The Variation Analysis of DNA Methylation in Wheat Carrying Gametocidal Chromosome 3C from Aegilops triuncialis

Abstract
:1. Introduction
2. Results
2.1. Increased Level of Cytosine Methylation in the CS–3C Gametocidal Chromosome Addition Lines
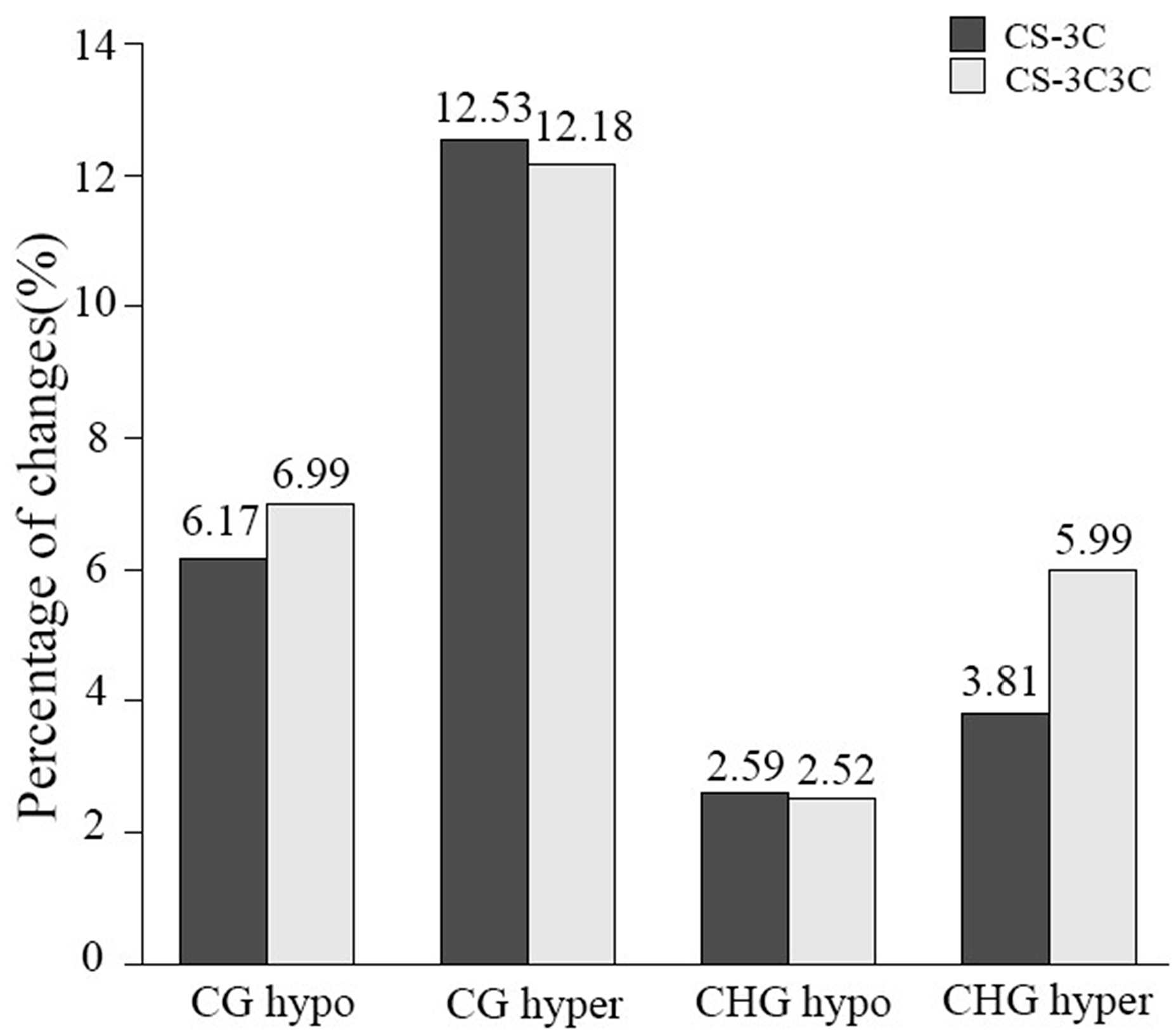
2.2. Alteration of Locus-Specific Cytosine Methylation Patters
2.3. Sequencing Analysis of the Differentially Methylated DNA Sequences
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. DNA Extraction
4.3. Enzyme Cleavage and Adaptor Ligation of MSAP Analysis
4.4. Preselective Amplification
4.5. Selective Amplification
4.6. MSAP Band Scoring and Data Analysis
4.7. Cloning and Sequencing of MSAP Fragments
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Endo, T. Gametocidal chromosomes and their induction of chromosome mutations in wheat. Jpn. J. Genet. 1990, 65, 135–152. [Google Scholar] [CrossRef]
- Endo, T.R.; Tsunewaki, K. Sterility of common wheat with Aegilops triuncialis cytoplasm. J. Hered. 1975, 66, 13–18. [Google Scholar] [CrossRef]
- Tsujimoto, H.; Tsunewaki, K. Gametocidal genes in wheat and its relatives. II. Suppressor of the chromosome 3c gametocidal gene of Aegilops triuncialis. Can. J. Genet. Cytol. 1985, 27, 178–185. [Google Scholar] [CrossRef]
- Nasuda, S.; Friebe, B.; Gill, B.S. Gametocidal genes induce chromosome breakage in the interphase prior to the first mitotic cell division of the male gametophyte in wheat. Genetics 1998, 149, 1115–1124. [Google Scholar] [PubMed]
- King, I.P.; Laurie, D.A. Chromosome damage in early embryo and endosperm development in crosses involving the preferentially transmitted 4Sl chromosome of Aegilops sharonensis. Heredity 1993, 70, 52–59. [Google Scholar] [CrossRef]
- Endo, T.R.; Yamamoto, M.; Mukai, Y. Structural changes of rye chromosome 1R induced by a gametocidal chromosome. Jpn. J. Genet. 1994, 69, 13–19. [Google Scholar] [CrossRef]
- Shi, F.; Endo, T.R. Production of wheat-barley disomic addition lines possessing an Aegilops cylindrica gametocidal chromosome. Genes Genet. Syst. 1997, 72, 243–248. [Google Scholar] [CrossRef]
- Li, J.; Xu, X.; Xu, P.; Guo, C. Inducing chromosome translocation and deletions by Chinese Spring-Agilops 2C disomic addition x Chinese Spring-Elytriga 5E disomic addition. Yi Chuan Xue Bao 2003, 30, 345–349. [Google Scholar] [PubMed]
- Liu, W.; Guo, Y.; Wu, J.; Wang, X.; Wang, R.; Yang, X.; Xu, X.; Li, J.; Li, L. Cytological characteristics of F1 hybrids between wheat-agropyron addition lines and wheat-gametocidal chromosome addition line. J. Artic. 2007, 33, 898–902. [Google Scholar]
- Endo, T. The gametocidal chromosome as a tool for chromosome manipulation in wheat. Chromosome Res. 2007, 15, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, H. Gametocidal genes in wheat as the inducer of chromosome breakage. Wheat Inf. Serv. 2005, 100, 33–48. [Google Scholar]
- Chan, S.W.; Henderson, I.R.; Jacobsen, S.E. Gardening the genome: DNA methylation in Arabidopsis thaliana. Nat. Rev. Genet. 2005, 6, 351–360. [Google Scholar] [CrossRef] [PubMed]
