The Future Challenge of Reactive Oxygen Species (ROS) in Hypertension: From Bench to Bed Side

Abstract
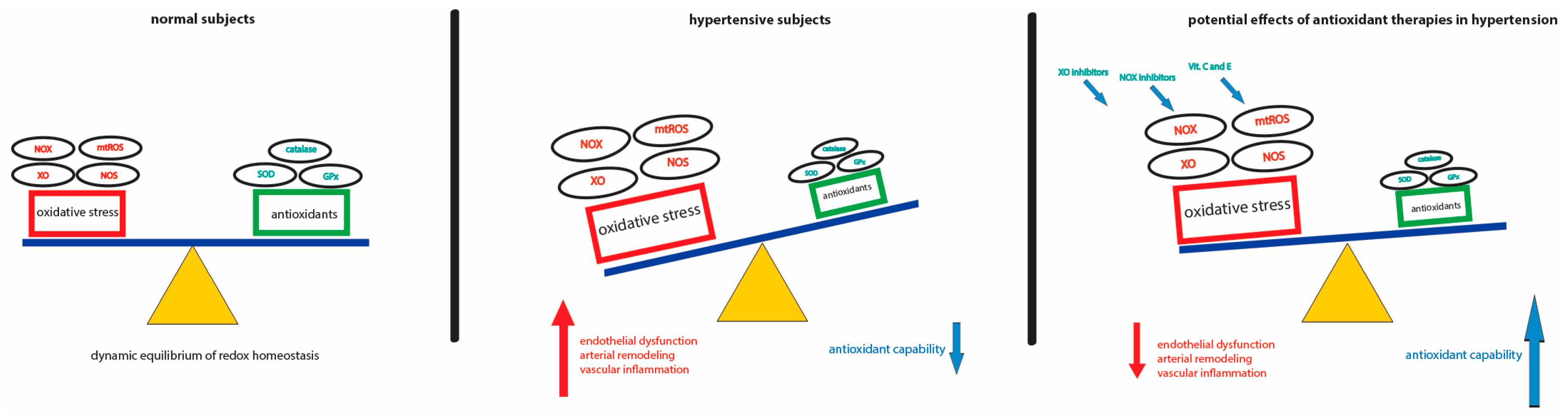
:1. Introduction
2. Redox Signaling
2.1. Xanthine Oxidase
2.2. Nitric Oxide Synthase and Arginase
2.3. NADPH Oxidases
2.4. Mitochondria
3. Oxidative Stress and Hypertension
4. Antioxidant Therapies in Hypertension
4.1. Clinical Studies
4.2. Future Therapeutic Challenges
5. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| GTx | Glutathione peroxidase |
| mtROS | Mitochondrial ROS |
| NOS | Nitric oxide synthases |
| NOX | NADPH oxidase |
| SOD | Superoxide dismutase |
| Vit. C and E | Vitamin C and E |
| XO | Xanthine oxidase |
References
- Sliwa, K.; Stewart, S.; Gersh, B.J. Hypertension: A global perspective. Circulation 2011, 123, 2892–2896. [Google Scholar] [CrossRef] [PubMed]
- Bauer, U.E.; Briss, P.A.; Goodman, R.A.; Bowman, B.A. Prevention of chronic disease in the 21st century: Elimination of the leading preventable causes of premature death and disability in the USA. Lancet 2014, 384, 45–52. [Google Scholar] [CrossRef]
- Montezano, A.C.; Touyz, R.M. Molecular mechanisms of hypertension—Reactive oxygen species and antioxidants: A basic science update for the clinician. Can. J. Cardiol. 2012, 28, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Mulvany, M.J.; Baumbach, G.L.; Aalkjaer, C.; Heagerty, A.M.; Korsgaard, N.; Schiffrin, E.L.; Heistad, D.D. Vascular remodeling. Hypertension 1996, 28, 505–506. [Google Scholar] [PubMed]
- Hayashi, K.; Naiki, T. Adaptation and remodeling of vascular wall; biomechanical response to hypertension. J. Mech. Behav. Biomed. Mater. 2009, 2, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.R.; Witting, P.K.; Drummond, G.R. Redox control of endothelial function and dysfunction: Molecular mechanisms and therapeutic opportunities. Antioxid. Redox Signal. 2008, 10, 1713–1765. [Google Scholar] [CrossRef] [PubMed]
- Schulz, E.; Anter, E.; Keaney, J.F., Jr. Oxidative stress, antioxidants, and endothelial function. Curr. Med. Chem. 2004, 11, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Taniyama, Y.; Griendling, K.K. Reactive oxygen species in the vasculature: Molecular and cellular mechanisms. Hypertension 2003, 42, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P. Radical-free biology of oxidative stress. Am. J. Physiol. Cell Physiol. 2008, 295, C849–C868. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.D.; Ridnour, L.A.; Isenberg, J.S.; Flores-Santana, W.; Switzer, C.H.; Donzelli, S.; Hussain, P.; Vecoli, C.; Paolocci, N.; Ambs, S.; et al. The chemical biology of nitric oxide: Implications in cellular signaling. Free Radic. Biol. Med. 2008, 45, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Redón, J.; Oliva, M.R.; Tormos, C.; Giner, V.; Chaves, J.; Iradi, A.; Sáez, G.T. Antioxidant activities and oxidative stress byproducts in human hypertension. Hypertension 2003, 41, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Sasaki, S.; Nakagawa, K.; Matsuura, H.; Oshima, T.; Chayama, K. Endothelial function and oxidative stress in renovascular hypertension. N. Engl. J. Med. 2002, 346, 1954–1962. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, T.; Wenzel, U.; Hink, U.; Krollner, D.; Skatchkov, M.; Stahl, R.A.; MacHarzina, R.; Bräsen, J.H.; Meinertz, T.; Münzel, T. Increased NAD(P)H oxidase-mediated superoxide production in renovascular hypertension: Evidence for an involvement of protein kinase C. Kidney Int. 1999, 55, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Jung, O.; Schreiber, J.G.; Geiger, H.; Pedrazzini, T.; Busse, R.; Brandes, R.P. gp91phox-containing NADPH oxidase mediates endothelial dysfunction in renovascular hypertension. Circulation 2004, 109, 1795–1801. [Google Scholar] [CrossRef] [PubMed]
- Laursen, J.B.; Rajagopalan, S.; Galis, Z.; Tarpey, M.; Freeman, B.A.; Harrison, D.G. Role of superoxide in angiotensin II-induced but not catecholamine-induced hypertension. Circulation 1997, 95, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Kurz, S.; Münzel, T.; Tarpey, M.; Freeman, B.A.; Griendling, K.K.; Harrison, D.G. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J. Clin. Investig. 1996, 97, 1916–1923. [Google Scholar] [CrossRef] [PubMed]
- Lacy, F.; O’Connor, D.T.; Schmid-Schönbein, G.W. Plasma hydrogen peroxide production in hypertensives and normotensive subjects at genetic risk of hypertension. J. Hypertens. 1998, 16, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Lacy, F.; Kailasam, M.T.; O’Connor, D.T.; Schmid-Schönbein, G.W.; Parmer, R.J. Plasma hydrogen peroxide production in human essential hypertension: Role of heredity, gender, and ethnicity. Hypertension 2000, 36, 878–884. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.Y.; Wong, W.T.; Leung, F.P.; Zhang, Y.; Wang, Y.X.; Lee, H.K.; Ng, C.F.; Chen, Z.Y.; Yao, X.; Au, C.L.; et al. Oxidative stress-dependent cyclooxygenase-2-derived prostaglandin f(2α) impairs endothelial function in renovascular hypertensive rats. Antioxid. Redox Signal. 2012, 16, 363–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungvari, Z.; Csiszar, A.; Kaminski, P.M.; Wolin, M.S.; Koller, A. Chronic high pressure-induced arterial oxidative stress: Involvement of protein kinase C-dependent NAD(P)H oxidase and local renin-angiotensin system. Am. J. Pathol. 2004, 165, 219–226. [Google Scholar] [CrossRef]
