High-Mobility Group Box 1 Mediates Fibroblast Activity via RAGE-MAPK and NF-κB Signaling in Keloid Scar Formation


Abstract
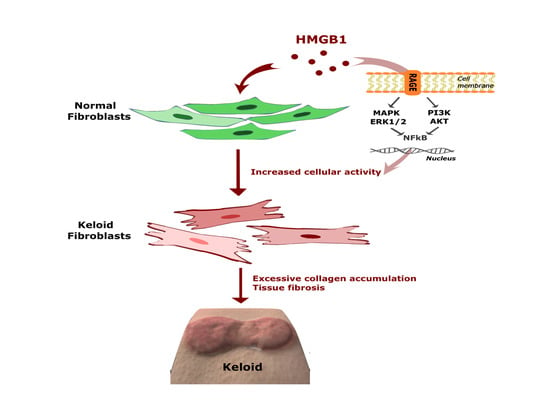
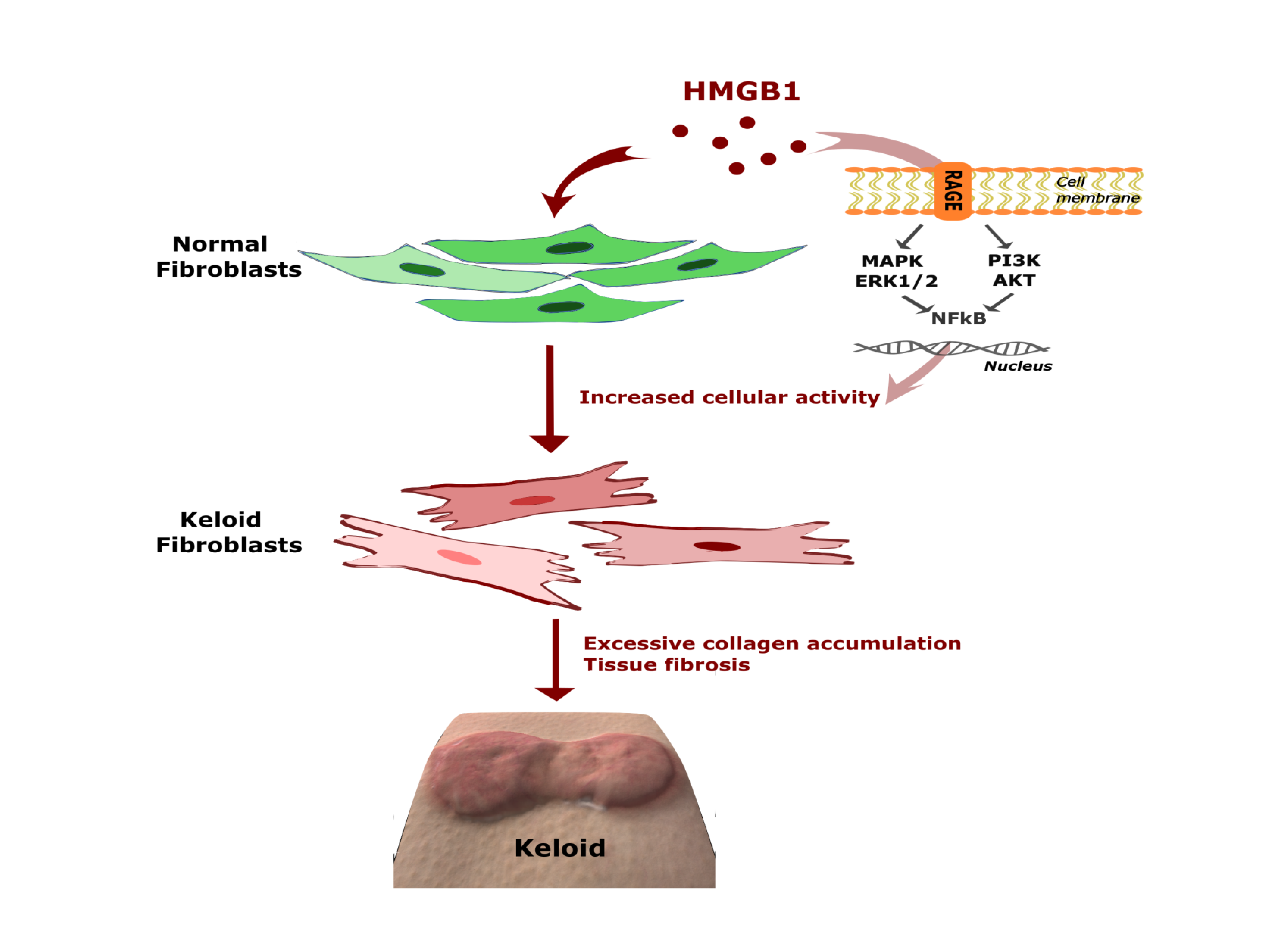
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
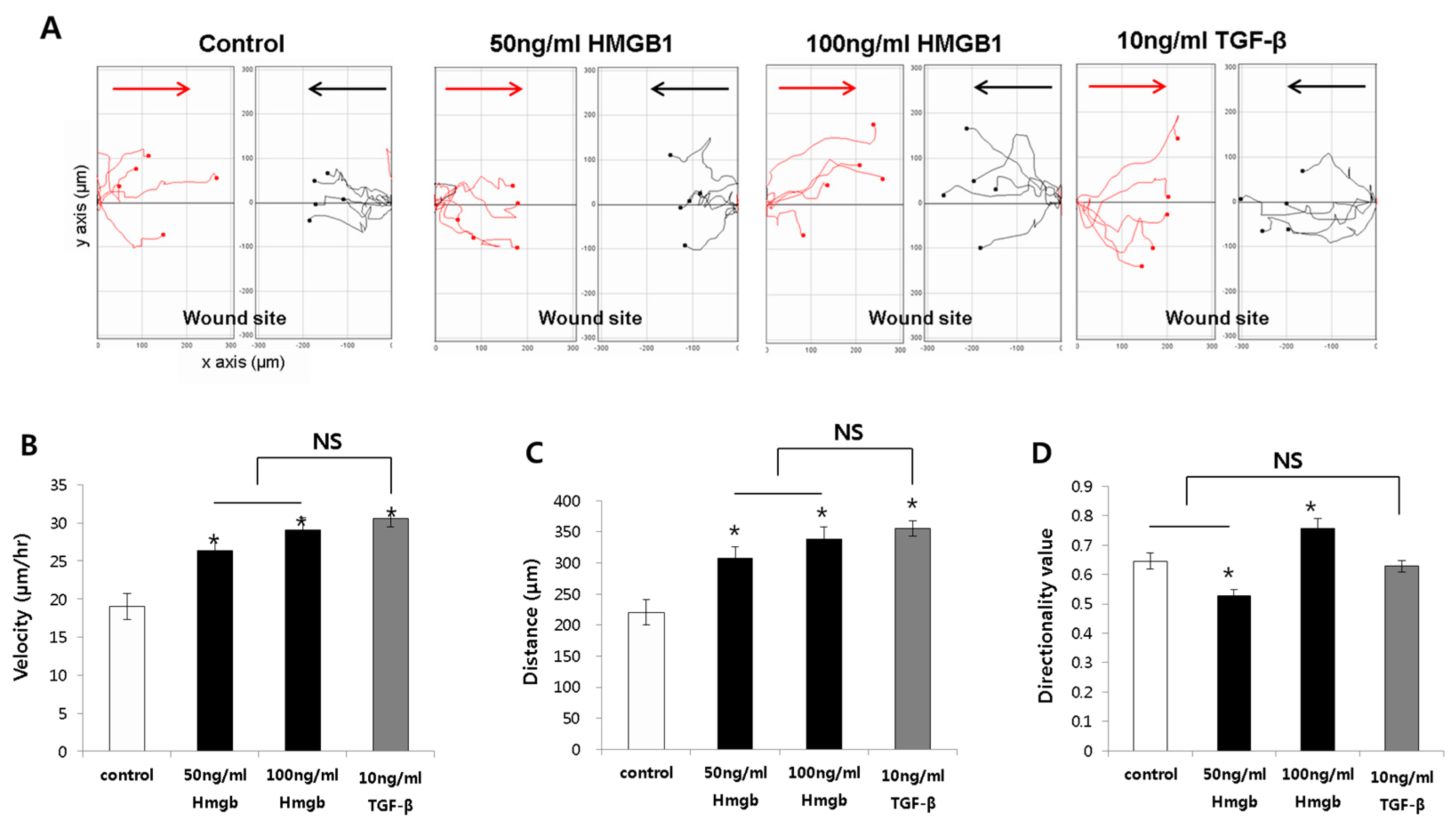
2.1. HMGB1 Administration Promoted Fibroblast Migration
2.2. HMGB1 Administration Increased the Migration Speed and Distance Traveled in Normal Fibroblasts
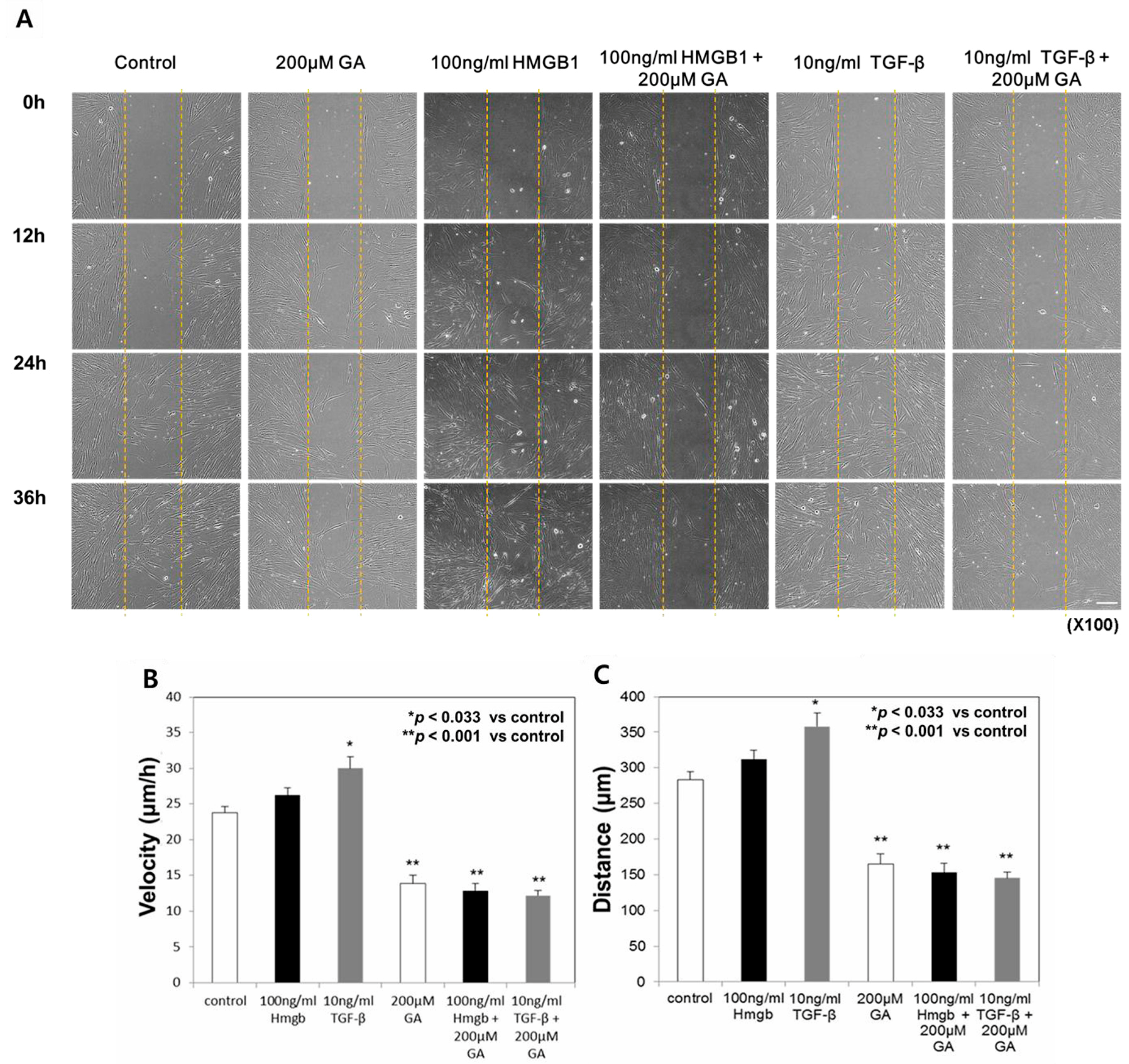
2.3. Treatment with Glycyrrhizic Acid (GA), an HMGB1 Inhibitor, Decreased the Migration Speed and Distance Traveled in Normal Fibroblasts
2.4. GA Treatment Decreased the Migration Speed and Distance Traveled in Keloid Fibroblasts
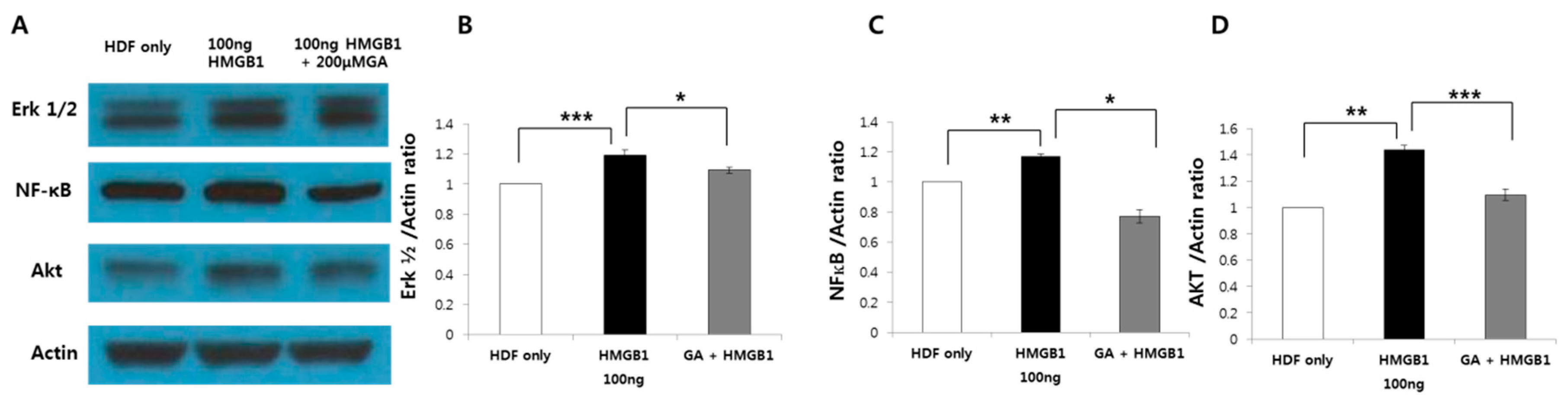
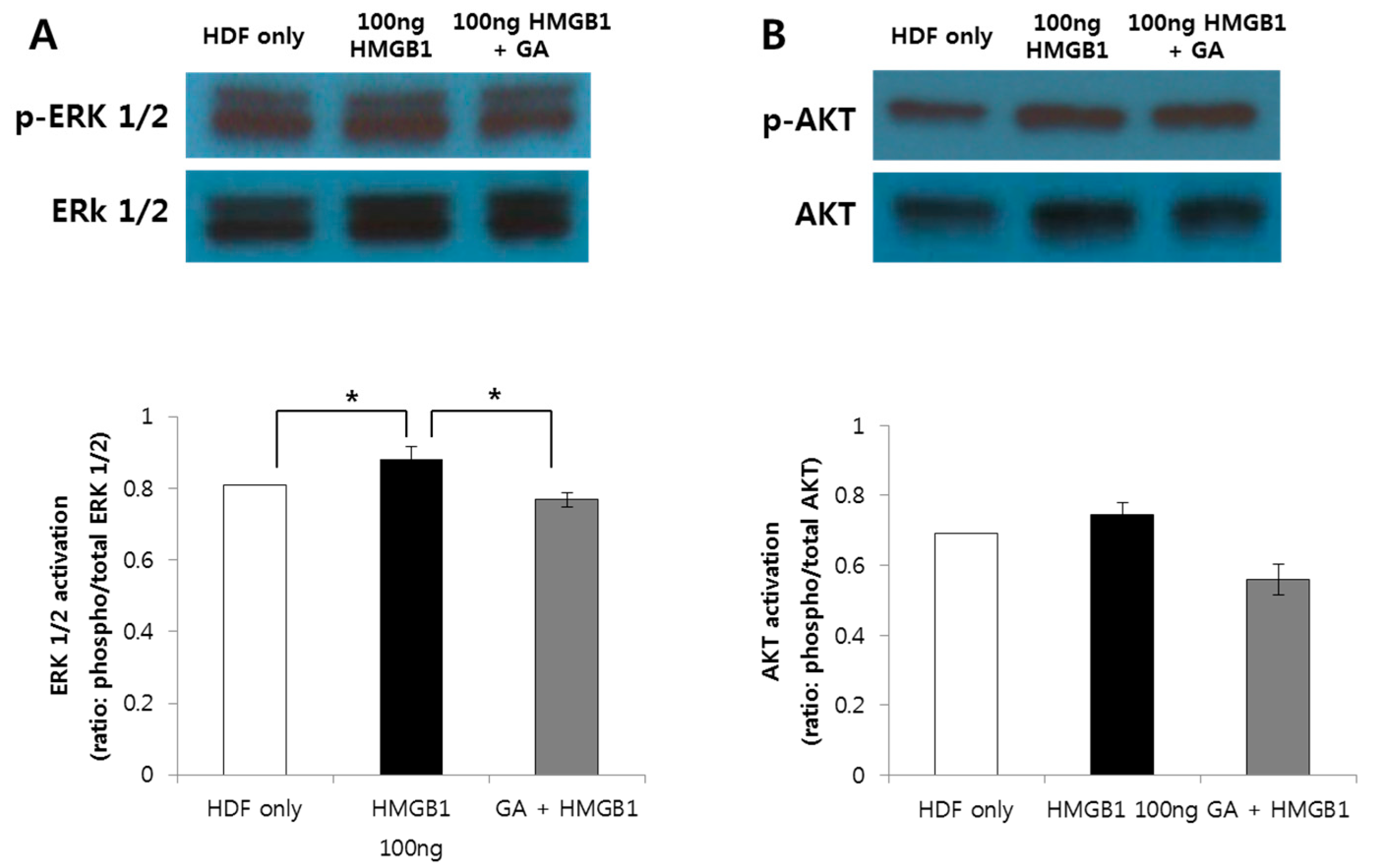
2.5. HMGB1-Induced ERK1/2, AKT, and NF-κB Protein Expression was Suppressed by GA Treatment in Human Normal Fibroblasts
3. Discussion
4. Materials and Methods
4.1. Keloid Tissues, Keloid-Derived Fibroblasts, and Human Dermal Fibroblast Cell Culture
4.2. In Vitro Wound Healing Assay and Cell Tracking System for Cell Migration
4.3. Western Blotting Analysis
4.4. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AKT | Protein kinase B |
| CXCL12 | C-X-C motif chemokine 12 |
| CXCR4 | Chemokine Receptor type 4 |
| ECM | Extracellular matrix |
| EMT | Epithelial-mesenchymal transition |
| ERK | Extracellular signal-regulated kinases |
| GA | Glycyrrhizic acid |
| HMGB1 | High-mobility group box 1 |
| MAPK | Mitogen-activated protein kinase |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| PBS | Phosphate-buffered saline |
| PI3K | Phosphatidylinositol-4,5-bisphosphate 3-kinase |
