M.; Imamura, Y.; Matsuda, H.; Hiramatsu, K.; Nemoto, T.; Nakamoto, Y. Effect of green tea catechins on gastric mucosal dysplasia in insulin-gastrin mice. Oncol. Rep. 2016, 35, 3241–3247. [Google Scholar] [CrossRef] [PubMed]
- Marone, P.; Bono, L.; Leone, E.; Bona, S.; Carretto, E.; Perversi, L. Bactericidal activity of pistacia lentiscus mastic gum against Helicobacter pylori. J. Chemother. 2001, 13, 611–614. [Google Scholar] [CrossRef] [PubMed]
- Bebb, J.R.; Bailey-Flitter, N.; Ala’Aldeen, D.; Atherton, J.C. Mastic gum has no effect on Helicobacter pylori load in vivo. J. Antimicrob. Chemother. 2003, 52, 522–523. [Google Scholar] [CrossRef] [PubMed]
- Dabos, K.J.; Sfika, E.; Vlatta, L.J.; Giannikopoulos, G. The effect of mastic gum on Helicobacter pylori: A randomized pilot study. Phytomedicine 2010, 17, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Loughlin, M.F.; Ala’Aldeen, D.A.; Jenks, P.J. Monotherapy with mastic does not eradicate Helicobacter pylori infection from mice. J. Antimicrob. Chemother. 2003, 51, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Paraschos, S.; Magiatis, P.; Mitakou, S.; Petraki, K.; Kalliaropoulos, A.; Maragkoudakis, P.; Mentis, A.; Sgouras, D.; Skaltsounis, A.L. In vitro and in vivo activities of Chios mastic gum extracts and constituents against Helicobacter pylori. Antimicrob. Agents Chemother. 2007, 51, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Bae, M.; Jang, S.; Lim, J.W.; Kang, J.; Bak, E.J.; Cha, J.H.; Kim, H. Protective effect of korean red ginseng extract against Helicobacter pylori-induced gastric inflammation in Mongolian gerbils. J. Ginseng Res. 2014, 38, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.W.; Baek, Y.M.; Jang, I.S.; Yang, K.E.; Lee, D.G.; Yoon, S.J.; Rho, J.; Cho, C.K.; Lee, Y.W.; Kwon, K.R.; et al. An enzymatically fortified ginseng extract inhibits proliferation and induces apoptosis of KATO3 human gastric cancer cells via modulation of Bax, mTOR, PKB and IκBα. Mol. Med. Rep. 2015, 11, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Yun, T.K.; Choi, S.Y. Preventive effect of ginseng intake against various human cancers: A case-control study on 1987 pairs. Cancer Epidemiol. Biomarkers Prev. 1995, 4, 401–408. [Google Scholar] [PubMed]
- Kamangar, F.; Gao, Y.T.; Shu, X.O.; Kahkeshani, K.; Ji, B.T.; Yang, G.; Li, H.L.; Rothman, N.; Chow, W.H.; Zheng, W. Ginseng intake and gastric cancer risk in the Shanghai Women’s Health Study cohort. Cancer Epidemiol. Biomarkers Prev. 2007, 16, 629–630. [Google Scholar] [CrossRef] [PubMed]
- Miwa, H.; Go, M.F.; Sato, N. H. pylori and gastric cancer: The Asian enigma. Am. J. Gastroenterol. 2002, 97, 1106–1112. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, T.; Shi, L.; Takasu, S.; Cho, Y.M.; Kiriyama, Y.; Nishikawa, A.; Ogawa, K.; Tatematsu, M.; Tsukamoto, T. Anti-inflammatory effects of capsaicin and piperine on Helicobacter pylori-induced chronic gastritis in mongolian gerbils. Helicobacter 2016, 21, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Okubo, M.; Tahara, T.; Shibata, T.; Nakamura, M.; Yoshioka, D.; Maeda, Y.; Yonemura, J.; Ishizuka, T.; Arisawa, T.; Hirata, I. Changes in gastric mucosal patterns seen by magnifying NBI during H. pylori eradication. J. Gastroenterol. 2011, 46, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Kiriyama, Y.; Tahara, T.; Shibata, T.; Okubo, M.; Nakagawa, M.; Okabe, A.; Ohmiya, N.; Kuroda, M.; Sugioka, A.; Ichinose, M.; et al. Gastric-and-intestinal mixed intestinal metaplasia is irreversible point with eradication of Helicobacter pylori. Open J. Pathol. 2016, 6, 93–104. [Google Scholar] [CrossRef]