- Callera, G.E.; Tostes, R.C.; Yogi, A.; Montezano, A.C.; Touyz, R.M. Endothelin-1-induced oxidative stress in DOCA-salt hypertension involves NADPH-oxidase-independent mechanisms. Clin. Sci. Lond. 2006, 110, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Nguyen Dinh Cat, A.; Montezano, A.C.; Burger, D.; Touyz, R.M. Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid. Redox Signal. 2013, 19, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.I.; Ungvari, Z. Role of mitochondrial oxidative stress in hypertension. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Araujo, M.; Wilcox, C.S. Oxidative stress in hypertension: Role of the kidney. Antioxid. Redox Signal. 2014, 20, 74–101. [Google Scholar] [CrossRef] [PubMed]
- Egea, J.; Fabregat, I.; Frapart, Y.M.; Ghezzi, P.; Görlach, A.; Kietzmann, T.; Kubaichuk, K.; Knaus, U.G.; Lopez, M.G.; Olaso-Gonzalez, G.; et al. European contribution to the study of ROS: A summary of the findings and prospects for the future from the COST action BM1203 (EU-ROS). Redox Biol. 2017, 13, 94–162. [Google Scholar] [CrossRef] [PubMed]
- Mancia, G.; De Backer, G.; Dominiczak, A.; Cifkova, R.; Fagard, R.; Germano, G.; Grassi, G.; Heagerty, A.M.; Kjeldsen, S.E.; Laurent, S.; et al. Management of Arterial Hypertension of the European Society of Hypertension; European Society of Cardiology. 2007 Guidelines for the Management of Arterial Hypertension: The Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J. Hypertens. 2007, 25, 1105–1187. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Schiffrin, E.L. Reactive oxygen species in vascular biology: Implications in hypertension. Histochem. Cell Biol. 2004, 122, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Rodriguez-Iturbe, B. Mechanisms of disease: Oxidative stress and inflammation in the pathogenesis of hypertension. Nat. Clin. Pract. Nephrol. 2006, 2, 582–593. [Google Scholar] [CrossRef] [PubMed]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, H.; Li, H.; Kalyanaraman, B.; Nicolosi, A.C.; Gutterman, D.D. Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circ. Res. 2003, 93, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Matoba, T.; Shimokawa, H.; Nakashima, M.; Hirakawa, Y.; Mukai, Y.; Hirano, K.; Kanaide, H.; Takeshita, A. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J. Clin. Investig. 2000, 106, 1521–1530. [Google Scholar] [CrossRef] [PubMed]
- Nagler, L.G.; Vartanyan, L.S. Subunit structure of bovine milk xanthine oxidase. Biochim. Biophys. Acta 1976, 427, 78–90. [Google Scholar] [CrossRef]
- McCord, J.M. Oxygen-derived free radicals in postischemic tissue injury. N. Engl. J. Med. 1985, 312, 159–163. [Google Scholar] [PubMed]
- Harrison, R. Structure and function of xanthine oxidoreductase: Where are we now? Free Radic. Biol. Med. 2002, 33, 774–797. [Google Scholar] [CrossRef]
- Berry, C.E.; Hare, J.M. Xanthine oxidoreductase and cardiovascular disease: Molecular mechanisms and pathophysiological implications. J. Physiol. 2004, 555, 589–606. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; DeLano, F.A.; Parks, D.A.; Jamshidi, N.; Granger, D.N.; Ishii, H.; Suematsu, M.; Zweifach, B.W.; Schmid-Schönbein, G.W. Xanthine oxidase activity associated with arterial blood pressure in spontaneously hypertensive rats. Proc. Natl. Acad. Sci. USA 1998, 95, 4754–4759. [Google Scholar] [CrossRef] [PubMed]
- Laakso, J.T.; Teräväinen, T.L.; Martelin, E.; Vaskonen, T.; Lapatto, R. Renal xanthine oxidoreductase activity during development of hypertension in spontaneously hypertensive rats. J. Hypertens. 2004, 22, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- Landmesser, U.; Spiekermann, S.; Preuss, C.; Sorrentino, S.; Fischer, D.; Manes, C.; Mueller, M.; Drexler, H. Angiotensin II induces endothelial xanthine oxidase activation: Role for endothelial dysfunction in patients with coronary disease. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Ohara, Y.; Peterson, T.E.; Harrison, D.G. Hypercholesterolemia increases endothelial superoxide anion production. J. Clin. Investig. 1993, 91, 2546–2551. [Google Scholar] [CrossRef] [PubMed]
- White, C.R.; Darley-Usmar, V.; Berrington, W.R.; McAdams, M.; Gore, J.Z.; Thompson, J.A.; Parks, D.A.; Tarpey, M.M.; Freeman, B.A. Circulating plasma xanthine oxidase contributes to vascular dysfunction in hypercholesterolemic rabbits. Proc. Natl. Acad. Sci. USA 1996, 93, 8745–8749. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Sadowski, J.; Guzik, B.; Jopek, A.; Kapelak, B.; Przybylowski, P.; Wierzbicki, K.; Korbut, R.; Harrison, D.G.; Channon, K.M. Coronary artery superoxide production and nox isoform expression in human coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Patetsios, P.; Song, M.; Shutze, W.P.; Pappas, C.; Rodino, W.; Ramirez, J.A.; Panetta, T.F. Identification of uric acid and xanthine oxidase in atherosclerotic plaque. Am. J. Cardiol. 2001, 88, 188–191. [Google Scholar] [CrossRef]
- Riegersperger, M.; Covic, A.; Goldsmith, D. Allopurinol, uric acid, and oxidative stress in cardiorenal disease. Int. Urol. Nephrol. 2011, 43, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Beattie, C.J.; Fulton, R.L.; Higgins, P.; Padmanabhan, S.; McCallum, L.; Walters, M.R.; Dominiczak, A.F.; Touyz, R.M.; Dawson, J. Allopurinol initiation and change in blood pressure in older adults with hypertension. Hypertension 2014, 64, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef] [PubMed]