| RAGE | Receptor for advanced glycation end product |
| TGF-β | Transforming growth factor-β |
| TLR | Toll-like receptor |
References
- Huang, C.; Akaishi, S.; Ogawa, R. Mechanosignaling pathways in cutaneous scarring. Arch. Dermatol. Res. 2012, 304, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Akaishi, S.; Hyakusoku, H.; Ogawa, R. Are keloid and hypertrophic scar different forms of the same disorder? A fibroproliferative skin disorder hypothesis based on keloid findings. Int. Wound J. 2014, 11, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Wu, X.; Gao, Z.; Zhou, G.; Zhang, W.J.; Liu, W. Enhanced expression of membrane transporter and drug resistance in keloid fibroblasts. Hum. Pathol. 2012, 43, 2024–2032. [Google Scholar] [CrossRef] [PubMed]
- Qu, M.; Song, N.; Chai, G.; Wu, X.; Liu, W. Pathological niche environment transforms dermal stem cells to keloid stem cells: A hypothesis of keloid formation and development. Med. Hypotheses 2013, 81, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Gauglitz, G.G.; Korting, H.C.; Pavicic, T.; Ruzicka, T.; Jeschke, M.G. Hypertrophic scarring and keloids: Pathomechanisms and current and emerging treatment strategies. Mol. Med. 2011, 17, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Tuan, T.L.; Nichter, L.S. The molecular basis of keloid and hypertrophic scar formation. Mol. Med. Today 1998, 4, 19–24. [Google Scholar] [CrossRef]
- Bianchi, M.E.; Beltrame, M.; Paonessa, G. Specific recognition of cruciform DNA by nuclear protein HMG1. Science 1989, 243 Pt 1, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- Ellerman, J.E.; Brown, C.K.; de Vera, M.; Zeh, H.J.; Billiar, T.; Rubartelli, A.; Lotze, M.T. Masquerader: High mobility group box-1 and cancer. Clin. Cancer Res. 2007, 13, 2836–2848. [Google Scholar] [CrossRef] [PubMed]