- Lee, Y.C.; Chen, T.H.; Chiu, H.M.; Shun, C.T.; Chiang, H.; Liu, T.Y.; Wu, M.S.; Lin, J.T. The benefit of mass eradication of Helicobacter pylori infection: A community-based study of gastric cancer prevention. Gut 2013, 62, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Annibale, B.; Aprile, M.R.; D’Ambra, G.; Caruana, P.; Bordi, C.; delle Fave, G. Cure of Helicobacter pylori infection in atrophic body gastritis patients does not improve mucosal atrophy but reduces hypergastrinemia and its related effects on body ECL-cell hyperplasia. Aliment. Pharmacol. Ther. 2000, 14, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Forbes, G.M.; Warren, J.R.; Glaser, M.E.; Cullen, D.J.; Marshall, B.J.; Collins, B.J. Long-term follow-up of gastric histology after Helicobacter pylori eradication. J. Gastroenterol. Hepatol. 1996, 11, 670–673. [Google Scholar] [CrossRef] [PubMed]
- Kodama, M.; Murakami, K.; Okimoto, T.; Sato, R.; Uchida, M.; Abe, T.; Shiota, S.; Nakagawa, Y.; Mizukami, K.; Fujioka, T. Ten-year prospective follow-up of histological changes at five points on the gastric mucosa as recommended by the updated Sydney system after Helicobacter pylori eradication. J. Gastroenterol. 2012, 47, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Haruma, K.; Kamada, T.; Mihara, M.; Kim, S.; Kitadai, Y.; Sumii, M.; Tanaka, S.; Yoshihara, M.; Chayama, K. Helicobacter pylori eradication therapy improves atrophic gastritis and intestinal metaplasia: A 5-year prospective study of patients with atrophic gastritis. Aliment. Pharmacol. Ther. 2002, 16, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Toyokawa, T.; Suwaki, K.; Miyake, Y.; Nakatsu, M.; Ando, M. Eradication of Helicobacter pylori infection improved gastric mucosal atrophy and prevented progression of intestinal metaplasia, especially in the elderly population: A long-term prospective cohort study. J. Gastroenterol. Hepatol. 2010, 25, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Fujioka, T.; Kodama, R.; Kubota, T.; Tokieda, M.; Nasu, M. Helicobacter pylori infection accelerates human gastric mucosal cell proliferation. J. Gastroenterol. 1997, 32, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Erkan, G.; Gonul, I.I.; Kandilci, U.; Dursun, A. Evaluation of apoptosis along with BCL-2 and Ki-67 expression in patients with intestinal metaplasia. Pathol. Res. Pract. 2012, 208, 89–93. [Google Scholar] [CrossRef] [PubMed]
- You, W.C.; Brown, L.M.; Zhang, L.; Li, J.Y.; Jin, M.L.; Chang, Y.S.; Ma, J.L.; Pan, K.F.; Liu, W.D.; Hu, Y.; et al. Randomized double-blind factorial trial of three treatments to reduce the prevalence of precancerous gastric lesions. J. Natl. Cancer Inst. 2006, 98, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Mera, R.; Fontham, E.T.; Bravo, L.E.; Bravo, J.C.; Piazuelo, M.B.; Camargo, M.C.; Correa, P. Long term follow up of patients treated for Helicobacter pylori infection. Gut 2005, 54, 1536–1540. [Google Scholar] [CrossRef] [PubMed]
- Leung, W.K.; Lin, S.R.; Ching, J.Y.; To, K.F.; Ng, E.K.; Chan, F.K.; Lau, J.Y.; Sung, J.J. Factors predicting progression of gastric intestinal metaplasia: Results of a randomised trial on Helicobacter pylori eradication. Gut 2004, 53, 1244–1249. [Google Scholar] [CrossRef] [PubMed]
- Uemura, N.; Mukai, T.; Okamoto, S.; Yamaguchi, S.; Mashiba, H.; Taniyama, K.; Sasaki, N.; Haruma, K.; Sumii, K.; Kajiyama, G. Effect of Helicobacter pylori eradication on subsequent development of cancer after endoscopic resection of early gastric cancer. Cancer Epidemiol. Biomarkers Prev. 1997, 6, 639–642. [Google Scholar] [CrossRef]
- Fukase, K.; Kato, M.; Kikuchi, S.; Inoue, K.; Uemura, N.; Okamoto, S.; Terao, S.; Amagai, K.; Hayashi, S.; Asaka, M. Effect of eradication of Helicobacter pylori on incidence of metachronous gastric carcinoma after endoscopic resection of early gastric cancer: An open-label, randomised controlled trial. Lancet 2008, 372, 392–397. [Google Scholar] [CrossRef]