- Balligand, J.L.; Feron, O.; Dessy, C. eNOS activation by physical forces: From short-term regulation of contraction to chronic remodeling of cardiovascular tissues. Physiol. Rev. 2009, 89, 481–534. [Google Scholar] [CrossRef] [PubMed]
- Fleming, I.; Busse, R. Signal transduction of eNOS activation. Cardiovasc. Res. 1999, 43, 532–541. [Google Scholar] [CrossRef]
- Nathan, C. Inducible nitric oxide synthase: What difference does it make? J. Clin. Investig. 1997, 100, 2417–2423. [Google Scholar] [CrossRef] [PubMed]
- Arstall, M.A.; Sawyer, D.B.; Fukazawa, R.; Kelly, R.A. Cytokine-mediated apoptosis in cardiac myocytes: The role of inducible nitric oxide synthase induction and peroxynitrite generation. Circ. Res. 1999, 85, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Mungrue, I.N.; Gros, R.; You, X.; Pirani, A.; Azad, A.; Csont, T.; Schulz, R.; Butany, J.; Stewart, D.J.; Husain, M. Cardiomyocyte overexpression of iNOS in mice results in peroxynitrite generation, heart block, and sudden death. J. Clin. Investig. 2002, 109, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Heger, J.; Gödecke, A.; Flögel, U.; Merx, M.W.; Molojavyi, A.; Kühn-Velten, W.N.; Schrader, J. Cardiac-specific overexpression of inducible nitric oxide synthase does not result in severe cardiac dysfunction. Circ. Res. 2002, 90, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Flögel, U.; Merx, M.W.; Godecke, A.; Decking, U.K.; Schrader, J. Myoglobin: A scavenger of bioactive NO. Proc. Natl. Acad. Sci. USA 2001, 98, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Gödecke, A.; Molojavyi, A.; Heger, J.; Flögel, U.; Ding, Z.; Jacoby, C.; Schrader, J. Myoglobin protects the heart from inducible nitric-oxide synthase (iNOS)-mediated nitrosative stress. J. Biol. Chem. 2003, 278, 21761–21766. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi, H.; Kubota, T.; Kawamura, N.; Machida, Y.; Feldman, A.M.; Tsutsui, H.; Shimokawa, H.; Takeshita, A. Disruption of inducible nitric oxide synthase improves beta-adrenergic inotropic responsiveness but not the survival of mice with cytokine-induced cardiomyopathy. Circ. Res. 2002, 90, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Meininger, C.J.; Hawker, J.R., Jr.; Haynes, T.E.; Kepka-Lenhart, D.; Mistry, S.K.; Morris, S.M., Jr.; Wu, G. Regulatory role of arginase I and II in nitric oxide, polyamine, and proline syntheses in endothelial cells. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E75–E82. [Google Scholar] [PubMed]
- Odenlund, M.; Holmqvist, B.; Baldetorp, B.; Hellstrand, P.; Nilsson, B.O. Polyamine synthesis inhibition induces S phase cell cycle arrest in vascular smooth muscle cells. Amino Acids 2009, 36, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Orlando, G.F.; Wolf, G.; Engelmann, M. Role of neuronal nitric oxide synthase in the regulation of the neuroendocrine stress response in rodents: Insights from mutant mice. Amino Acids 2008, 35, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Johnson, F.K.; Johnson, R.A.; Peyton, K.J.; Durante, W. Arginase inhibition restores arteriolar endothelial function in Dahl rats with salt-induced hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R1057–R1062. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, S.; Richert, L.; Berthelot, A. Increased arginase activity in aorta of mineralocorticoid-salt hypertensive rats. Clin. Exp. Hypertens. 2000, 22, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Hein, T.W.; Wang, W.; Miller, M.W.; Fossum, T.W.; McDonald, M.M.; Humphrey, J.D.; Kuo, L. Upregulation of vascular arginase in hypertension decreases nitric oxide-mediated dilation of coronary arterioles. Hypertension 2004, 44, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Demougeot, C.; Prigent-Tesssier, A.; Marie, C.; Berthelot, A. Arginase inhibition reduced endothelial dysfunction and blood pressure rising in spontaneously hypertensive rats. J. Hypertens. 2005, 23, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Durante, W.; Liao, L.; Reyna, S.V.; Peyton, K.J.; Schafer, A.I. Physiologic cyclic stretch directs L-arginine transport and metabolism to collagen synthesis in vascular smooth muscle cells. FASEB J. 2000, 14, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Taddei, S.; Virdis, A.; Ghiadoni, L.; Sudano, I.; Salvetti, A. Endothelial dysfunction in hypertension. J. Cardiovasc. Pharmacol. 2001, 38, S11–S14. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed]
- Berka, V.; Wu, G.; Yeh, H.C.; Palmer, G.; Tsai, A.L. Three different oxygen-induced radical species in endothelial nitric oxide synthase oxygenase domain under regulation by l-arginine and tetrahydrobiopterin. J. Biol. Chem. 2004, 279, 32243–32251. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, C.S. Oxidative stress and nitric oxide deficiency in the kidney: A critical link to hypertension? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R913–R935. [Google Scholar] [CrossRef] [PubMed]
