Mast Cells: Key Contributors to Cardiac Fibrosis


Abstract
:1. Introduction
2. Studies Associating Mast Cells with Cardiac Fibrosis
3. Evidence for the Causal Involvement of Mast Cells in Cardiac Fibrosis
3.1. Studies with Mast Cell Stabilizers
3.2. Studies with Mast Cell-Deficient Rodents
3.2.1. Types of Mast Cell-Deficient Rodents
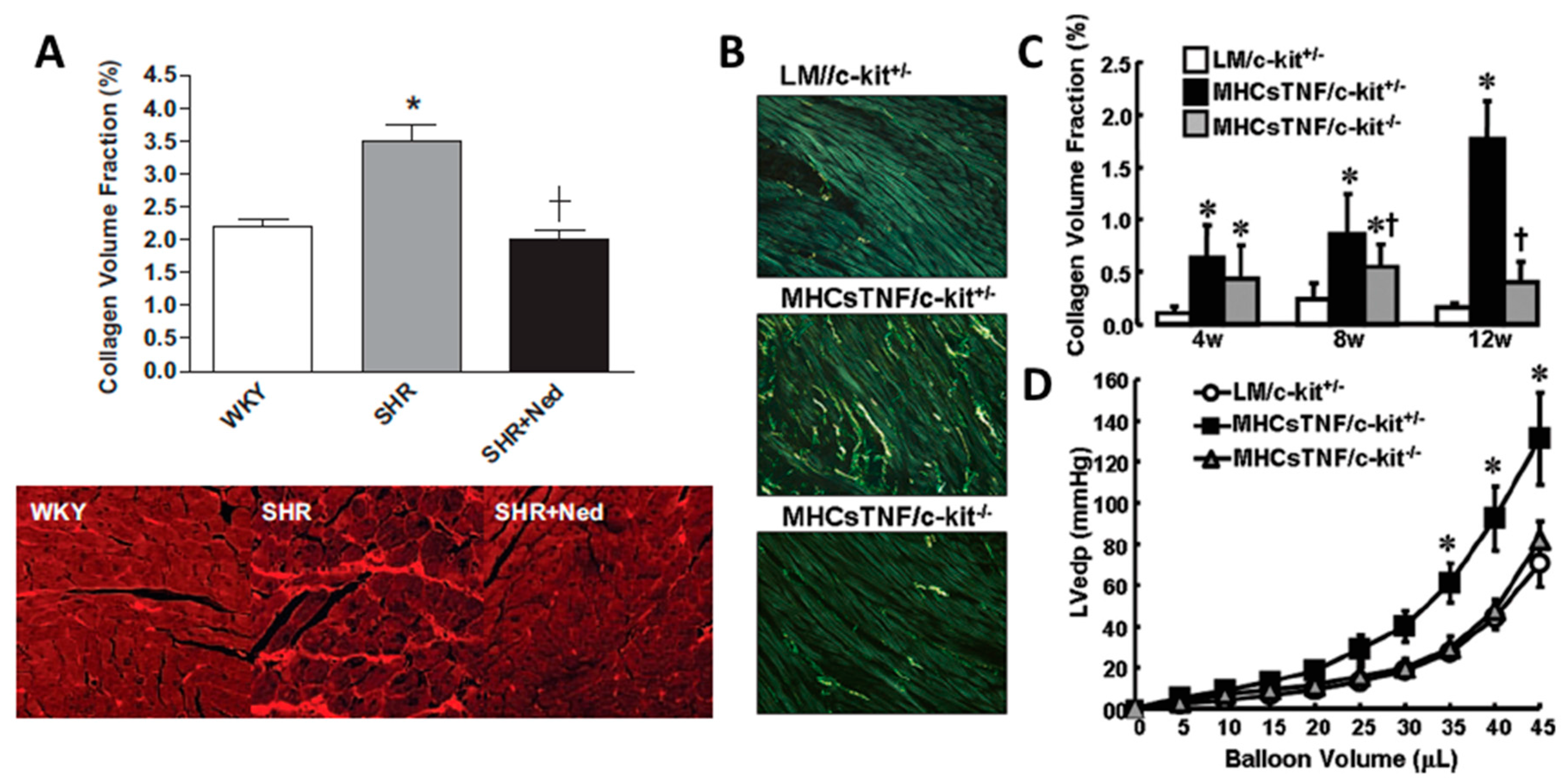
3.2.2. Pro-Fibrotic Role for Mast Cells
3.2.3. Anti-Fibrotic Role of Mast Cells
4. MC products
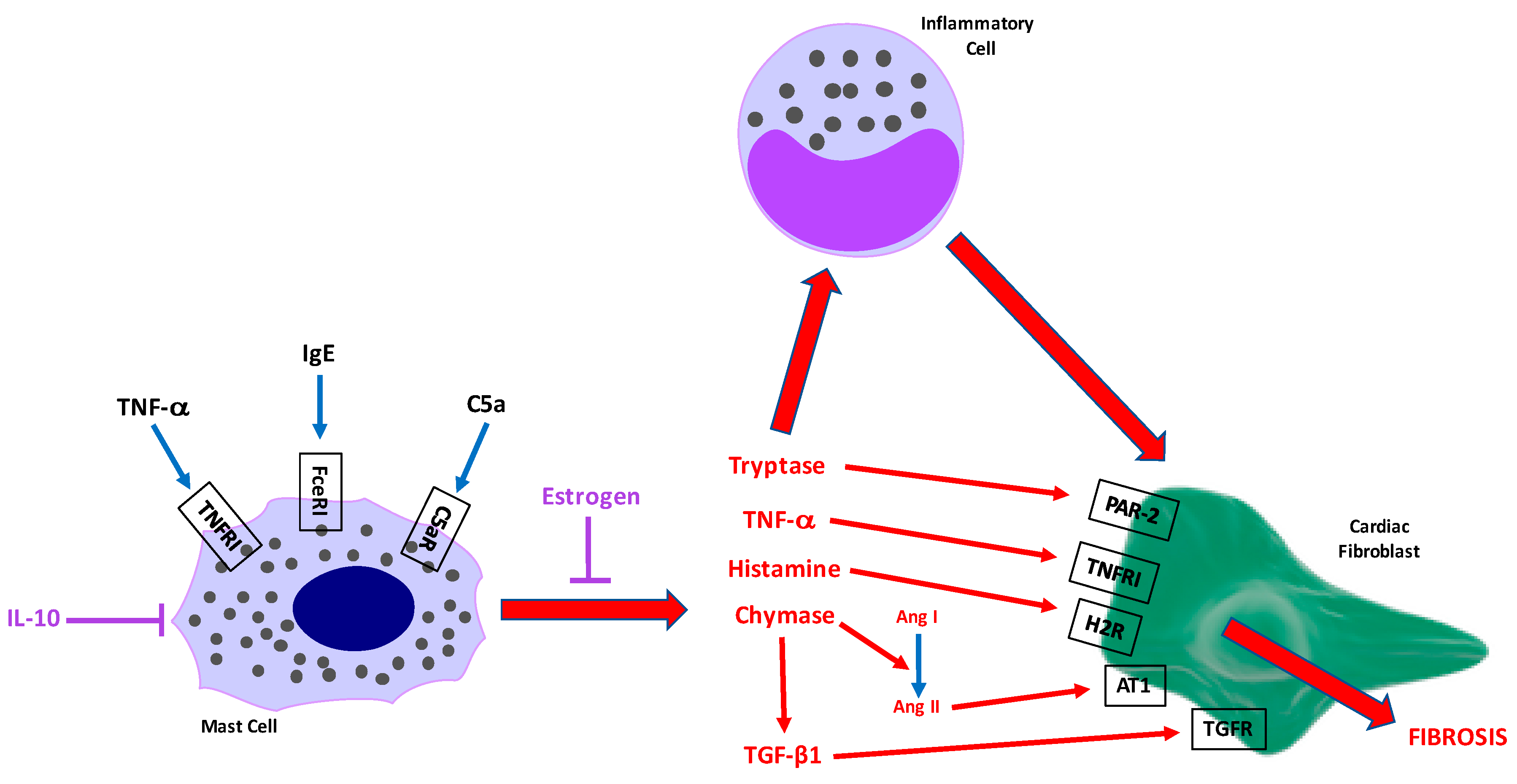
4.1. Proteases
4.1.1. Chymase
4.1.2. Tryptase
4.2. Other Mast Cell Products
4.2.1. Histamine
4.2.2. Components of the Renin Angiotensin System
4.2.3. TNF-α
4.2.4. TGF-β
4.2.5. Matrix Metalloproteinases
5. Important Questions
5.1. What Activates Cardiac Mast Cells?
5.1.1. Immunoglobulin E
5.1.2. TNF-α
5.1.3. Complement 5a
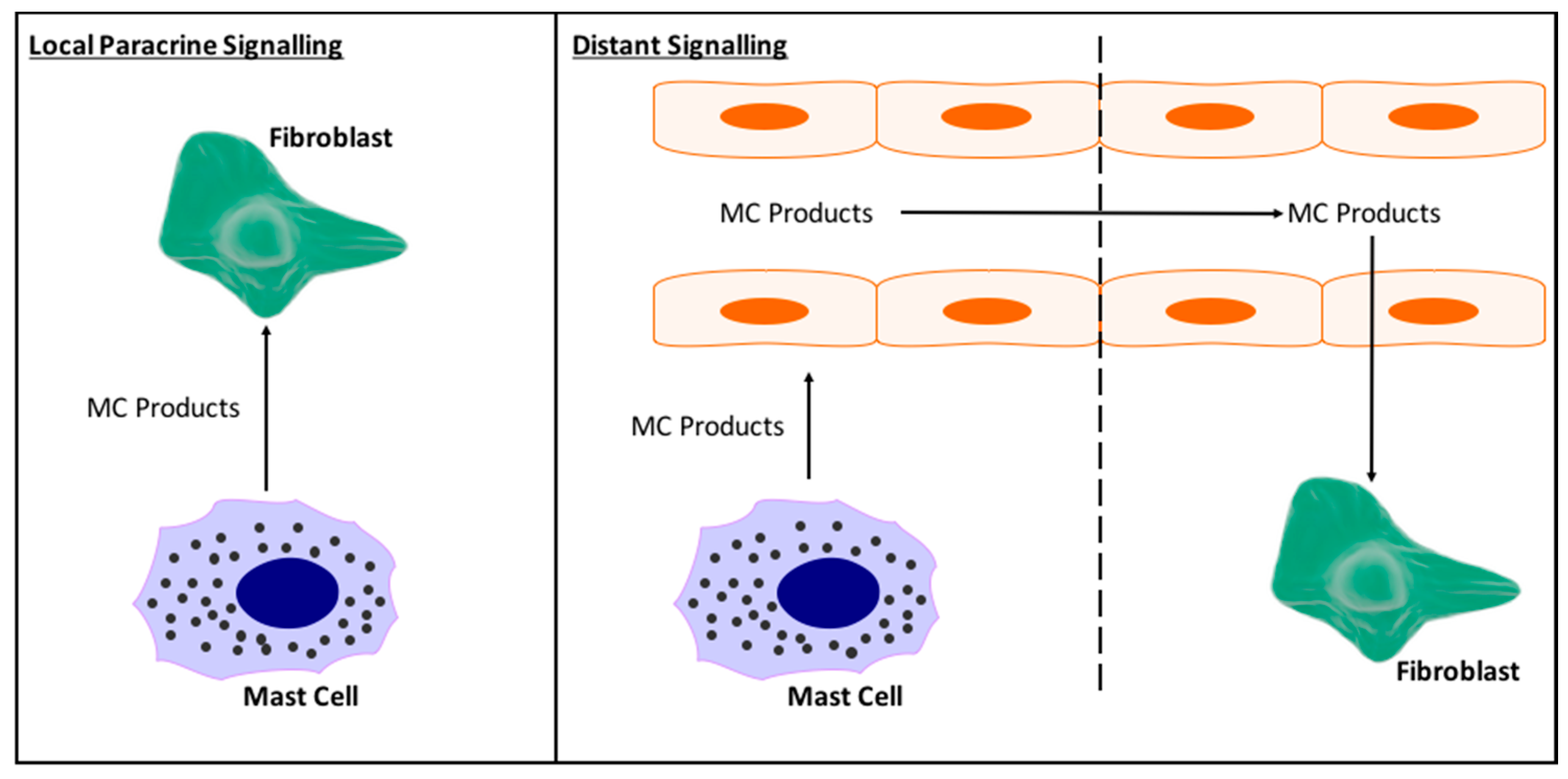
5.2. What Are the Specific Mechanisms by Which Mast Cells Cause Cardiac Fibrosis?
5.3. What Is the Cardiac Mast Cell Phenotype and Are There Gender Differences?
6. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| MC | Mast cell |
| MI | Myocardial Infarction |
| RV | Right Ventricle |
| LV | Left Ventricle |
| SHR | Spontaneously Hypertensive Rat |
| ECM | Extracellular Matrix |
| Sld | Steel Dickie |
| ACE | Angiotensin Converting Enzyme |
| PAR-2 | Protease Activated Receptor 2 |
| H1R | Histamine Receptor 1 |
| H2R | Histamine Receptor 2 |
| H3R | Histamine Receptor 3 |
| H4R | Histamine Receptor 4 |
| RAS | Renin Angiotensin System |
| HMC-1 | Human Mast Cell Line 1 |
| MMP | Matrix Metalloproteinase |
| TNFRI | Tumor Necrosis Factor Receptor I |
| TNFRII | Tumor Necrosis Factor Receptor II |
| C5a | Complement Factor 5a |
| Ovx | Ovariectomised |
References
- Levick, S.P.; Melendez, G.C.; Plante, E.; McLarty, J.L.; Brower, G.L.; Janicki, J.S. Cardiac mast cells: The centrepiece in adverse myocardial remodelling. Cardiovasc. Res. 2011, 89, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Engels, W.; Reiters, P.H.; Daemen, M.J.; Smits, J.F.; van der Vusse, G.J. Transmural changes in mast cell density in rat heart after infarct induction in vivo. J. Pathol. 1995, 177, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G.; Perrard, J.L.; Mendoza, L.H.; Burns, A.R.; Lindsey, M.L.; Ballantyne, C.M.; Michael, L.H.; Smith, C.W.; Entman, M.L. Stem cell factor induction is associated with mast cell accumulation after canine myocardial ischemia and reperfusion. Circulation 1998, 98, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, P.; Ren, G.; Nagar, H.; Kraemer, D.; Mendoza, L.; Michael, L.H.; Caughey, G.H.; Entman, M.L.; Frangogiannis, N.G. Mast cell tryptase may modulate endothelial cell phenotype in healing myocardial infarcts. J. Pathol. 2005, 205, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Panizo, A.; Mindán, F.J.; Galindo, M.F.; Cenarruzabeitia, E.; Hernández, M.; Díez, J. Are mast cells involved in hypertensive heart disease? J. Hypertens. 1995, 13, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Olivetti, G.; Lagrasta, C.; Ricci, R.; Sonnenblick, E.H.; Capasso, J.M.; Anversa, P. Long-term pressure-induced cardiac hypertrophy: Capillary and mast cell proliferation. AJP-Heart Circ. Phys. 1989, 257, H1766–H1772. [Google Scholar] [CrossRef] [PubMed]
- Shiota, N.; Rysä, J.; Kovanen, P.T.; Ruskoaho, H.; Kokkonen, J.O.; Lindstedt, K.A. A role for cardiac mast cells in the pathogenesis of hypertensive heart disease. J. Hypertens. 2003, 21, 1823–1825. [Google Scholar] [CrossRef]
- Li, Q.Y.; Raza-Ahmad, A.; MacAulay, M.A.; Lalonde, L.D.; Rowden, G.; Trethewey, E.; Dean, S. The relationship of mast cells and their secreted products to the volume of fibrosis in posttransplant hearts. Transplantation 1992, 53, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Estensen, R.D. Eosinophilic myocarditis: A role for mast cells? Arch. Pathol. Lab. Med. 1984, 108, 358–359. [Google Scholar] [PubMed]
