InSiDDe: A Server for Designing Artificial Disordered Proteins



 and
and 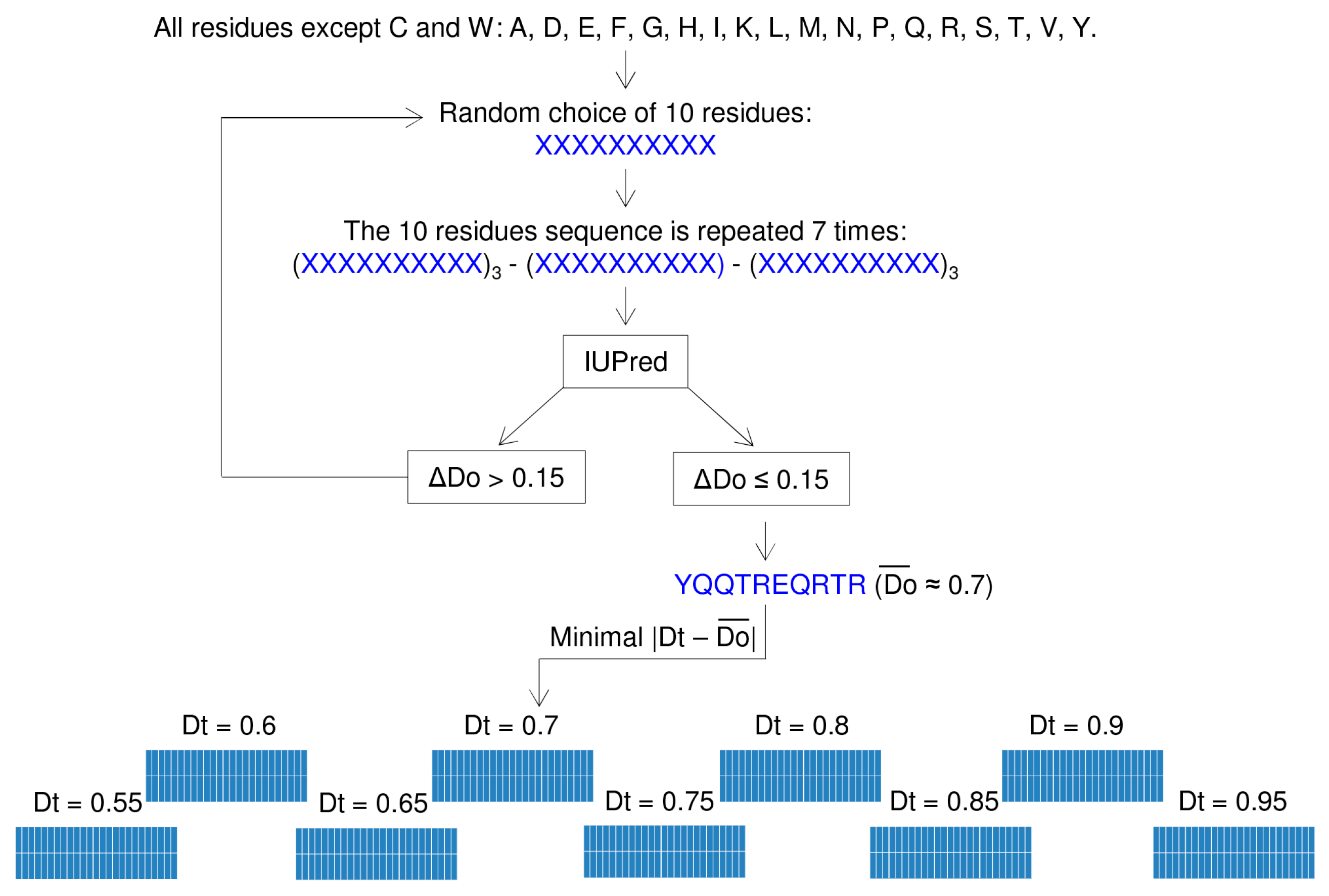{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Rationale and Implementation of In Silico Disorder Design (InSiDDe)
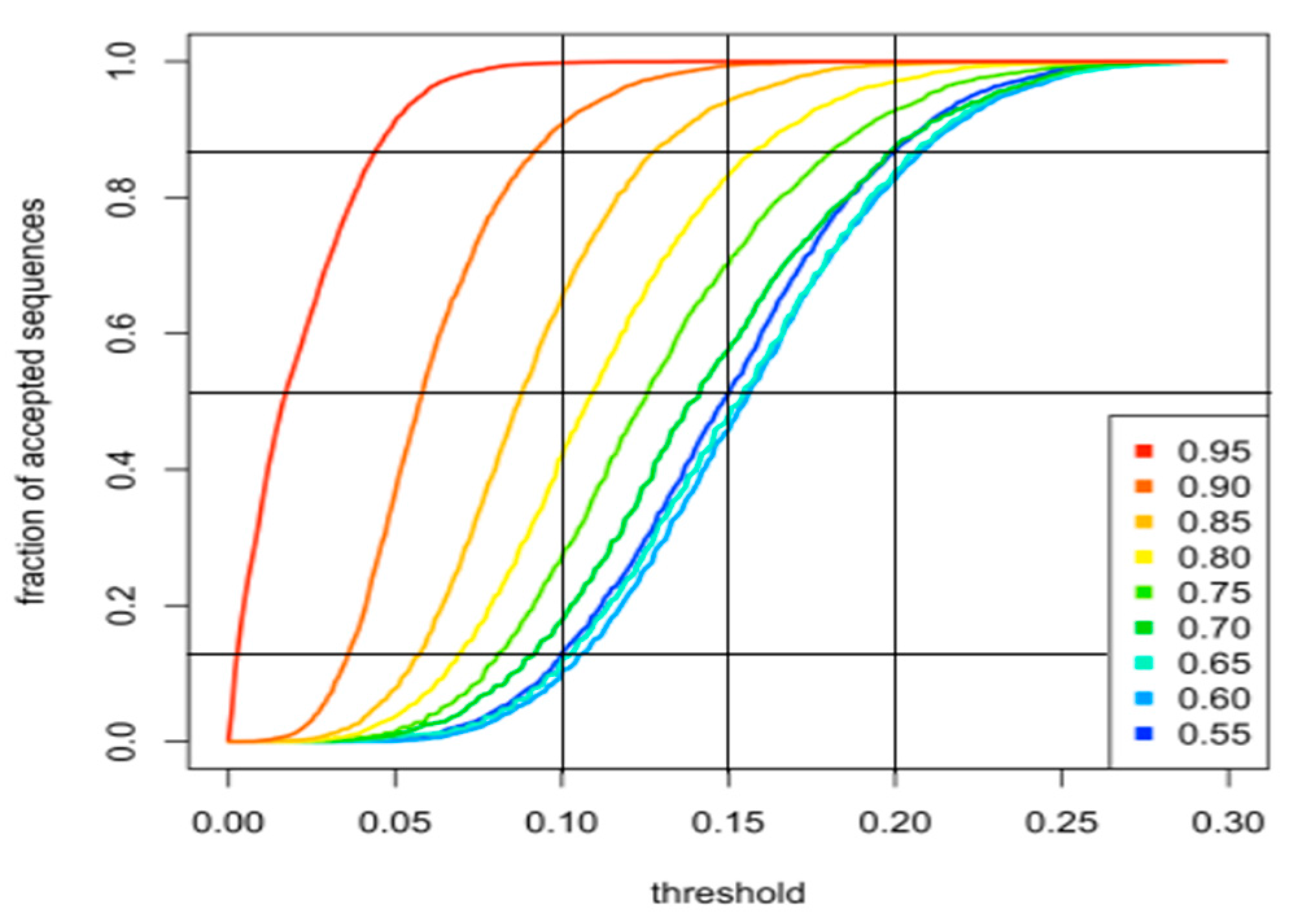
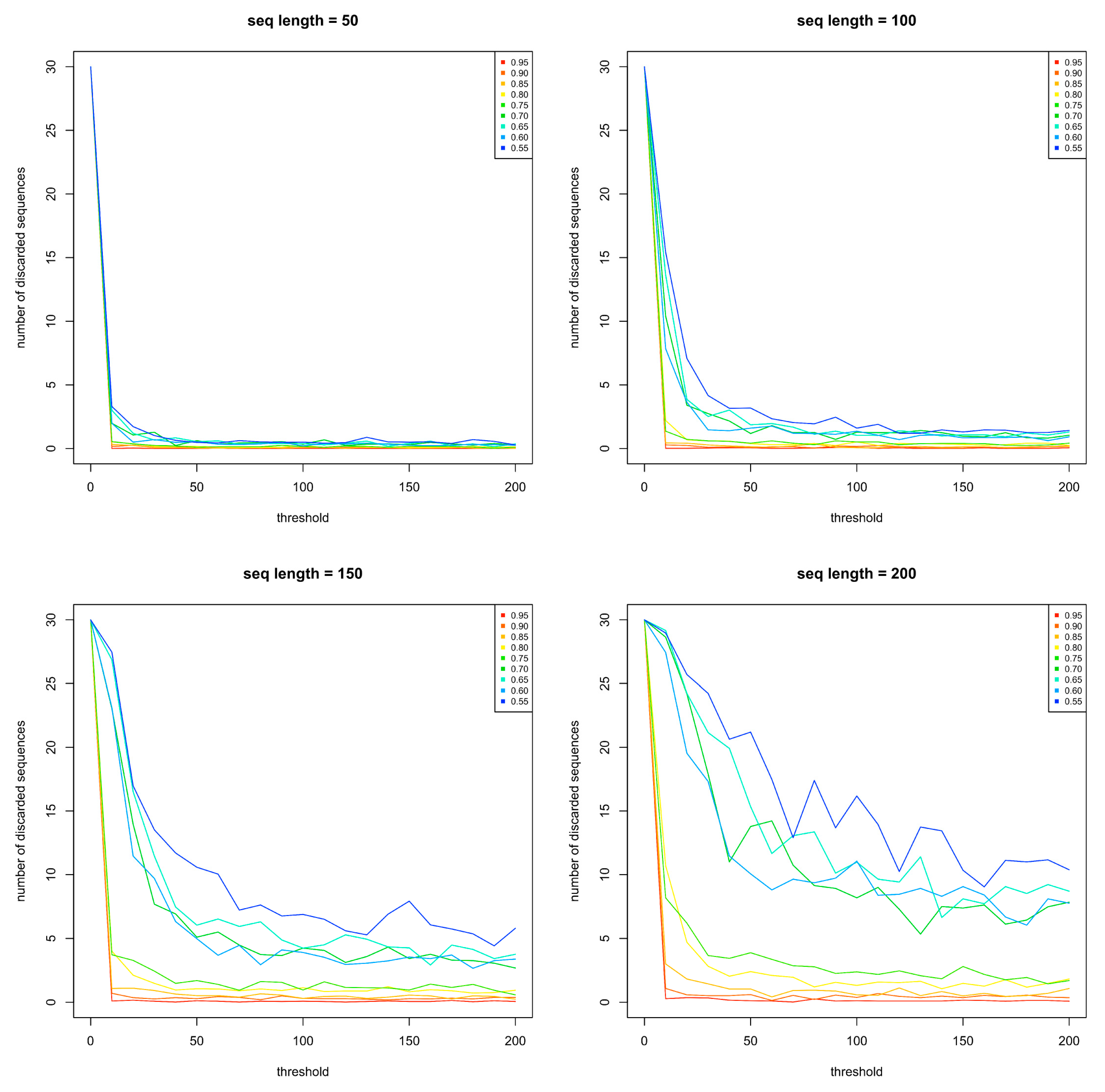
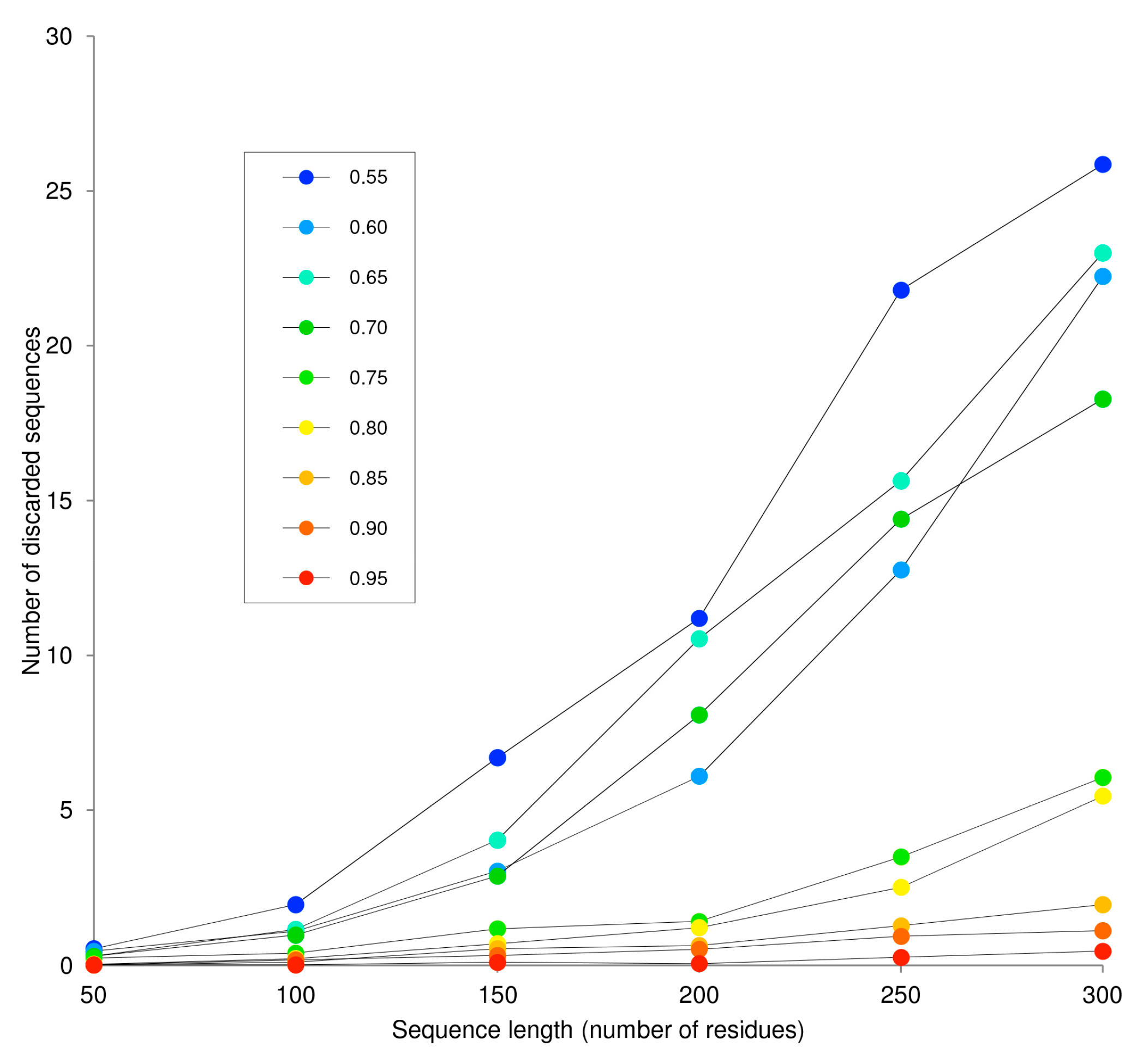
2.1.1. Creation of the Sequence Library
2.1.2. Generation of Artificial Sequences
2.2. Example of Use
3. Materials and Methods
3.1. Set-Up of InSiDDE
3.2. Availability and Requirements of InSiDDE
3.3. DNA Constructs
3.3.1. Split-GFP Constructs
3.3.2. Thioredoxin-His-Tev-100-0.6-MoRE Construct
3.4. Split-GFP Reassembly Assay and Purification of NGFP Fusions
3.5. Expression and Purification of Thioredoxin-His-Tev-100-0.6-MoRE
3.6. Hydrodynamic Analysis
3.7. Far-UV Circular Dichroism (CD) Measurement
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dunker, A.K.; Garner, E.; Guilliot, S.; Romero, P.; Albrecht, K.; Hart, J.; Obradovic, Z.; Kissinger, C.; Villafranca, J.E. Protein disorder and the evolution of molecular recognition: Theory, predictions and observations. Pac. Symp. Biocomput. 