- Taddei, S.; Virdis, A.; Mattei, P.; Salvetti, A. Vasodilation to acetylcholine in primary and secondary forms of human hypertension. Hypertension 1993, 21, 929–933. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Sasaki, S.; Nakagawa, K.; Fukuda, Y.; Matsuura, H.; Oshima, T.; Chayama, K. Tetrahydrobiopterin enhances forearm vascular response to acetylcholine in both normotensive and hypertensive individuals. Am. J. Hypertens. 2002, 15, 326–332. [Google Scholar] [CrossRef]
- Heitzer, T.; Krohn, K.; Albers, S.; Meinertz, T. Tetrahydrobiopterin improves endothelium-dependent vasodilation by increasing nitric oxide activity in patients with Type II diabetes mellitus. Diabetologia 2000, 43, 1435–1438. [Google Scholar] [CrossRef] [PubMed]
- Stroes, E.; Kastelein, J.; Cosentino, F.; Erkelens, W.; Wever, R.; Koomans, H.; Lüscher, T.; Rabelink, T. Tetrahydrobiopterin restores endothelial function in hypercholesterolemia. J. Clin. Investig. 1997, 99, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, T.; Brockhoff, C.; Mayer, B.; Warnholtz, A.; Mollnau, H.; Henne, S.; Meinertz, T.; Münzel, T. Tetrahydrobiopterin improves endothelium-dependent vasodilation in chronic smokers: Evidence for a dysfunctional nitric oxide synthase. Circ. Res. 2000, 86, E36–E41. [Google Scholar] [CrossRef] [PubMed]
- Kerr, S.; Brosnan, M.J.; McIntyre, M.; Reid, J.L.; Dominiczak, A.F.; Hamilton, C.A. Superoxide anion production is increased in a model of genetic hypertension: Role of the endothelium. Hypertension 1999, 33, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Witte, K.; August, M.; Brausch, I.; Gödtel-Armbrust, U.; Habermeier, A.; Closs, E.I.; Oelze, M.; Münzel, T.; Förstermann, U. Reversal of endothelial nitric oxide synthase uncoupling and up-regulation of endothelial nitric oxide synthase expression lowers blood pressure in hypertensive rats. J. Am. Coll. Cardiol. 2006, 47, 2536–2544. [Google Scholar] [CrossRef] [PubMed]
- Hink, U.; Li, H.; Mollnau, H.; Oelze, M.; Matheis, E.; Hartmann, M.; Skatchkov, M.; Thaiss, F.; Stahl, R.A.; Warnholtz, A.; et al. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ. Res. 2001, 88, E14–E22. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, M.; Shimokawa, H.; Otsuji, Y.; Yanagihara, N. Pathophysiological relevance of NO signaling in the cardiovascular system: Novel insight from mice lacking all NO synthases. Pharmacol. Ther. 2010, 128, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Hyndman, K.A.; Boesen, E.I.; Elmarakby, A.A.; Brands, M.W.; Huang, P.; Kohan, D.E.; Pollock, D.M.; Pollock, J.S. Renal collecting duct NOS1 maintains fluid-electrolyte homeostasis and blood pressure. Hypertension 2013, 62, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Cowley, A.W., Jr.; Yang, C.; Zheleznova, N.N.; Staruschenko, A.; Kurth, T.; Rein, L.; Kumar, V.; Sadovnikov, K.; Dayton, A.; Hoffman, M.; et al. Evidence of the Importance of Nox4 in Production of Hypertension in Dahl Salt-Sensitive Rats. Hypertension 2016, 67, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.L.; Garvin, J.L.; Carretero, O.A. Role of macula densa nitric oxide and cGMP in the regulation of tubuloglomerular feedback. Kidney Int. 2000, 58, 2053–2060. [Google Scholar] [CrossRef] [PubMed]
- Welch, W.J.; Tojo, A.; Lee, J.U.; Kang, D.G.; Schnackenberg, C.G.; Wilcox, C.S. Nitric oxide synthase in the JGA of the SHR: Expression and role in tubuloglomerular feedback. Am. J. Physiol. 1999, 277, F130–F138. [Google Scholar] [PubMed]
- Liu, R.; Carretero, O.A.; Ren, Y.; Garvin, J.L. Increased intracellular pH at the macula densa activates nNOS during tubuloglomerular feedback. Kidney Int. 2005, 67, 1837–1843. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chandrashekar, K.; Wang, L.; Lai, E.Y.; Wei, J.; Zhang, G.; Wang, S.; Zhang, J.; Juncos, L.A.; Liu, R. Inhibition of Nitric Oxide Synthase 1 Induces Salt-Sensitive Hypertension in Nitric Oxide Synthase 1α Knockout and Wild-Type Mice. Hypertension 2016, 67, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wei, J.; Stec, D.E.; Roman, R.J.; Ge, Y.; Cheng, L.; Liu, E.Y.; Zhang, J.; Hansen, P.B.; Fan, F.; et al. Macula Densa Nitric Oxide Synthase 1β Protects against Salt-Sensitive Hypertension. Am. Soc. Nephrol. 2016, 27, 2346–2356. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Kreuzer, J. Vascular NADPH oxidases: Molecular mechanisms of activation. Cardiovasc. Res. 2005, 65, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Lassegue, B.; Griendling, K.K. NADPH oxidases: Functions and pathologies in the vasculature. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Ago, T.; Kuroda, J.; Kamouchi, M.; Sadoshima, J.; Kitazono, T. Pathophysiological roles of NADPH oxidase/Nox family proteins in the vascular system review and perspective. Circ. J. 2011, 75, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.P.; Di Marco, E.; Okabe, J.; Szyndralewiez, C.; Heitz, F.; Montezano, A.C.; de Haan, J.B.; Koulis, C.; El-Osta, A.; Andrews, K.L.; et al. NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation 2013, 127, 1888–1902. [Google Scholar] [CrossRef] [PubMed]