- Bae, S.E.; Jung, H.Y.; Kang, J.; Park, Y.S.; Baek, S.; Jung, J.H.; Choi, J.Y.; Kim, M.Y.; Ahn, J.Y.; Choi, K.S.; et al. Effect of Helicobacter pylori eradication on metachronous recurrence after endoscopic resection of gastric neoplasm. Am. J. Gastroenterol. 2014, 109, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.C.; Lam, S.K.; Wong, W.M.; Chen, J.S.; Zheng, T.T.; Feng, R.E.; Lai, K.C.; Hu, W.H.; Yuen, S.T.; Leung, S.Y.; et al. Helicobacter pylori eradication to prevent gastric cancer in a high-risk region of China: A randomized controlled trial. Jama 2004, 291, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.N.; Wang, Z.; Li, X.; Zhou, Z.G. Helicobacter pylori eradication cannot reduce the risk of gastric cancer in patients with intestinal metaplasia and dysplasia: Evidence from a meta-analysis. Gastric Cancer 2016, 19, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Yanaoka, K.; Oka, M.; Ohata, H.; Yoshimura, N.; Deguchi, H.; Mukoubayashi, C.; Enomoto, S.; Inoue, I.; Iguchi, M.; Maekita, T.; et al. Eradication of Helicobacter pylori prevents cancer development in subjects with mild gastric atrophy identified by serum pepsinogen levels. Int. J. Cancer 2009, 125, 2697–2703. [Google Scholar] [CrossRef] [PubMed]
- Schlemper, R.J.; Kato, Y.; Stolte, M. Diagnostic criteria for gastrointestinal carcinomas in Japan and Western countries: Proposal for a new classification system of gastrointestinal epithelial neoplasia. J. Gastroenterol. Hepatol. 2000, 15, G49–G57. [Google Scholar] [CrossRef] [PubMed]
- Japanese Gastric Cancer Association. Japanese classification of gastric carcinoma: 3rd English edition. Gastric Cancer 2011, 14, 101–112. [Google Scholar]
- Ito, M.; Tanaka, S.; Takata, S.; Oka, S.; Imagawa, S.; Ueda, H.; Egi, Y.; Kitadai, Y.; Yasui, W.; Yoshihara, M.; et al. Morphological changes in human gastric tumours after eradication therapy of Helicobacter pylori in a short-term follow-up. Aliment. Pharmacol. Ther. 2005, 21, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Ito, M.; Matsuo, T.; Boda, T.; Oka, S.; Yoshihara, M.; Tanaka, S.; Chayama, K. Characteristic epithelium with low-grade atypia appears on the surface of gastric cancer after successful Helicobacter pylori eradication therapy. Helicobacter 2014, 19, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, N.; Ikehara, Y.; Inada, K.; Nakanishi, H.; Tsukamoto, T.; Nozaki, K.; Kaminishi, M.; Kuramoto, S.; Sugiyama, A.; Katsuyama, T.; et al. Eradication diminishes enhancing effects of Helicobacter pylori infection on glandular stomach carcinogenesis in Mongolian gerbils. Cancer Res. 2000, 60, 1512–1514. [Google Scholar] [PubMed]
- Nozaki, K.; Shimizu, N.; Ikehara, Y.; Inoue, M.; Tsukamoto, T.; Inada, K.; Tanaka, H.; Kumagai, T.; Kaminishi, M.; Tatematsu, M. Effect of early eradication on Helicobacter pylori-related gastric carcinogenesis in Mongolian gerbils. Cancer Sci. 2003, 94, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Tsugane, S.; Sasazuki, S. Diet and the risk of gastric cancer: Review of epidemiological evidence. Gastric Cancer 2007, 10, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Naito, Y.; Yoshikawa, T. Molecular and cellular mechanisms involved in Helicobacter pylori-induced inflammation and oxidative stress. Free Radic. Biol. Med. 2002, 33, 323–336. [Google Scholar] [CrossRef]
- Cao, X.; Tsukamoto, T.; Nozaki, K.; Tanaka, H.; Cao, L.; Toyoda, T.; Takasu, S.; Ban, H.; Kumagai, T.; Tatematsu, M. Severity of gastritis determines glandular stomach carcinogenesis in Helicobacter pylori-infected Mongolian gerbils. Cancer Sci. 2007, 98, 478–483. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Tsukamoto, T.; Seki, T.; Tanaka, H.; Morimura, S.; Cao, L.; Mizoshita, T.; Ban, H.; Toyoda, T.; Maeda, H.; et al. 4-Vinyl-2,6-dimethoxyphenol (canolol) suppresses oxidative stress and gastric carcinogenesis in Helicobacter pylori-infected carcinogen-treated Mongolian gerbils. Int. J. Cancer 2008, 122, 1445–1454. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Jiang, J.; Tsukamoto, T.; Liu, R.; Ma, L.; Jia, Z.; Kong, F.; Oshima, M.; Cao, X. Canolol inhibits gastric tumors initiation and progression through COX-2/PGE2 pathway in K19-C2mE transgenic mice. PLoS ONE 2015, 10, e0120938. [Google Scholar] [CrossRef] [PubMed]