- Brower, G.L.; Chancey, A.L.; Thanigaraj, S.; Matsubara, B.B.; Janicki, J.S. Cause and effect relationship between myocardial mast cell number and matrix metalloproteinase activity. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H518–H525. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.A.; Wei, C.C.; Brower, G.L.; Rynders, P.E.; Hankes, G.H.; Dillon, A.R.; Lucchesi, P.A.; Janicki, J.S.; Dell’Italia, L.J. Cardiac mast cell- and chymase-mediated matrix metalloproteinase activity and left ventricular remodeling in mitral regurgitation in the dog. J. Mol. Cell. Cardiol. 2003, 35, 311–319. [Google Scholar] [CrossRef]
- Batlle, M.; Roig, E.; Perez-Villa, F.; Lario, S.; Cejudo-Martin, P.; Garcia-Pras, E.; Ortiz, J.; Roque, M.; Orus, J.; Rigol, M.; et al. Increased expression of the renin-angiotensin system and mast cell density but not of angiotensin-converting enzyme II in late stages of human heart failure. J. Heart Lung Transplant. 2006, 25, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Fernex, M.; Sternby, N.H. Mast cells and coronary heart disease. Relationship between number of mast cells in the myocardium, severity of coronary atherosclerosis and myocardial infarction in an autopsy series of 672 cases. Acta Pathol. Microbiol. Scand. 1964, 62, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Turlington, B.S.; Edwards, W.D. Quantitation of mast cells in 100 normal and 92 diseased human hearts. Implications for interpretation of endomyocardial biopsy specimens. Am. J. Cardiovasc. Pathol. 1988, 2, 151–157. [Google Scholar] [PubMed]
- Pires, J.G.; Milanez, M.C.; Pereira, F.E. Histamine levels in tissues of Trypanosoma cruzi-infected mice. Agents Actions Suppl. 1992, 36, 96–98. [Google Scholar] [PubMed]
- Postan, M.; Correa, R.; Ferrans, V.J.; Tarleton, R.L. In vitro culture of cardiac mast cells from mice experimentally infected with Trypanosoma cruzi. Int. Arch. Allergy Immunol. 1994, 105, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Fairweather, D.; Frisancho-Kiss, S.; Gatewood, S.; Njoku, D.; Steele, R.; Barrett, M.; Rose, N.R. Mast cells and innate cytokines are associated with susceptibility to autoimmune heart disease following coxsackievirus B3 infection. Autoimmunity 2004, 37, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Kitaura-Inenaga, K.; Hara, M.; Higuchi, K.; Yamamoto, K.; Yamaki, A.; Ono, K.; Nakano, A.; Kinoshita, M.; Sasayama, S.; Matsumori, A. Gene expression of cardiac mast cell chymase and tryptase in a murine model of heart failure caused by viral myocarditis. Circ. J. 2003, 67, 881–884. [Google Scholar] [CrossRef] [PubMed]
- Palaniyandi, S.S.; Watanabe, K.; Ma, M.; Tachikawa, H.; Kodama, M.; Aizawa, Y. Involvement of mast cells in the development of fibrosis in rats with postmyocarditis dilated cardiomyopathy. Biol. Pharm. Bull. 2005, 28, 2128–2132. [Google Scholar] [CrossRef]
- Helske, S.; Syväranta, S.; Kupari, M.; Lappalainen, J.; Laine, M.; Lommi, J.; Turto, H.; Mayranpaa, M.; Werkkala, K.; Kovanen, P.T.; et al. Possible role for mast cell-derived cathepsin G in the adverse remodelling of stenotic aortic valves. Eur. Heart J. 2006, 27, 1495–1504. [Google Scholar] [CrossRef] [PubMed]
- Luitel, H.; Sydykov, A.; Schymura, Y.; Mamazhakypov, A.; Janssen, W.; Pradhan, K.; Wietelmann, A.; Kosanovic, D.; Dahal, B.K.; Weissmann, N.; et al. Pressure overload leads to an increased accumulation and activity of mast cells in the right ventricle. Physiol. Rep. 2017, 5, e13146. [Google Scholar] [CrossRef] [PubMed]