- Fukui, T.; Ishizaka, N.; Rajagopalan, S.; Laursen, J.B.; Capers, Q., 4th; Taylor, W.R.; Harrison, D.G.; de Leon, H.; Wilcox, J.N.; Griendling, K.K. p22phox mRNA expression and NADPH oxidase activity are increased in aortas from hypertensive rats. Circ. Res. 1997, 80, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Rey, F.E.; Cifuentes, M.E.; Kiarash, A.; Quinn, M.T.; Pagano, P.J. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O2− and systolic blood pressure in mice. Circ. Res. 2001, 89, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Landmesser, U.; Cai, H.; Dikalov, S.; McCann, L.; Hwang, J.; Jo, H.; Holland, S.M.; Harrison, D.G. Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension 2002, 40, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, K.; Yamada, H.; Iwata, K.; Jin, D.; Katsuyama, M.; Matsuki, M.; Takai, S.; Yamanishi, K.; Miyazaki, M.; Matsubara, H.; et al. Nox1 is involved in angiotensin II-mediated hypertension: A study in Nox1-deficient mice. Circulation 2005, 112, 2677–2685. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.; Clempus, R.; Lassègue, B.; Cheng, G.; McCoy, J.; Dikalov, S.; San Martin, A.; Lyle, A.; Weber, D.S.; Weiss, D.; et al. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation 2005, 112, 2668–2676. [Google Scholar] [CrossRef] [PubMed]
- Bendall, J.K.; Rinze, R.; Adlam, D.; Tatham, A.L.; de Bono, J.; Wilson, N.; Volpi, E.; Channon, K.M. Endothelial Nox2 overexpression potentiates vascular oxidative stress and hemodynamic response to angiotensin II: Studies in endothelial-targeted Nox2 transgenic mice. Circ. Res. 2007, 100, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.T.; Tian, X.Y.; Huang, Y. Endothelial dysfunction in diabetes and hypertension: Cross talk in RAS, BMP4, and ROS-dependent COX-2-derived prostanoids. J. Cardiovasc. Pharmacol. 2013, 61, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Hernanz, R.; Briones, A.M.; Salaices, M.; Alonso, M.J. New roles for old pathways? A circuitous relationship between reactive oxygen species and cyclo-oxygenase in hypertension. Clin. Sci. Lond. 2014, 126, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Revelles, S.; Avendaño, M.S.; García-Redondo, A.B.; Alvarez, Y.; Aguado, A.; Pérez-Girón, J.V.; García-Redondo, L.; Esteban, V.; Redondo, J.M.; Alonso, M.J.; et al. Reciprocal relationship between reactive oxygen species and cyclooxygenase-2 and vascular dysfunction in hypertension. Antioxid. Redox Signal. 2013, 18, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.Z.; Majid, D.S. High salt intake delayed angiotensin II-induced hypertension in mice with a genetic variant of NADPH oxidase. Am. J. Hypertens. 2011, 24, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.C.; Ryan, R.P.; Broadway, E.; Skelton, M.M.; Kurth, T.; Cowley, A.W., Jr. Null mutation of the nicotinamide adenine dinucleotide phosphate-oxidase subunit p67phox protects the Dahl-S rat from salt-induced reductions in medullary blood flow and glomerular filtration rate. Hypertension 2015, 65, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Vaziri, N.D. Salt-sensitive hypertension in mitochondrial superoxide dismutase deficiency is associated with intra-renal oxidative stress and inflammation. Clin. Exp. Nephrol. 2014, 18, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Beswick, R.A.; Dorrance, A.M.; Leite, R.; Webb, R.C. NADH/NADPH oxidase and enhanced superoxide production in the mineralocorticoid hypertensive rat. Hypertension 2001, 38, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R. Mitochondria in health and disease: Perspectives on a new mitochondrial biology. Mol. Asp. Med. 2004, 25, 365–451. [Google Scholar] [CrossRef] [PubMed]
- De Cavanagh, E.M.; Toblli, J.E.; Ferder, L.; Piotrkowski, B.; Stella, I.; Fraga, C.G.; Inserra, F. Angiotensin II blockade improves mitochondrial function in spontaneously hypertensive rats. Cell. Mol. Biol. 2005, 51, 573–578. [Google Scholar] [PubMed]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 2008, 102, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Xiang, W.; Potts, J.; Floyd, M.; Sharan, C.; Yang, H.; Ross, J.; Nyanda, A.M.; Guo, Z. Reduction in extracellular superoxide dismutase activity in African-American patients with hypertension. Free Radic. Biol. Med. 2006, 41, 1384–1391. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.; Harrison, D.G.; Dikalov, S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 2010, 107, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Itani, H.A.; Dikalova, A.E.; McMaster, W.G.; Nazarewicz, R.R.; Bikineyeva, A.T.; Harrison, D.G.; Dikalov, S.I. Mitochondrial Cyclophilin D in Vascular Oxidative Stress and Hypertension. Hypertension 2016, 67, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Widder, J.D.; Fraccarollo, D.; Galuppo, P.; Hansen, J.M.; Jones, D.P.; Ertl, G.; Bauersachs, J. Attenuation of angiotensin II-induced vascular dysfunction and hypertension by overexpression of Thioredoxin 2. Hypertension 2009, 54, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wang, Q.; Zhu, J.; Xiao, Q.; Zhang, L. Reactive Oxygen Species: Key Regulators in Vascular Health and Diseases. Br. J. Pharmacol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Nakazono, K.; Watanabe, N.; Matsuno, K.; Sasaki, J.; Sato, T.; Inoue, M. Does superoxide underlie the pathogenesis of hypertension? Proc. Natl. Acad. Sci. USA 1991, 88, 10045–10048. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Ma, X.; Xie, X.; Shen, G.X. Involvement of NADPH oxidase in oxidized LDL-induced upregulation of heat shock factor-1 and plasminogen activator inhibitor-1 in vascular endothelial cells. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E104–E111. [Google Scholar] [CrossRef] [PubMed]