- Ranzato, E.; Martinotti, S.; Pedrazzi, M.; Patrone, M. High mobility group box protein-1 in wound repair. Cells 2012, 1, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Ban, T.; Wang, Z.; Choi, M.K.; Kawamura, T.; Negishi, H.; Nakasato, M.; Lu, Y.; Hangai, S.; Koshiba, R.; et al. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature 2009, 462, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Taniguchi, T. Nucleic acid sensing and beyond: Virtues and vices of high-mobility group box 1. J. Intern. Med. 2014, 276, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.Y.; Lagares, D.; Tager, A.M.; Kapoor, M. Fibrosis—A lethal component of systemic sclerosis. Nat. Rev. Rheumatol. 2014, 10, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Li, L.C.; Gao, J.; Li, J. Emerging role of HMGB1 in fibrotic diseases. J. Cell. Mol. Med. 2014, 18, 2331–2339. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.; Nolan, S.; Slattery, C.; Feighery, R.; Ryan, M.P.; McMorrow, T. High-mobility group box protein 1: A novel mediator of inflammatory-induced renal epithelial-mesenchymal transition. Am. J. Nephrol. 2010, 32, 590–602. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.P.; Li, L.; Li, J.; Wang, J.Y.; Wang, L.Y.; Jiang, W. High mobility group box-1 promotes the proliferation and migration of hepatic stellate cells via TLR4-dependent signal pathways of PI3K/Akt and JNK. PLoS ONE 2013, 8, e64373. [Google Scholar] [CrossRef] [PubMed]
- Straino, S.; Di Carlo, A.; Mangoni, A.; De Mori, R.; Guerra, L.; Maurelli, R.; Panacchia, L.; Di Giacomo, F.; Palumbo, R.; Di Campli, C.; et al. High-mobility group box 1 protein in human and murine skin: Involvement in wound healing. J. Investig. Dermatol. 2008, 128, 1545–1553. [Google Scholar] [CrossRef] [PubMed]