- Mina, Y.; Rinkevich-Shop, S.; Konen, E.; Goitein, O.; Kushnir, T.; Epstein, F.H.; Feinberg, M.S.; Leor, J.; Landa-Rouben, N. Mast cell inhibition attenuates myocardial damage, adverse remodeling, and dysfunction during fulminant myocarditis in the rat. J. Cardiovasc. Pharmacol. Ther. 2013, 18, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Levick, S.P.; McLarty, J.L.; Murray, D.B.; Freeman, R.M.; Carver, W.E.; Brower, G.L. Cardiac mast cells mediate left ventricular fibrosis in the hypertensive rat heart. Hypertension 2009, 53, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Palaniyandi, S.S.; Watanabe, K.; Ma, M.; Tachikawa, H.; Kodama, M.; Aizawa, Y. Inhibition of mast cells by interleukin-10 gene transfer contributes to protection against acute myocarditis in rats. Eur. J. Immunol. 2004, 34, 3508–3515. [Google Scholar] [CrossRef] [PubMed]
- Kanellakis, P.; Ditiatkovski, M.; Kostolias, G.; Bobik, A. A pro-fibrotic role for interleukin-4 in cardiac pressure overload. Cardiovasc. Res. 2012, 95, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.H.; Akazawa, H.; Tamagawa, M.; Ito, K.; Yasuda, N.; Kudo, Y.; Yamamoto, R.; Ozasa, Y.; Fujimoto, M.; Wang, P.; et al. Cardiac mast cells cause atrial fibrillation through PDGF-A-mediated fibrosis in pressure-overloaded mouse hearts. J. Clin. Investig. 2010, 120, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.G.; Jin, Q.; Fan, M.; Cong, X.L.; Han, S.F.; Gao, H.; Shan, Y. Myocardial remodeling in diabetic cardiomyopathy associated with cardiac mast cell activation. PLoS ONE 2013, 8, e60827. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jubair, S.; Levick, S.P.; Janicki, J.S. The autocrine role of tryptase in pressure overload-induced mast cell activation, chymase release and cardiac fibrosis. IJC Metab. Endocr. 2016, 10, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.; Ono, K.; Hwang, M.W.; Iwasaki, A.; Okada, M.; Nakatani, K.; Sasayama, S.; Matsumori, A. Evidence for a role of mast cells in the evolution to congestive heart failure. J. Exp. Med. 2002, 195, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chancey, A.L.; Tzeng, H.P.; Zhou, Z.; Lavine, K.J.; Gao, F.; Sivasubramanian, N.; Barger, P.M.; Mann, D.L. The development of myocardial fibrosis in transgenic mice with targeted overexpression of tumor necrosis factor requires mast cell-fibroblast interactions. Circulation 2011, 124, 2106–2116. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.; Kennedy, R.H.; Devi, S.; Wang, J.; Joseph, L.; Hauer-Jensen, M. Protective role of mast cells in homocysteine-induced cardiac remodeling. AJP-Heart Circ. Phys. 2005, 288, H2541–H2545. [Google Scholar] [CrossRef] [PubMed]
- Boerma, M.; Wang, J.; Wondergem, J.; Joseph, J.; Qiu, X.; Kennedy, R.H.; Hauer-Jensen, M. Influence of mast cells on structural and functional manifestations of radiation-induced heart disease. Cancer Res. 2005, 65, 3100–3107. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Wada, A.; Tsutamoto, T.; Ohnishi, M.; Isono, T.; Kinoshita, M. Chymase inhibition prevents cardiac fibrosis and improves diastolic dysfunction in the progression of heart failure. Circulation 2003, 107, 2555–2558. [Google Scholar] [CrossRef] [PubMed]
- Kanemitsu, H.; Takai, S.; Tsuneyoshi, H.; Nishina, T.; Yoshikawa, K.; Miyazaki, M.; Ikeda, T.; Komeda, M. Chymase inhibition prevents cardiac fibrosis and dysfunction after myocardial infarction in rats. Hypertens. Res. 2006, 29, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, C.; Hayashi, T.; Kitada, K.; Yamashita, C.; Miyamura, M.; Mori, T.; Ukimura, A.; Ohkita, M.; Jin, D.; Takai, S.; et al. Chymase plays an important role in left ventricular remodeling induced by intermittent hypoxia in mice. Hypertension 2009, 54, 164–171. [Google Scholar] [CrossRef] [PubMed]