- Gulati, P.; Klöhn, P.C.; Krug, H.; Göttlicher, M.; Markova, B.; Böhmer, F.D.; Herrlich, P. Redox regulation in mammalian signal transduction. IUBMB Life 2001, 52, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Stojiljkovic, M.P.; Lopes, H.F.; Zhang, D.; Morrow, J.D.; Goodfriend, T.L.; Egan, B.M. Increasing plasma fatty acids elevates F2-isoprostanes in humans: Implications for the cardiovascular risk factor cluster. J. Hypertens. 2002, 20, 1215–1221. [Google Scholar] [CrossRef] [PubMed]
- San José, G.; Fortuño, A.; Moreno, M.U.; Robador, P.A.; Bidegain, J.; Varo, N.; Beloqui, O.; Díez, J.; Zalba, G. The angiotensin-converting enzyme insertion/deletion polymorphism is associated with phagocytic NADPH oxidase-dependent superoxide generation: Potential implication in hypertension. Clin. Sci. Lond. 2009, 116, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, T.; Dohi, Y.; Yamashita, S.; Hirowatari, Y.; Fujii, S.; Ohte, N. Serotonin in peripheral blood reflects oxidative stress and plays a crucial role in atherosclerosis: Novel insights toward holistic anti-atherothrombotic strategy. Atherosclerosis 2016, 246, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Yao, G.; Viel, E.; Amiri, F.; Schiffrin, E.L. Angiotensin II and endothelin-1 regulate MAP kinases through different redox-dependent mechanisms in human vascular smooth muscle cells. J. Hypertens. 2004, 22, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Basset, O.; Deffert, C.; Foti, M.; Bedard, K.; Jaquet, V.; Ogier-Denis, E.; Krause, K.H. NADPH oxidase 1 deficiency alters caveolin phosphorylation and angiotensin II-receptor localization in vascular smooth muscle. Antioxid. Redox Signal. 2009, 11, 2371–2384. [Google Scholar] [CrossRef] [PubMed]
- Lai, E.Y.; Solis, G.; Luo, Z.; Carlstrom, M.; Sandberg, K.; Holland, S.; Wellstein, A.; Welch, W.J.; Wilcox, C.S. p47(phox) is required for afferent arteriolar contractile responses to angiotensin II and perfusion pressure in mice. Hypertension 2012, 59, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.; Lee, H.B. Oxidative stress in diabetic nephropathy: Basic and clinical information. Curr. Diabetes Rep. 2001, 1, 282–287. [Google Scholar] [CrossRef]
- Dentelli, P.; Rosso, A.; Zeoli, A.; Gambino, R.; Pegoraro, L.; Pagano, G.; Falcioni, R.; Brizzi, M.F. Oxidative stress-mediated mesangial cell proliferation requires RAC-1/reactive oxygen species production and beta4 integrin expression. J. Biol. Chem. 2007, 282, 26101–26110. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, M.C.; Sanabria, E.; Manriquez, M.C.; Romero, J.C.; Juncos, L.A. Role of endothelin and isoprostanes in slow pressor responses to angiotensin II. Hypertension 2001, 37, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Minuz, P.; Patrignani, P.; Gaino, S.; Degan, M.; Menapace, L.; Tommasoli, R.; Seta, F.; Capone, M.L.; Tacconelli, S.; Palatresi, S.; et al. Increased oxidative stress and platelet activation in patients with hypertension and renovascular disease. Circulation 2002, 106, 2800–2805. [Google Scholar] [CrossRef] [PubMed]
- Rawat, D.K.; Alzoubi, A.; Gupte, R.; Chettimada, S.; Watanabe, M.; Kahn, A.G.; Okada, T.; McMurtry, I.F.; Gupte, S.A. Increased reactive oxygen species, metabolic maladaptation, and autophagy contribute to pulmonary arterial hypertension-induced ventricular hypertrophy and diastolic heart failure. Hypertension 2014, 64, 1266–1274. [Google Scholar] [CrossRef] [PubMed]
- Al Ghouleh, I.; Sahoo, S.; Meijles, D.N.; Amaral, J.H.; de Jesus, D.S.; Sembrat, J.; Rojas, M.; Goncharov, D.A.; Goncharova, E.A.; Pagano, P.J. Endothelial Nox1 Oxidase Assembly in Human Pulmonary Arterial Hypertension; Driver of Gremlin1-Mediated Proliferation. Clin. Sci. Lond. 2017, 131, 2019–2035. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Luo, X.J.; Yang, Z.B.; Zhang, J.J.; Li, T.B.; Zhang, X.J.; Ma, Q.L.; Zhang, G.G.; Hu, C.P.; Peng, J. Inhibition of NOX/VPO1 pathway and inflammatory reaction by trimethoxystilbene in prevention of cardiovascular remodeling in hypoxia-induced pulmonary hypertensive rats. J. Cardiovasc. Pharmacol. 2014, 63, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Roth, M.; Konig, P.; Hofmann, S.; Dony, E.; Goyal, P.; Selbitz, A.C.; Schermuly, R.T.; Ghofrani, H.A.; Kwapiszewska, G.; et al. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ. Res. 2007, 101, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.E.; Hong, K.W.; Jin, H.S.; Oh, B. Association analysis of reactive oxygen species-hypertension genes discovered by literature mining. Genom. Inform. 2012, 10, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Jialal, I.; Grundy, S.M. Preservation of the endogenous antioxidants in low density lipoprotein by ascorbate but not probucol during oxidative modification. J. Clin. Investig. 1991, 87, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.R.; Stocker, R. Molecular action of vitamin E in lipoprotein oxidation: Implications for atherosclerosis. Free Radic. Biol. Med. 2000, 28, 1795–1805. [Google Scholar] [CrossRef]
- Fraga, C.G.; Oteiza, P.I.; Galleano, M. In vitro measurements and interpretation of total antioxidant capacity. Biochim. Biophys. Acta 2014, 1840, 931–934. [Google Scholar] [CrossRef] [PubMed]