- Dardenne, A.D.; Wulff, B.C.; Wilgus, T.A. The alarmin HMGB-1 influences healing outcomes in fetal skin wounds. Wound Repair Regen. 2013, 21, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Erlandsson Harris, H.; Andersson, U. Mini-review: The nuclear protein HMGB1 as a proinflammatory mediator. Eur. J. Immunol. 2004, 34, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.; Werner, S. Cancer as an overhealing wound: An old hypothesis revisited. Nat. Rev. Mol. Cell Biol. 2008, 9, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.S.; Phan, T.T.; Mukhopadhyay, A.; Lim, H.Y.; Halliwell, B.; Wong, K.P. Human skin keloid fibroblasts display bioenergetics of cancer cells. J. Investig. Dermatol. 2008, 128, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Yun, I.S.; Lee, M.H.; Rah, D.K.; Lew, D.H.; Park, J.C.; Lee, W.J. Heat Shock Protein 90 Inhibitor (17-AAG) Induces Apoptosis and Decreases Cell Migration/Motility of Keloid Fibroblasts. Plast. Reconstr. Surg. 2015, 136, 44e–53e. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Grimm, W.A.; Garner, W.L.; Qin, L.; Travis, T.; Tan, N.; Han, Y.P. Epithelial to mesenchymal transition in human skin wound healing is induced by tumor necrosis factor-alpha through bone morphogenic protein-2. Am. J. Pathol. 2010, 176, 2247–2258. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Park, J.H.; Shin, J.U.; Noh, H.; Lew, D.H.; Yang, W.I.; Yun, C.O.; Lee, K.H.; Lee, J.H. Endothelial-to-mesenchymal transition induced by Wnt 3a in keloid pathogenesis. Wound Repair Regen. 