- McLarty, J.L.; Melendez, G.C.; Brower, G.L.; Janicki, J.S.; Levick, S.P. Tryptase/Protease-activated receptor 2 interactions induce selective mitogen-activated protein kinase signaling and collagen synthesis by cardiac fibroblasts. Hypertension 2011, 58, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Shen, L.; Li, X.; Luo, T.; Wei, X.; Zhang, J.; Cao, S.; Huang, X.; Fukushima, Y.; Bin, J.; et al. Disruption of histamine H2 receptor slows heart failure progression through reducing myocardial apoptosis and fibrosis. Clin. Sci. 2014, 127, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Geissler, E.N.; McFarland, E.C.; Russell, E.S. Analysis of pleiotropism at the dominant white-spotting (W) locus of the house mouse: A description of ten new W alleles. Genetics 1981, 97, 337–361. [Google Scholar] [PubMed]
- Kitamura, Y.; Go, S.; Hatanaka, K. Decrease of mast cells in W/Wv mice and their increase by bone marrow transplantation. Blood 1978, 52, 447–452. [Google Scholar] [PubMed]
- Lyon, M.F.; Glenister, P.H. A new allele sash (Wsh) at the W-locus and a spontaneous recessive lethal in mice. Genet. Res. 1982, 39, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Niwa, Y.; Kasugai, T.; Ohno, K.; Morimoto, M.; Yamazaki, M.; Dohmae, K.; Nishimune, Y.; Kondo, K.; Kitamura, Y. Anemia and mast cell depletion in mutant rats that are homozygous at “white spotting (Ws)” locus. Blood 1991, 78, 1936–1941. [Google Scholar] [PubMed]
- Kitamura, Y. Heterogeneity of mast cells and phenotypic change between subpopulations. Annu. Rev. Immunol. 1989, 7, 59–76. [Google Scholar] [CrossRef] [PubMed]
- Nocka, K.; Tan, J.C.; Chiu, E.; Chu, T.Y.; Ray, P.; Traktman, P.; Besmer, P. Molecular bases of dominant negative and loss of function mutations at the murine c-kit/white spotting locus: W37, Wv, W41 and W. EMBO J. 1990, 9, 1805–1813. [Google Scholar] [PubMed]
- Reith, A.D.; Rottapel, R.; Giddens, E.; Brady, C.; Forrester, L.; Bernstein, A. W mutant mice with mild or severe developmental defects contain distinct point mutations in the kinase domain of the c-kit receptor. Genes Dev. 1990, 4, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Huizinga, J.D.; Thuneberg, L.; Kluppel, M.; Malysz, J.; Mikkelsen, H.B.; Bernstein, A. W/kit gene required for interstitial cells of Cajal and for intestinal pacemaker activity. Nature 1995, 373, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Waki, N.; Asai, H.; Kitamura, Y. Different repopulation profile between erythroid and nonerythroid progenitor cells in genetically anemic W/Wv mice after bone marrow transplantation. Blood 1989, 74, 1552–1556. [Google Scholar] [PubMed]
- Puddington, L.; Olson, S.; Lefrancois, L. Interactions between stem cell factor and c-Kit are required for intestinal immune system homeostasis. Immunity 1994, 1, 733–739. [Google Scholar] [CrossRef]
- Tsai, M.; Tam, S.Y.; Wedemeyer, J.; Galli, S.J. Mast cells derived from embryonic stem cells: A model system for studying the effects of genetic manipulations on mast cell development, phenotype, and function in vitro and in vivo. Int. J. Hematol. 2002, 75, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Galli, S.J.; Arizono, N.; Murakami, T.; Dvorak, A.M.; Fox, J.G. Development of large numbers of mast cells at sites of idiopathic chronic dermatitis in genetically mast cell-deficient WBB6F1-W/Wv mice. Blood 1987, 69, 1661–1666. [Google Scholar] [PubMed]
- Galli, S.J.; Zsebo, K.M.; Geissler, E.N. The kit ligand, stem cell factor. Adv. Immunol. 1994, 55, 1–96. [Google Scholar] [PubMed]
- Shimada, M.; Kitamura, Y.; Yokoyama, M.; Miyano, Y.; Maeyama, K.; Yamatodani, A.; Takahashi, Y.; Tatsuta, M. Spontaneous stomach ulcer in genetically mast-cell depleted W/Wv mice. Nature 1980, 283, 662–664. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, M.; Tatsuta, M.; Baba, M.; Kitamura, Y. Bile reflux: A possible cause of stomach ulcer in nontreated mutant mice of W/WV genotype. Gastroenterology 1982, 82 Pt 1, 857–863. [Google Scholar] [PubMed]