- Pinchuk, I.; Shoval, H.; Dotan, Y.; Lichtenberg, D. Evaluation of antioxidants: Scope, limitations and relevance of assays. Chem. Phys. Lipids 2012, 165, 638–647. [Google Scholar] [CrossRef] [PubMed]
- González, J.; Valls, N.; Brito, R.; Rodrigo, R. Essential hypertension and oxidative stress: New insights. World J. Cardiol. 2014, 6, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Simic, D.V.; Mimic-Oka, J.; Pljesa-Ercegovac, M.; Savic-Radojevic, A.; Opacic, M.; Matic, D.; Ivanovic, B.; Simic, T. Byproducts of oxidative protein damage and antioxidant enzyme activities in plasma of patients with different degrees of essential hypertension. J. Hum. Hypertens. 2006, 20, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Mullan, B.A.; Young, I.S.; Fee, H.; McCance, D.R. Ascorbic acid reduces blood pressure and arterial stiffness in type 2 diabetes. Hypertension 2002, 40, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Takac, I.; Schröder, K.; Brandes, R.P. The Nox family of NADPH oxidases: Friend or foe of the vascular system? Curr. Hypertens. Rep. 2012, 14, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Lassègue, B.; Clempus, R.E. Vascular NAD(P)H oxidases: Specific features, expression, and regulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, R277–R297. [Google Scholar] [CrossRef] [PubMed]
- Briones, A.M.; Touyz, R.M. Oxidative stress and hypertension: Current concepts. Curr. Hypertens. Rep. 2010, 12, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, R.; Prat, H.; Passalacqua, W.; Araya, J.; Bächler, J.P. Decrease in oxidative stress through supplementation of vitamins C and E is associated with a reduction in blood pressure in patients with essential hypertension. Clin. Sci. Lond. 2008, 114, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Newberry, S.J. What is the evidence that vitamin C supplements lower blood pressure? Am. J. Clin. Nutr. 2012, 95, 997–998. [Google Scholar] [CrossRef] [PubMed]
- MacIsaac, R.L.; Salatzki, J.; Higgins, P.; Walters, M.R.; Padmanabhan, S.; Dominiczak, A.F.; Touyz, R.M.; Dawson, J. Allopurinol and cardiovascular outcomes in adults with hypertension. Hypertension 2016, 67, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Vivekananthan, D.P.; Penn, M.S.; Sapp, S.K.; Hsu, A.; Topol, E.J. Use of antioxidant vitamins for the prevention of cardiovascular disease: Meta-analysis of randomised trials. Lancet 2003, 361, 2017–2023. [Google Scholar] [CrossRef]
- Montezano, A.C.; Touyz, R.M. Oxidative stress, Noxs, and hypertension: Experimental evidence and clinical controversies. Ann. Med. 2012, 44, S2–S16. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.R.; Lyon, C.J.; Xia, X.; Liu, J.Z.; Tangirala, R.K.; Yin, F.; Boyadjian, R.; Bikineyeva, A.; Praticò, D.; Harrison, D.G.; et al. Age-accelerated atherosclerosis correlates with failure to upregulate antioxidant genes. Circ. Res. 2009, 104, e42–e54. [Google Scholar] [CrossRef] [PubMed]
- Czernichow, S.; Bertrais, S.; Blacher, J.; Galan, P.; Briançon, S.; Favier, A.; Safar, M.; Hercberg, S. Effect of supplementation with antioxidants upon long-term risk of hypertension in the SU.VI.MAX study: Association with plasma antioxidant levels. J. Hypertens. 2005, 23, 2013–2018. [Google Scholar] [CrossRef] [PubMed]
- Sesso, H.D.; Buring, J.E.; Christen, W.G.; Kurth, T.; Belanger, C.; MacFadyen, J.; Bubes, V.; Manson, J.E.; Glynn, R.J.; Gaziano, J.M. Vitamins E and C in the prevention of cardiovascular disease in men: The Physicians’ Health Study II randomized controlled trial. JAMA 2008, 300, 2123–2133. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.R.; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.A.; Appel, L.J.; Guallar, E. Meta-analysis: High-dosage vitamin E supplementation may increase all-cause mortality. Ann. Intern. Med. 2005, 142, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Drummond, G.R.; Selemidis, S.; Griendling, K.K.; Sobey, C.G. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Wind, S.; Beuerlein, K.; Eucker, T.; Müller, H.; Scheurer, P.; Armitage, M.E.; Ho, H.; Schmidt, H.H.; Wingler, K. Comparative pharmacology of chemically distinct NADPH oxidase inhibitors. Br. J. Pharmacol. 2010, 161, 885–898. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Wang, H.D.; McNeill, J.R. Role of oxidative stress and nitric oxide in regulation of spontaneous tone in aorta of DOCA-salt hypertensive rats. Br. J. Pharmacol. 2004, 141, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Jaquet, V.; Scapozza, L.; Clark, R.A.; Krause, K.H.; Lambeth, J.D. Small-molecule NOX inhibitors: ROS-generating NADPH oxidases as therapeutic targets. Antioxid. Redox Signal. 2009, 11, 2535–2552. [Google Scholar] [CrossRef] [PubMed]
- Spychalowicz, A.; Wilk, G.; Śliwa, T.; Ludew, D.; Guzik, T.J. Novel therapeutic approaches in limiting oxidative stress and inflammation. Curr. Pharm. Biotechnol. 2012, 13, 2456–2466. [Google Scholar] [CrossRef] [PubMed]