- Echizen, K.; Hirose, O.; Maeda, Y.; Oshima, M. Inflammation in gastric cancer: Interplay of the COX-2/prostaglandin E2 and Toll-like receptor/MyD88 pathways. Cancer Sci. 2016, 107, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Leung, W.K.; Sung, J.J. Chemoprevention of gastric cancer. Eur. J. Gastroenterol. Hepatol. 2006, 18, 867–871. [Google Scholar] [CrossRef] [PubMed]
- Prescott, S.M.; Fitzpatrick, F.A. Cyclooxygenase-2 and carcinogenesis. Biochim. Biophys. Acta 2000, 1470, M69–M78. [Google Scholar] [CrossRef]
- Futagami, S.; Suzuki, K.; Hiratsuka, T.; Shindo, T.; Hamamoto, T.; Tatsuguchi, A.; Ueki, N.; Shinji, Y.; Kusunoki, M.; Wada, K.; et al. Celecoxib inhibits Cdx2 expression and prevents gastric cancer in Helicobacter pylori-infected Mongolian gerbils. Digestion 2006, 74, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Magari, H.; Shimizu, Y.; Inada, K.; Enomoto, S.; Tomeki, T.; Yanaoka, K.; Tamai, H.; Arii, K.; Nakata, H.; Oka, M.; et al. Inhibitory effect of etodolac, a selective cyclooxygenase-2 inhibitor, on stomach carcinogenesis in Helicobacter pylori-infected Mongolian gerbils. Biochem. Biophys. Res. Commun. 2005, 334, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.H.; McEntee, M.F.; Whelan, J. Sulindac causes rapid regression of preexisting tumors in Min/+ mice independent of prostaglandin biosynthesis. Cancer Res. 1997, 57, 4267–4273. [Google Scholar] [PubMed]
- Reddy, B.S.; Maruyama, H.; Kelloff, G. Dose-related inhibition of colon carcinogenesis by dietary piroxicam, a nonsteroidal antiinflammatory drug, during different stages of rat colon tumor development. Cancer Res. 1987, 47, 5340–5346. [Google Scholar] [PubMed]
- Yanaoka, K.; Oka, M.; Yoshimura, N.; Deguchi, H.; Mukoubayashi, C.; Enomoto, S.; Maekita, T.; Inoue, I.; Ueda, K.; Utsunomiya, H.; et al. Preventive effects of etodolac, a selective cyclooxygenase-2 inhibitor, on cancer development in extensive metaplastic gastritis, a Helicobacter pylori-negative precancerous lesion. Int. J. Cancer 2010, 126, 1467–1473. [Google Scholar] [PubMed]
- Zhang, Y.; Pan, K.F.; Zhang, L.; Ma, J.L.; Zhou, T.; Li, J.Y.; Shen, L.; You, W.C. Helicobacter pylori, cyclooxygenase-2 and evolution of gastric lesions: Results from an intervention trial in China. Carcinogenesis 2015, 36, 1572–1579. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsukamoto, T.; Nakagawa, M.; Kiriyama, Y.; Toyoda, T.; Cao, X. Prevention of Gastric Cancer: Eradication of Helicobacter Pylori and Beyond. Int. J. Mol. Sci. 2017, 18, 1699. https://doi.org/10.3390/ijms18081699
Tsukamoto T, Nakagawa M, Kiriyama Y, Toyoda T, Cao X. Prevention of Gastric Cancer: Eradication of Helicobacter Pylori and Beyond. International Journal of Molecular Sciences. 2017; 18(8):1699. https://doi.org/10.3390/ijms18081699
Chicago/Turabian StyleTsukamoto, Tetsuya, Mitsuru Nakagawa, Yuka Kiriyama, Takeshi Toyoda, and Xueyuan Cao. 2017. "Prevention of Gastric Cancer: Eradication of Helicobacter Pylori and Beyond" International Journal of Molecular Sciences 18, no. 8: 1699. https://doi.org/10.3390/ijms18081699
APA StyleTsukamoto, T., Nakagawa, M., Kiriyama, Y., Toyoda, T., & Cao, X. (2017). Prevention of Gastric Cancer: Eradication of Helicobacter Pylori and Beyond. International Journal of Molecular Sciences, 18(8), 1699. https://doi.org/10.3390/ijms18081699