1998, 3, 473–484. [Google Scholar]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. Flexible nets. FEBS J. 2005, 272, 5129–5148. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Showing your ID: Intrinsic disorder as an ID for recognition, regulation and cell signaling. J. Mol. Recognit. 2005, 18, 343–384. [Google Scholar] [CrossRef] [PubMed]
- Dyson, H.J.; Wright, P.E. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 2002, 12, 54–60. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing Protein Intrinsic Disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohan, A.; Oldfield, C.J.; Radivojac, P.; Vacic, V.; Cortese, M.S.; Dunker, A.K.; Uversky, V.N. Analysis of molecular recognition features (MoRFs). J. Mol. Biol. 2006, 362, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P.; Fuxreiter, M. Fuzzy complexes: Polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 2008, 33, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Gruet, A.; Dosnon, M.; Blocquel, D.; Brunel, J.; Gerlier, D.; Das, R.K.; Bonetti, D.; Gianni, S.; Fuxreiter, M.; Longhi, S.; et al. Fuzzy regions in an intrinsically disordered protein impair protein-protein interactions. FEBS J. 2016, 283, 576–594. [Google Scholar] [CrossRef] [PubMed]
- Dosztanyi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef] [PubMed]
- Otieno, S.; Kriwacki, R. Probing the role of nascent helicity in p27 function as a cell cycle regulator. PLoS ONE 2012, 7, e47177. [Google Scholar] [CrossRef] [PubMed]
- Bignon, C.; Troilo, F.; Gianni, S.; Longhi, S. Partner-mediated polymorphism of an intrinsically disordered protein. J. Mol. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, M.; Kragelund, B.B.; Papaleo, E.; Lindorff-Larsen, K. An Efficient Method for Estimating the Hydrodynamic Radius of Disordered Protein Conformations. Biophys. J. 2017, 113, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Harmon, T.S.; Crabtree, M.D.; Shammas, S.L.; Posey, A.E.; Clarke, J.; Pappu, R.V. GADIS: Algorithm for designing sequences to achieve target secondary structure profiles of intrinsically disordered proteins. Protein Eng. Des. Sel. 2016, 29, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Campen, A.; Williams, R.M.; Brown, C.J.; Meng, J.; Uversky, V.N.; Dunker, A.K. TOP-IDP-scale: A new amino acid scale measuring propensity for intrinsic disorder. Protein Pept. Lett. 2008, 15, 956–963. [Google Scholar] [CrossRef] [PubMed]