- Kitamura, Y.; Yokoyama, M.; Matsuda, H.; Shimada, M. Coincidental development of forestomach papilloma and prepyloric ulcer in nontreated mutant mice of W/Wv and SI/SId genotypes. Cancer Res. 1980, 40, 3392–3397. [Google Scholar] [PubMed]
- Nagle, D.L.; Kozak, C.A.; Mano, H.; Chapman, V.M.; Bucan, M. Physical mapping of the Tec and Gabrb1 loci reveals that the Wsh mutation on mouse chromosome 5 is associated with an inversion. Hum. Mol. Genet. 1995, 4, 2073–2079. [Google Scholar] [CrossRef] [PubMed]
- Mallen-St Clair, J.; Pham, C.T.; Villalta, S.A.; Caughey, G.H.; Wolters, P.J. Mast cell dipeptidyl peptidase I mediates survival from sepsis. J. Clin. Investig. 2004, 113, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Wolters, P.J.; Mallen-St Clair, J.; Lewis, C.C.; Villalta, S.A.; Baluk, P.; Erle, D.J.; Caughey, G.H. Tissue-selective mast cell reconstitution and differential lung gene expression in mast cell-deficient Kit(W-sh)/Kit(W-sh) sash mice. Clin. Exp. Allergy 2005, 35, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Tono, T.; Tsujimura, T.; Koshimizu, U.; Kasugai, T.; Adachi, S.; Isozaki, K.; Nishikawa, S.; Morimoto, M.; Nishimune, Y.; Nomura, S.; et al. c-kit Gene was not transcribed in cultured mast cells of mast cell-deficient Wsh/Wsh mice that have a normal number of erythrocytes and a normal c-kit coding region. Blood 1992, 80, 1448–1453. [Google Scholar] [PubMed]
- Grimbaldeston, M.A.; Chen, C.C.; Piliponsky, A.M.; Tsai, M.; Tam, S.Y.; Galli, S.J. Mast cell-deficient W-sash c-kit mutant kitW-sh/W-sh mice as a model for investigating mast cell biology in vivo. Am. J. Pathol. 2005, 167, 835–848. [Google Scholar] [CrossRef]
- Brannan, C.I.; Lyman, S.D.; Williams, D.E.; Eisenman, J.; Anderson, D.M.; Cosman, D.; Bedell, M.A.; Jenkins, N.A.; Copeland, N.G. Steel-Dickie mutation encodes a c-kit ligand lacking transmembrane and cytoplasmic domains. Proc. Natl. Acad. Sci. USA 1991, 88, 4671–4674. [Google Scholar] [CrossRef] [PubMed]
- Copeland, N.G.; Gilbert, D.J.; Cho, B.C.; Donovan, P.J.; Jenkins, N.A.; Cosman, D.; Anderson, D.; Lyman, S.D.; Williams, D.E. Mast cell growth factor maps near the steel locus on mouse chromosome 10 and is deleted in a number of steel alleles. Cell 1990, 63, 175–183. [Google Scholar] [CrossRef]
- Huang, E.; Nocka, K.; Beier, D.R.; Chu, T.Y.; Buck, J.; Lahm, H.W.; Wellner, D.; Leder, P.; Besmer, P. The hematopoietic growth factor KL is encoded by the Sl locus and is the ligand of the c-kit receptor, the gene product of the W locus. Cell 1990, 63, 225–233. [Google Scholar] [CrossRef]
- Zsebo, K.M.; Williams, D.A.; Geissler, E.N.; Broudy, V.C.; Martin, F.H.; Atkins, H.L.; Hsu, R.Y.; Birkett, N.C.; Okino, K.H.; Murdock, D.C.; et al. Stem cell factor is encoded at the Sl locus of the mouse and is the ligand for the c-kit tyrosine kinase receptor. Cell 1990, 63, 213–224. [Google Scholar] [CrossRef]
- Bernstein, S. Steel Dickie. Mouse News Lett. 1960, 23, 33–34. [Google Scholar]
- Shiota, N.; Jin, D.; Takai, S.; Kawamura, T.; Koyama, M.; Nakamura, N.; Miyazaki, M. Chymase is activated in the hamster heart following ventricular fibrosis during the chronic stage of hypertension. FEBS Lett. 1997, 406, 301–304. [Google Scholar] [CrossRef]
- Helske, S.; Lindstedt, K.A.; Laine, M.; Mäyränpää, M.; Werkkala, K.; Lommi, J.; Turto, H.; Kupari, M.; Kovanen, P.T. Induction of local angiotensin II-producing systems in stenotic aortic valves. J. Am. Coll. Cardiol. 2004, 44, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Tanaka, R.; Fukuyama, T.; Aoki, R.; Orito, K.; Yamane, Y. Cardiac remodeling and angiotensin II-forming enzyme activity of the left ventricle in hamsters with chronic pressure overload induced by ascending aortic stenosis. J. Vet. Med. Sci. 2006, 68, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Varagic, J.; Westwood, B.M.; Chappell, M.C.; Ferrario, C.M. Uptake and metabolism of the novel peptide angiotensin-(1-12) by neonatal cardiac myocytes. PLoS ONE 2011, 6, e15759. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Simmons, T.; Varagic, J.; Moniwa, N.; Chappell, M.C.; Ferrario, C.M. Chymase-dependent generation of angiotensin II from angiotensin-(1-12) in human atrial tissue. PLoS ONE 2011, 6, e28501. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Wei, C.C.; Tallaj, J.; Dell’Italia, L.J.; Moniwa, N.; Varagic, J.; Ferrario, C.M. Chymase mediates angiotensin-(1-12) metabolism in normal human hearts. J. Am. Soc. Hypertens. 2013, 7, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Balcells, E.; Meng, Q.C.; Johnson, W.H., Jr.; Oparil, S.; Dell’Italia, L.J. Angiotensin II formation from ACE and chymase in human and animal hearts: Methods and species considerations. Am. J. Physiol. 1997, 273 Pt 2, H1769–H1774. [Google Scholar] [CrossRef]
- Akasu, M.; Urata, H.; Kinoshita, A.; Sasaguri, M.; Ideishi, M.; Arakawa, K. Differences in tissue angiotensin II-forming pathways by species and organs in vitro. Hypertension 1998, 32, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Y.; Zhao, L.Y.; Zheng, Q.S.; Su, J.L.; Guan, H.; Shang, F.J.; Niu, X.L.; He, Y.P.; Lu, X.L. Chymase induces profibrotic response via transforming growth factor-β1/Smad activation in rat cardiac fibroblasts. Mol. Cell. Biochem. 2008, 310, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Gristwood, R.W.; Lincoln, J.C.; Owen, D.A.; Smith, I.R. Histamine release from human right atrium. Br. J. Pharmacol. 1981, 74, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, H.; Minamino, T.; Ogai, A.; Kim, J.; Asakura, M.; Komamura, K.; Sanada, S.; Fujita, M.; Hirata, A.; Wakeno, M.; et al. Blockade of histamine H2 receptors protects the heart against ischemia and reperfusion injury in dogs. J. Mol. Cell. Cardiol. 2006, 40, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Jesmin, S.; Takahashi, Y.; Hatta, E.; Kobayashi, M.; Matsuyama, K.; Kawakami, N.; Sakuma, I.; Gando, S.; Fukui, H.; et al. Histamine H1 and H2 receptor gene and protein levels are differentially expressed in the hearts of rodents and humans. J. Pharmacol. Exp. Ther. 2004, 309, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Powers, M.J.; Peterson, B.A.; Hardwick, J.C. Regulation of parasympathetic neurons by mast cells and histamine in the guinea pig heart. Auton. Neurosci. 2001, 87, 37–45. [Google Scholar] [CrossRef]
- Hardwick, J.C.; Kotarski, A.F.; Powers, M.J. Ionic mechanisms of histamine-induced responses in guinea pig intracardiac neurons. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R241–R250. [Google Scholar] [CrossRef] [PubMed]
- Fuder, H.; Ries, P.; Schwarz, P. Histamine and serotonin released from the rat perfused heart by compound 48/80 or by allergen challenge influence noradrenaline or acetylcholine exocytotic release. Fundam. Clin. Pharmacol. 1994, 8, 477–490. [Google Scholar] [CrossRef] [PubMed]
- Leary, P.J.; Tedford, R.J.; Bluemke, D.A.; Bristow, M.R.; Heckbert, S.R.; Kawut, S.M.; Krieger, E.V.; Lima, J.A.; Masri, C.S.; Ralph, D.D.; et al. Histamine H2 receptor antagonists, left ventricular morphology, and heart failure risk: The MESA study. J. Am. Coll. Cardiol. 2016, 67, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Dehlin, H.M.; Manteufel, E.J.; Monroe, A.L.; Reimer, M.H., Jr.; Levick, S.P. Substance P acting via the neurokinin-1 receptor regulates adverse myocardial remodeling in a rat model of hypertension. Int. J. Cardiol. 2013, 168, 4643–4651. [Google Scholar] [CrossRef] [PubMed]