- De Las Heras, J.I.; King, I.P.; Parker, J.S. 5-azacytidine induces chromosomal breakage in the root tips of wheat carrying the cuckoo chromosome 4Sl from Aegilops sharonensis. Heredity 2001, 87, 474–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, W.; Cong, W.; Shu, Y.; Wang, D.; Xu, G.; Guo, C. Gametocidal chromosomes enhancing chromosome aberration in common wheat induced by 5-azacytidine. Genet. Mol. Res. GMR 2013, 12, 2227. [Google Scholar] [CrossRef] [PubMed]
- Goll, M.G.; Bestor, T.H. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005, 74, 481–514. [Google Scholar] [CrossRef] [PubMed]
- Le, T.N.; Schumann, U.; Smith, N.A.; Tiwari, S.; Au, P.C.; Zhu, Q.H.; Taylor, J.M.; Kazan, K.; Llewellyn, D.J.; Zhang, R.; et al. DNA demethylases target promoter transposable elements to positively regulate stress responsive genes in Arabidopsis. Genome Biol. 2014, 15, 458. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P. DNA methylation systems and targets in plants. FEBS Lett. 2011, 585, 2008–2015. [Google Scholar] [CrossRef] [PubMed]
- Henderson, I.R.; Jacobsen, S.E. Epigenetic inheritance in plants. Nature 2007, 447, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Paszkowski, J.; Whitham, S.A. Gene silencing and DNA methylation processes. Curr. Opin. Plant. Biol. 2001, 4, 123–129. [Google Scholar] [CrossRef]
- Tycko, B. DNA methylation in genomic imprinting. Mutat. Res. 1997, 386, 131–140. [Google Scholar] [CrossRef]
- Furner, I.J.; Matzke, M. Methylation and demethylation of the Arabidopsis genome. Curr. Opin. Plant Biol. 2011, 14, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-García, L.; Cervera, M.T.; Martínez-Zapater, J.M. DNA methylation increases throughout Arabidopsis development. Planta 2005, 222, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hu, J.; Zhang, H.; Ding, Y. DNA methylation changes in photoperiod-thermo-sensitive male sterile rice pa64s under two different conditions. Gene 2014, 537, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Xu, Y.-H.; Wang, J.-B. DNA-methylation changes induced by salt stress in wheat Triticum Aestivum. Afr. J. Biotechnol. 2009, 8, 6201–6207. [Google Scholar]
- Bednarek, P.T.; Orlowska, R.; Niedziela, A. A relative quantitative methylation-sensitive amplified polymorphism (MSAP) method for the analysis of abiotic stress. BMC Plant Biol. 2017, 17, 79. [Google Scholar] [CrossRef] [PubMed]
- Ci, D.; Song, Y.; Du, Q.; Tian, M.; Han, S.; Zhang, D. Variation in genomic methylation in natural populations of populus simonii is associated with leaf shape and photosynthetic traits. J. Exp. Bot. 2016, 67, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Fan, G.; Deng, M.; Zhao, Z.; Dong, Y. Identification of genes related to paulownia witches’ broom by AFLP and MSAP. Int. J. Mol. Sci. 2014, 15, 14669–14683. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Wang, H.; Dong, B.; Yang, X.; Chen, S.; Jiang, J.; Zhang, Z.; Liu, C.; Zhao, N.; Chen, F. Morphological, genome and gene expression changes in newly induced autopolyploid Chrysanthemum lavandulifolium (fisch. Ex trautv.) makino. Int. J. Mol. Sci. 2016, 17, 1690. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Figueroa, A. Msap: A tool for the statistical analysis of methylation-sensitive amplified polymorphism data. Mol. Ecol. Resour. 2013, 13, 522–527. [Google Scholar] [CrossRef] [PubMed]
- McClelland, M.; Nelson, M.; Raschke, E. Effect of site-specific modification on restriction endonucleases and DNA modification methyltransferases. Nucleic Acids Res. 1994, 22, 3640–3659. [Google Scholar] [CrossRef] [PubMed]
- Colot, V.; Rossignol, J.L. Eukaryotic DNA methylation as an evolutionary device. Bioessays 1999, 21, 402–411. [Google Scholar] [CrossRef]
- Rangwala, S.H.; Richards, E.J. The value-added genome: Building and maintaining genomic cytosine methylation landscapes. Curr. Opin. Genet. Dev. 2004, 14, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Paszkowski, J. DNA methylation and epigenetic inheritance during plant gametogenesis. Chromosoma 2006, 115, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Portis, E.; Acquadro, A.; Comino, C.; Lanteri, S. Analysis of DNA methylation during germination of pepper (Capsicum annuum L.) seeds using methylation-sensitive amplification polymorphism (MSAP). Plant Sci. 2004, 166, 169–178. [Google Scholar] [CrossRef]
- Xiong, L.; Xu, C.; Maroof, M.S.; Zhang, Q. Patterns of cytosine methylation in an elite rice hybrid and its parental lines, detected by a methylation-sensitive amplification polymorphism technique. Mol. Gen. Genet. MGG 1999, 261, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Ashikawa, I. Surveying CpG methylation at 5′-CCGG in the genomes of rice cultivars. Plant Mol. Biol. 2001, 45, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Cervera, M.-T.; Ruiz-Garcia, L.; Martinez-Zapater, J. Analysis of DNA methylation in Arabidopsis thaliana based on methylation-sensitive AFLP markers. Mol. Genet. Genom. 2002, 268, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Cantu, D.; Vanzetti, L.S.; Sumner, A.; Dubcovsky, M.; Matvienko, M.; Distelfeld, A.; Michelmore, R.W.; Dubcovsky, J. Small RNAs, DNA methylation and transposable elements in wheat. BMC Genom. 2010, 11, 408. [Google Scholar] [CrossRef] [PubMed]
- Sabot, F.; Simon, D.; Bernard, M. Plant transposable elements, with an emphasis on grass species. Euphytica 2004, 139, 227–247. [Google Scholar] [CrossRef]
- Flavell, R. Repetitive DNA and chromosome evolution in plants. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1986, 312, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Feschotte, C.; Jiang, N.; Wessler, S.R. Plant transposable elements: Where genetics meets genomics. Nat. Rev. Genet. 2002, 3, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-López, M.; García-Pérez, J.L. DNA transposons: Nature and applications in genomics. Curr. Genom. 2010, 11, 115. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, R.K.; Martienssen, R. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 2007, 8, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.G.; Pérez-Escuredo, J.; Castro-Santos, P.; Marcos, C.Á.; Pendás, J.L.L.; Fraga, M.F.; Hermsen, M.A. Hypomethylation of LINE-1, and not centromeric SAT-α, is associated with centromeric instability in head and neck squamous cell carcinoma. Cell. Oncol. 2012, 35, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Marconi, G.; Pace, R.; Traini, A.; Raggi, L.; Lutts, S.; Chiusano, M.; Guiducci, M.; Falcinelli, M.; Benincasa, P.; Albertini, E. Use of MSAP markers to analyse the effects of salt stress on DNA methylation in rapeseed (Brassica napus var. Oleifera). PLoS ONE 2013, 8, e75597. [Google Scholar] [CrossRef] [PubMed]
- Bocchini, M.; Bartucca, M.L.; Ciancaleoni, S.; Mimmo, T.; Cesco, S.; Pii, Y.; Albertini, E.; del Buono, D. Iron deficiency in barley plants: Phytosiderophore release, iron translocation, and DNA methylation. Front. Plant Sci. 2015, 6, 514. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Primer/Adapter | EcoR I (E) 5′-3′ | Hpa II/Msp I (H/M) 5′-3′ |
|---|---|---|
| Adapter-1 | CTCGTAGACTGCGTACC | GACGATGAGTCTAGAA |
| Adapter-2 | AATTGGTACGCAGTCTAC | CGTTCTAGACTCATC |
| Pre-selective primers | GACTGCGTACCAATTCA | GATGAGTCTAGAACGGT |
| Selective amplification primers | GACTGCGTACCAATTC AAC(Ea) | GATGAGTCTAGAACGG TAC(H/M1) |
| GACTGCGTACCAATTC AAG(Eb) | GATGAGTCTAGAACGG TAG(H/M2) | |
| GACTGCGTACCAATTC ACA(Ec) | GATGAGTCTAGAACGG TCT(H/M3) | |
| GACTGCGTACCAATTC ACT(Ed) | GATGAGTCTAGAACGG TCG(H/M4) | |
| GACTGCGTACCAATTC ACC(Ee) | GATGAGTCTAGAACGG TTC(H/M5) | |
| GACTGCGTACCAATTC ACG(Ef) | GATGAGTCTAGAACGG TTG(H/M6) | |
| GACTGCGTACCAATTC AGC(Eg) | GATGAGTCTAGAACGG TTA(H/M7) | |
| GACTGCGTACCAATTC AGG(Eh) | GATGAGTCTAGAACGG TGA(H/M8) | |
| GACTGCGTACCAATTC AGA(Ei) | GATGAGTCTAGAACGG TGC(H/M9) | |
| GACTGCGTACCAATTC ATC(Ej) | GATGAGTCTAGAACGG TGT(H/M10) |
| Cultivars | Total Amplified Sites | Unmethylated Sites and Ratio | Methylated Sites | ||
|---|---|---|---|---|---|
| Full Methylated Sites (CG) and Ratio | Hemimethylated Sites (CHG) and Ratio | Total Methylated Sites and MSAP Ratio | |||
| CS | 2956 | 1735 (58.69%) | 950 (32.14%) | 271 (9.17%) | 1221 (41.31%)b |
| CS–3C | 2956 | 1517 (51.32%) | 1149 (38.87%) | 290 (9.81%) | 1439 (48.68%)a |
| CS–3C3C | 2956 | 1518 (51.35%) | 1129 (38.19%) | 309 (10.45%) | 1438 (48.65%)a |
| Patterns | Common Wheat | Gc Addition Line | Number and Frequency of Sites | Sataus | |||
|---|---|---|---|---|---|---|---|
| H | M | H | M | CS–3C | CS–3C3C | ||
| MA1 | + | + | + | + | 1324 (44.79%) | 1304 (44.11%) | → |
| MA2 | + | + | + | - | 68 (2.30%) | 86 (2.91%) | ↑ |
| MA3 | + | + | - | + | 306 (10.35%) | 273 (9.24%) | ↑ |
| MA4 | + | + | - | - | 37 (1.25%) | 72 (2.44%) | ↑ |
| MB1 | + | - | + | - | 164 (5.55%) | 171 (5.78%) | → |
| MB2 | + | - | + | + | 37 (1.25%) | 28 (0.95%) | ↓ |