- Streeter, J.; Thiel, W.; Brieger, K.; Miller, F.J., Jr. Opportunity nox: The future of NADPH oxidases as therapeutic targets in cardiovascular disease. Cardiovasc. Ther. 2013, 31, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Gianni, D.; Taulet, N.; Zhang, H.; DerMardirossian, C.; Kister, J.; Martinez, L.; Roush, W.R.; Brown, S.J.; Bokoch, G.M.; Rosen, H. A novel and specific NADPH oxidase-1 (Nox1) small-molecule inhibitor blocks the formation of functional invadopodia in human colon cancer cells. ACS Chem. Biol. 2010, 5, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Neupane, G.P.; Lee, E.S.; Jeong, B.S.; Park, B.C.; Thapa, P. NADPH oxidase inhibitors: A patent review. Expert Opin. Ther. Pat. 2011, 21, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Altenhöfer, S.; Radermacher, K.A.; Kleikers, P.W.; Wingler, K.; Schmidt, H.H. Evolution of NADPH Oxidase Inhibitors: Selectivity and Mechanisms for Target Engagement. Antioxid. Redox Signal. 2015, 23, 406–427. [Google Scholar] [CrossRef] [PubMed]
- Ranayhossaini, D.J.; Rodriguez, A.I.; Sahoo, S.; Chen, B.B.; Mallampalli, R.K.; Kelley, E.E.; Csanyi, G.; Gladwin, M.T.; Romero, G.; Pagano, P.J. Selective recapitulation of conserved and nonconserved regions of putative NOXA1 protein activation domain confers isoform-specific inhibition of Nox1 oxidase and attenuation of endothelial cell migration. J. Biol. Chem. 2013, 288, 36437–36450. [Google Scholar] [CrossRef] [PubMed]
- Somanna, N.K.; Valente, A.J.; Krenz, M.; Fay, W.P.; Delafontaine, P.; Chandrasekar, B. The Nox1/4 Dual Inhibitor GKT137831 or Nox4 Knockdown Inhibits Angiotensin-II-Induced Adult Mouse Cardiac Fibroblast Proliferation and Migration. AT1 Physically Associates With Nox4. J. Cell. Physiol. 2016, 231, 1130–1141. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.P.; Jha, J.C.; Kennedy, K.; van Bommel, E.; Chew, P.; Szyndralewiez, C.; Touyz, R.M.; Schmidt, H.H.; Cooper, M.E.; Jandeleit-Dahm, K.A. Combined NOX1/4 inhibition with GKT137831 in mice provides dose-dependent reno- and atheroprotection even in established micro- and macrovascular disease. Diabetologia 2017, 60, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Marcos, M.A.; Blázquez-Medela, A.M.; Gamella-Pozuelo, L.; Recio-Rodriguez, J.I.; García-Ortiz, L.; Martínez-Salgado, C. Serum Superoxide Dismutase Is Associated with Vascular Structure and Function in Hypertensive and Diabetic Patients. Oxid. Med. Cell. Longev. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Togliatto, G.; Trombetta, A.; Dentelli, P.; Baragli, A.; Rosso, A.; Granata, R.; Ghigo, D.; Pegoraro, L.; Ghigo, E.; Brizzi, M.F. Unacylated ghrelin rescues endothelial progenitor cell function in individuals with type 2 diabetes. Diabetes 2010, 59, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Togliatto, G.; Trombetta, A.; Dentelli, P.; Cotogni, P.; Rosso, A.; Tschöp, M.H.; Granata, R.; Ghigo, E.; Brizzi, M.F. Unacylated ghrelin promotes skeletal muscle regeneration following hindlimb ischemia via SOD-2-mediated miR-221/222 expression. J. Am. Heart Assoc. 2013, 2, e000376. [Google Scholar] [CrossRef] [PubMed]
- Togliatto, G.; Trombetta, A.; Dentelli, P.; Gallo, S.; Rosso, A.; Cotogni, P.; Granata, R.; Falcioni, R.; Delale, T.; Ghigo, E.; et al. Unacylated ghrelin induces oxidative stress resistance in a glucose intolerance and peripheral artery disease mouse model by restoring endothelial cell miR-126 expression. Diabetes 2015, 64, 1370–1382. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Biomarkers | In Vitro/Pre-Clinical/Clinical Sudies | Results Obtained | Reference |
|---|---|---|---|
| Allopurinol | clinical | Lower rates of stroke and cardiac events | [145] |
| diphenyliodonium (DPI) | in vitro | Abolished NADPH oxidase-mediated ROS formation, but also inhibited other flavo-enzymes such as NO synthase (NOS) and xanthine oxidase (XOD) | [153] |
| Apocynin | in vitro | Interfered with ROS detection but varied in efficacy and potency | [153] |
| Apocynin | pre-clinical | Completely abolished the development of spontaneous tone in endothelium-intact aortic rings (DOCA-salt hypertensive rats vs. SHAM-control rats) | [154] |
| Gp91 ds-tat | pre-clinical | Reduces Ang II–induced hypertension | [94,157] |
| ML171 | in vitro | Blocks ROS-dependent formation | [157,158] |
| NoxA1ds | in vitro | Selective inhibitor of Nox1 activity and hypoxia-induced human pulmonary artery endothelial cell O2− production | [161] |
| GKT137831 | in vitro | Therapeutic potential in chronic hypertension-induced adverse cardiac remodeling | [162] |
| GKT137831 | pre-clinical | Athero- and renoprotection in micro- and macrovascular complications | [163] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Togliatto, G.; Lombardo, G.; Brizzi, M.F. The Future Challenge of Reactive Oxygen Species (ROS) in Hypertension: From Bench to Bed Side. Int. J. Mol. Sci. 2017, 18, 1988. https://doi.org/10.3390/ijms18091988
Togliatto G, Lombardo G, Brizzi MF. The Future Challenge of Reactive Oxygen Species (ROS) in Hypertension: From Bench to Bed Side. International Journal of Molecular Sciences. 2017; 18(9):1988. https://doi.org/10.3390/ijms18091988
Chicago/Turabian StyleTogliatto, Gabriele, Giusy Lombardo, and Maria Felice Brizzi. 2017. "The Future Challenge of Reactive Oxygen Species (ROS) in Hypertension: From Bench to Bed Side" International Journal of Molecular Sciences 18, no. 9: 1988. https://doi.org/10.3390/ijms18091988
APA StyleTogliatto, G., Lombardo, G., & Brizzi, M. F. (2017). The Future Challenge of Reactive Oxygen Species (ROS) in Hypertension: From Bench to Bed Side. International Journal of Molecular Sciences, 18(9), 1988. https://doi.org/10.3390/ijms18091988