2015, 23, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.W.; Jang, S.; Kim, H.; Lim, J.B. Combined targeting of high-mobility group box-1 and interleukin-8 to control micrometastasis potential in gastric cancer. Int. J. Cancer 2015, 137, 1598–1609. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Li, X.; Chen, Y.; Fang, J.; Ge, Z. High-mobility group box 1: A novel inducer of the epithelial-mesenchymal transition in colorectal carcinoma. Cancer Lett. 2015, 357, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.U.; Noh, J.Y.; Lee, J.H.; Lee, W.J.; Yoo, J.S.; Kim, J.Y.; Kim, H.; Jung, I.; Jin, S.; Lee, K.H. In vivo relative quantitative proteomics reveals HMGB1 as a downstream mediator of oestrogen-stimulated keratinocyte migration. Exp. Dermatol. 2015, 24, 478–480. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Yin, J.; Wang, T.; Sun, Y.; Ni, P.; Ma, R.; Zhu, H.; Zheng, D.; Shen, H.; Xu, W.; et al. Up-regulated HMGB1 in EAM directly led to collagen deposition by a PKCbeta/Erk1/2-dependent pathway: Cardiac fibroblast/myofibroblast might be another source of HMGB1. J. Cell. Mol. Med. 2014, 18, 1740–1751. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Anzai, T.; Naito, K.; Miyasho, T.; Okamoto, M.; Yokota, H.; Yamada, S.; Maekawa, Y.; Takahashi, T.; Yoshikawa, T.; et al. Role of high-mobility group box 1 protein in post-infarction healing process and left ventricular remodelling. Cardiovasc. Res. 2009, 81, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Ojo, O.O.; Ryu, M.H.; Jha, A.; Unruh, H.; Halayko, A.J. High-mobility group box 1 promotes extracellular matrix synthesis and wound repair in human bronchial epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L1354–L1366. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Wang, H.; Ding, A.; Golenbock, D.T.; Latz, E.; Czura, C.J.; Fenton, M.J.; Tracey, K.J.; Yang, H. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock 2006, 26, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Tsung, A.; Sahai, R.; Tanaka, H.; Nakao, A.; Fink, M.P.; Lotze, M.T.; Yang, H.; Li, J.; Tracey, K.J.; Geller, D.A.; et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J. Exp. Med. 2005, 201, 1135–1143. [Google Scholar] [CrossRef]
- Park, J.S.; Svetkauskaite, D.; He, Q.; Kim, J.Y.; Strassheim, D.; Ishizaka, A.; Abraham, E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J. Biol. Chem. 2004, 279, 7370–7377. [Google Scholar] [CrossRef]
- Kokkola, R.; Andersson, A.; Mullins, G.; Ostberg, T.; Treutiger, C.J.; Arnold, B.; Nawroth, P.; Andersson, U.; Harris, R.A.; Harris, H.E. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand. J. Immunol. 2005, 61, 1–9. [Google Scholar] [CrossRef]
- Ibrahim, Z.A.; Armour, C.L.; Phipps, S.; Sukkar, M.B. RAGE and TLRs: Relatives, friends or neighbours? Mol. Immunol. 2013, 56, 739–744. [Google Scholar] [CrossRef]
- Zhang, Z.; Nie, F.; Kang, C.; Chen, B.; Qin, Z.; Ma, J.; Ma, Y.; Zhao, X. Increased periostin expression affects the proliferation, collagen synthesis, migration and invasion of keloid fibroblasts under hypoxic conditions. Int. J. Mol. Med. 2014, 34, 253–261. [Google Scholar] [CrossRef]
- Lim, I.J.; Phan, T.T.; Tan, E.K.; Nguyen, T.T.; Tran, E.; Longaker, M.T.; Song, C.; Lee, S.T.; Huynh, H.T. Synchronous activation of ERK and phosphatidylinositol 3-kinase pathways is required for collagen and extracellular matrix production in keloids. J. Biol. Chem. 2003, 278, 40851–40858. [Google Scholar] [CrossRef] [PubMed]