| MB3 | + | - | - | + | 24 (0.81%) | 21 (0.71%) | others |
| MB4 | + | - | - | - | 46 (1.56%) | 51 (1.73%) | ↑ |
| MC1 | - | + | - | + | 652 (22.06%) | 587 (19.86%) | → |
| MC2 | - | + | + | + | 139 (4.70%) | 173 (5.85%) | ↓ |
| MC3 | - | + | + | - | 23 (0.78%) | 25 (0.85%) | others |
| MC4 | - | + | - | - | 26 (0.88%) | 55 (1.86%) | ↑ |
| MD1 | - | - | + | + | 17 (0.58%) | 13 (0.44%) | ↓ |
| MD2 | - | - | + | - | 35 (1.18%) | 27 (0.91%) | ↓ |
| MD3 | - | - | - | + | 31 (1.05%) | 40 (1.35%) | ↓ |
| MD4 | - | - | - | - | 27 (0.91%) | 30 (1.01%) | → |
| MSAP Fragment | Primer Combination | Length (bp) | Methylation Pattern | Accession No. | e Value | Sequence Homology |
|---|---|---|---|---|---|---|
| P1 | Ef/HM1 | 240 | Demethylated | HG670306.1 | 1×10−74 | Triticum aestivum chromosome 3B, genomic scaffold, cultivar Chinese Spring |
| P2 | Ee/HM4 | 247 | Demethylated | AK375691.1 | 6×10−32 | Hordeum vulgare subsp. vulgare mRNA for predicted protein, complete cds, clone: NIASHv3101H12 |
| P5 | Ee/HM4 | 140 | Demethylated | AY534123.1 | 3×10−32 | Aegilops tauschii transposons Stowaway MITE, transposons XJ1, Jody, Angela, and XJ and Stowaway MITE, complete sequence |
| P12 | Ee/HM4 | 103 | Methylated | DQ537335.1 | 2×10−25 | Triticum aestivum clones BAC 1031P08; BAC 754K10; BAC 1344C16, complete sequence (transposon:LTR-retrotransposon WIS-1p) |
| P14 | Ee/HM4 | 100 | Methylated | HG670306.1 | 4×10−25 | Triticum aestivum chromosome 3B, genomic scaffold, cultivar Chinese Spring |
| P16 | Ee/HM4 | 240 | Demethylated | HG670306.1 | 4×10−78 | Triticum aestivum chromosome 3B, genomic scaffold, cultivar Chinese Spring |
| P21 | Ee/HM4 | 137 | Demethylated | HG670306.1 | 3×10−23 | Triticum aestivum chromosome 3B, genomic scaffold, cultivar Chinese Spring |
| P28 | Ee/HM4 | 143 | Methylated | JF946485.1 | 1×10−5 | Triticum aestivum retrotransposons Gypsy TREP 3245_Sabrina, Copia TREP 3161_WIS, complete sequence |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Zhao, J.; Bai, Y.; Ao, Y.; Guo, C. The Variation Analysis of DNA Methylation in Wheat Carrying Gametocidal Chromosome 3C from Aegilops triuncialis. Int. J. Mol. Sci. 2017, 18, 1738. https://doi.org/10.3390/ijms18081738
Wang D, Zhao J, Bai Y, Ao Y, Guo C. The Variation Analysis of DNA Methylation in Wheat Carrying Gametocidal Chromosome 3C from Aegilops triuncialis. International Journal of Molecular Sciences. 2017; 18(8):1738. https://doi.org/10.3390/ijms18081738
Chicago/Turabian StyleWang, Dan, Jieyu Zhao, Yan Bai, You Ao, and Changhong Guo. 2017. "The Variation Analysis of DNA Methylation in Wheat Carrying Gametocidal Chromosome 3C from Aegilops triuncialis" International Journal of Molecular Sciences 18, no. 8: 1738. https://doi.org/10.3390/ijms18081738
APA StyleWang, D., Zhao, J., Bai, Y., Ao, Y., & Guo, C. (2017). The Variation Analysis of DNA Methylation in Wheat Carrying Gametocidal Chromosome 3C from Aegilops triuncialis. International Journal of Molecular Sciences, 18(8), 1738. https://doi.org/10.3390/ijms18081738