- Silver, R.B.; Reid, A.C.; Mackins, C.J.; Askwith, T.; Schaefer, U.; Herzlinger, D.; Levi, R. Mast cells: A unique source of renin. Proc. Natl. Acad. Sci. USA 2004, 101, 13607–13612. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.; Ono, K.; Wada, H.; Sasayama, S.; Matsumori, A. Preformed angiotensin II is present in human mast cells. Cardiovasc. Drugs Ther. 2004, 18, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Mackins, C.J.; Kano, S.; Seyedi, N.; Schafer, U.; Reid, A.C.; Machida, T.; Silver, R.B.; Levi, R. Cardiac mast cell-derived renin promotes local angiotensin formation, norepinephrine release, and arrhythmias in ischemia/reperfusion. J. Clin. Investig. 2006, 116, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Levick, S.P.; Gardner, J.D.; Holland, M.; Hauer-Jensen, M.; Janicki, J.S.; Brower, G.L. Protection from adverse myocardial remodeling secondary to chronic volume overload in mast cell deficient rats. J. Mol. Cell. Cardiol. 2008, 45, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G.; Lindsey, M.L.; Michael, L.H.; Youker, K.A.; Bressler, R.B.; Mendoza, L.H.; Spengler, R.N.; Smith, C.W.; Entman, M.L. Resident cardiac mast cells degranulate and release preformed TNF-{α}, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation 1998, 98, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Gilles, S.; Zahler, S.; Welsch, U.; Sommerhoff, C.P.; Becker, B.F. Release of TNF-α during myocardial reperfusion depends on oxidative stress and is prevented by mast cell stabilizers. Cardiovasc. Res. 2003, 60, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.R.; Galli, S.J. Promotion of mouse fibroblast collagen gene expression by mast cells stimulated via the Fc epsilon RI. Role for mast cell-derived transforming growth factor β and tumor necrosis factor alpha. J. Exp. Med. 1994, 180, 2027–2037. [Google Scholar] [CrossRef] [PubMed]
- Fang, K.C.; Wolters, P.J.; Steinhoff, M.; Bidgol, A.; Blount, J.L.; Caughey, G.H. Mast cell expression of gelatinases A and B is regulated by kit ligand and TGF-β. J. Immunol. 1999, 162, 5528–5535. [Google Scholar] [PubMed]
- Iyer, R.P.; Patterson, N.L.; Fields, G.B.; Lindsey, M.L. The history of matrix metalloproteinases: Milestones, myths, and misperceptions. Am. J. Physiol. Heart Circ. Phys. 2012, 303, H919–H930. [Google Scholar] [CrossRef] [PubMed]
- Chancey, A.L.; Brower, G.L.; Janicki, J.S. Cardiac mast cell-mediated activation of gelatinase and alteration of ventricular diastolic function. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H2152–H2158. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, A.; Mustin, D.; Forman, M.F.; Brower, G.L.; Janicki, J.S.; Carver, W. Effects of mast cells on the behavior of isolated heart fibroblasts: Modulation of collagen remodeling and gene expression. J. Cell. Physiol. 2002, 191, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Oyamada, S.; Bianchi, C.; Takai, S.; Chu, L.M.; Sellke, F.W. Chymase inhibition reduces infarction and matrix metalloproteinase-9 activation and attenuates inflammation and fibrosis after acute myocardial ischemia/reperfusion. J. Pharmacol. Exp. Ther. 2011, 339, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Patella, V.; Marino, I.; Lamparter, B.; Arbustini, E.; Adt, M.; Marone, G. Human heart mast cells. Isolation, purification, ultrastructure, and immunologic characterization. J. Immunol. 1995, 154, 2855–2865. [Google Scholar] [PubMed]
- Patella, V.; de Crescenzo, G.; Ciccarelli, A.; Marinò, I.; Adt, M.; Marone, G. Human heart mast cells: A definitive case of mast cell heterogeneity. Int. Arch. Allergy Immunol. 1995, 106, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Fureder, W.; Agis, H.; Willheim, M.; Bankl, H.C.; Maier, U.; Kishi, K.; Muller, M.R.; Czerwenka, K.; Radaszkiewicz, T.; Butterfield, J.H.; et al. Differential expression of complement receptors on human basophils and mast cells. Evidence for mast cell heterogeneity and CD88/C5aR expression on skin mast cells. J. Immunol. 1995, 155, 3152–3160. [Google Scholar] [PubMed]