- Washio, H.; Fukuda, N.; Matsuda, H.; Nagase, H.; Watanabe, T.; Matsumoto, Y.; Terui, T. Transcriptional inhibition of hypertrophic scars by a gene silencer, pyrrole-imidazole polyamide, targeting the TGF-beta1 promoter. J. Investig. Dermatol. 2011, 131, 1987–1995. [Google Scholar] [CrossRef]
- Seifert, O.; Mrowietz, U. Keloid scarring: Bench and bedside. Arch. Dermatol. Res. 2009, 301, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wang, D.R.; Cao, Y.L. TGF-beta: A fibrotic factor in wound scarring and a potential target for anti-scarring gene therapy. Curr. Gene Ther. 2004, 4, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.K.; Wang, B.; Lu, Q.H.; Zhang, W.; Qin, W.D.; Liu, X.J.; Liu, X.Q.; An, F.S.; Zhang, Y.; Zhang, M.X. Inhibition of high-mobility group box 1 improves myocardial fibrosis and dysfunction in diabetic cardiomyopathy. Int. J. Cardiol. 2014, 172, 202–212. [Google Scholar] [CrossRef]
- He, M.; Kubo, H.; Ishizawa, K.; Hegab, A.E.; Yamamoto, Y.; Yamamoto, H.; Yamaya, M. The role of the receptor for advanced glycation end-products in lung fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L1427–L1436. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Tang, H.; He, H.; Liu, J.; Mao, J.; Ji, H.; Lin, H.; Wu, T. Glycyrrhizic acid alleviates bleomycin-induced pulmonary fibrosis in rats. Front. Pharmacol. 2015, 6, 215. [Google Scholar] [CrossRef] [PubMed]
- Moro, T.; Shimoyama, Y.; Kushida, M.; Hong, Y.Y.; Nakao, S.; Higashiyama, R.; Sugioka, Y.; Inoue, H.; Okazaki, I.; Inagaki, Y. Glycyrrhizin and its metabolite inhibit Smad3-mediated type I collagen gene transcription and suppress experimental murine liver fibrosis. Life Sci. 2008, 83, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, R. Keloid and Hypertrophic Scars Are the Result of Chronic Inflammation in the Reticular Dermis. Int. J. Mol. Sci. 2017, 18, 606. [Google Scholar] [CrossRef] [PubMed]