- Ito, B.R.; Engler, R.L.; del Balzo, U. Role of cardiac mast cells in complement C5a-induced myocardial ischemia. Am. J. Physiol. 1993, 264 Pt 2, H1346–H1354. [Google Scholar] [CrossRef]
- Zhang, J.; Alcaide, P.; Liu, L.; Sun, J.; He, A.; Luscinskas, F.W.; Shi, G.P. Regulation of endothelial cell adhesion molecule expression by mast cells, macrophages, and neutrophils. PLoS ONE 2011, 6, e14525. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.; Matsumori, A.; Ono, K.; Kido, H.; Hwang, M.W.; Miyamoto, T.; Iwasaki, A.; Okada, M.; Nakatani, K.; Sasayama, S. Mast cells cause apoptosis of cardiomyocytes and proliferation of other intramyocardial cells in vitro. Circulation 1999, 100, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jubair, S.; Janicki, J.S. Estrogen inhibits mast cell chymase release to prevent pressure overload-induced adverse cardiac remodeling. Hypertension 2015, 65, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Marone, G.; Triggiani, M.; Cirillo, R.; Giacummo, A.; Hammarstrom, S.; Condorelli, M. IgE-mediated activation of human heart in vitro. Agents Actions 1986, 18, 194–196. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Model/Pathology | Outcome | Heart Chamber | References |
|---|---|---|---|---|
| Human | Fibrosis | ↑ MC | LV | [13,14] |
| Rat | Pulmonary hypertension | ↑ MC | RV | [6] |
| Human | Transplantation | MC number correlated with fibrosis | LV | [8] |
| Mouse | Myocarditis | ↑ histamine, ↑ MC correlated with fibrosis | LV | [15,19] |
| Rat | Hypertension | ↑ MC correlated with fibrosis | LV | [5] |
| Mouse | Pulmonary hypertension | ↑ MC | RV | [21] |
| Species | Intervention | Pathology | Outcome | Heart Chamber | Reference |
|---|---|---|---|---|---|
| MC Stabilizers | |||||
| Rat | Cromolyn | Myocarditis | ↓ fibrosis | LV | [19,22] |
| Rat | Nedocromil | Hypertension | ↓ fibrosis | LV | [23] |
| Mouse | Cromolyn | Transaortic constriction | ↓ fibrosis | LV | [25] |
| Mouse | Cromolyn | Transaortic constriction | ↓ fibrosis | Atria | [26] |
| Mouse | Nedocromil | STZ-induced diabetes | ↓ fibrosis | LV | [27] |
| Rat | Nedocromil | Transaortic constriction | ↓ fibrosis | LV | [28] |
| MC-deficient Rodents | |||||
| Mouse | KitW/Wv | Abdominal aortic banding | ↓ perivascular fibrosis | LV | [29] |
| Mouse | KitW/Wv | Transaortic constriction | ↓ fibrosis | Atria | [26] |
| Mouse | KitW/W-sh | TNF-α overexpression | ↓ fibrosis, ↓ diastolic dysfunction | LV | [30] |
| Rat | Ws/Ws | Hyperhomocysteinemia | ↑ fibrosis | LV | [31] |
| Rat | Ws/Ws | Radiation | ↑ fibrosis | LV | [32] |
| Targeting Proteases | |||||
| Canine | Chymase inhibitor (SUNC8257) | Pacing-induced heart failure | ↓ collagen I and III mRNA, ↓ fibrosis | LV | [33] |
| Rat | Chymase inhibitor (NK3201) | Myocardial infarction | ↓ collagen I and III mRNA, ↓ fibrosis, ↓ E/A | LV | [34] |
| Mouse | Chymase inhibitor (NK3201) | Intermittent hypoxia | ↓ perivascular fibrosis | LV | [35] |
| Rat | PAR-2 antagonist (FSLLRY, tryptase) | Hypertension | ↓ fibrosis | LV | [36] |
| Mouse | H2R−/− | Transaortic constriction | ↓ fibrosis, ↑ systolic function | LV | [37] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levick, S.P.; Widiapradja, A. Mast Cells: Key Contributors to Cardiac Fibrosis. Int. J. Mol. Sci. 2018, 19, 231. https://doi.org/10.3390/ijms19010231
Levick SP, Widiapradja A. Mast Cells: Key Contributors to Cardiac Fibrosis. International Journal of Molecular Sciences. 2018; 19(1):231. https://doi.org/10.3390/ijms19010231
Chicago/Turabian StyleLevick, Scott P., and Alexander Widiapradja. 2018. "Mast Cells: Key Contributors to Cardiac Fibrosis" International Journal of Molecular Sciences 19, no. 1: 231. https://doi.org/10.3390/ijms19010231
APA StyleLevick, S. P., & Widiapradja, A. (2018). Mast Cells: Key Contributors to Cardiac Fibrosis. International Journal of Molecular Sciences, 19(1), 231. https://doi.org/10.3390/ijms19010231