- Mollica, L.; De Marchis, F.; Spitaleri, A.; Dallacosta, C.; Pennacchini, D.; Zamai, M.; Agresti, A.; Trisciuoglio, L.; Musco, G.; Bianchi, M.E. Glycyrrhizin binds to high-mobility group box 1 protein and inhibits its cytokine activities. Chem. Biol. 2007, 14, 431–441. [Google Scholar] [CrossRef] [PubMed]






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Park, J.-C.; Lee, M.H.; Yang, C.E.; Lee, J.H.; Lee, W.J. High-Mobility Group Box 1 Mediates Fibroblast Activity via RAGE-MAPK and NF-κB Signaling in Keloid Scar Formation. Int. J. Mol. Sci. 2018, 19, 76. https://doi.org/10.3390/ijms19010076
Kim J, Park J-C, Lee MH, Yang CE, Lee JH, Lee WJ. High-Mobility Group Box 1 Mediates Fibroblast Activity via RAGE-MAPK and NF-κB Signaling in Keloid Scar Formation. International Journal of Molecular Sciences. 2018; 19(1):76. https://doi.org/10.3390/ijms19010076
Chicago/Turabian StyleKim, Jihee, Jong-Chul Park, Mi Hee Lee, Chae Eun Yang, Ju Hee Lee, and Won Jai Lee. 2018. "High-Mobility Group Box 1 Mediates Fibroblast Activity via RAGE-MAPK and NF-κB Signaling in Keloid Scar Formation" International Journal of Molecular Sciences 19, no. 1: 76. https://doi.org/10.3390/ijms19010076
APA StyleKim, J., Park, J. -C., Lee, M. H., Yang, C. E., Lee, J. H., & Lee, W. J. (2018). High-Mobility Group Box 1 Mediates Fibroblast Activity via RAGE-MAPK and NF-κB Signaling in Keloid Scar Formation. International Journal of Molecular Sciences, 19(1), 76. https://doi.org/10.3390/ijms19010076