- Borgi, I.; Gargouri, A. A spontaneous direct repeat deletion in the pGEX fusion vector decreases the expression level of recombinant proteins in Escherichia coli. Protein Expr. Purif. 2008, 60, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. What does it mean to be natively unfolded? Eur. J. Biochem. 2002, 269, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Marsh, J.A.; Forman-Kay, J.D. Sequence determinants of compaction in intrinsically disordered proteins. Biophys. J. 2010, 98, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.G.; Magliery, T.J.; Regan, L. Detecting protein-protein interactions with GFP-fragment reassembly. Nat. Methods 2004, 1, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Magliery, T.J.; Wilson, C.G.; Pan, W.; Mishler, D.; Ghosh, I.; Hamilton, A.D.; Regan, L. Detecting protein-protein interactions with a green fluorescent protein fragment reassembly trap: Scope and mechanism. J. Am. Chem. Soc. 2005, 127, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Gruet, A.; Dosnon, M.; Vassena, A.; Lombard, V.; Gerlier, D.; Bignon, C.; Longhi, S. Dissecting partner recognition by an intrinsically disordered protein using descriptive random mutagenesis. J. Mol. Biol. 2013, 425, 3495–3509. [Google Scholar] [CrossRef] [PubMed]
- Longhi, S.; Receveur-Brechot, V.; Karlin, D.; Johansson, K.; Darbon, H.; Bhella, D.; Yeo, R.; Finet, S.; Canard, B. The C-terminal domain of the measles virus nucleoprotein is intrinsically disordered and folds upon binding to the C-terminal moiety of the phosphoprotein. J. Biol. Chem. 2003, 278, 18638–18648. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Use of fast protein size-exclusion liquid chromatography to study the unfolding of proteins which denature through the molten globule. Biochemistry 1993, 32, 13288–13298. [Google Scholar] [CrossRef] [PubMed]









© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schramm, A.; Lieutaud, P.; Gianni, S.; Longhi, S.; Bignon, C. InSiDDe: A Server for Designing Artificial Disordered Proteins. Int. J. Mol. Sci. 2018, 19, 91. https://doi.org/10.3390/ijms19010091
Schramm A, Lieutaud P, Gianni S, Longhi S, Bignon C. InSiDDe: A Server for Designing Artificial Disordered Proteins. International Journal of Molecular Sciences. 2018; 19(1):91. https://doi.org/10.3390/ijms19010091
Chicago/Turabian StyleSchramm, Antoine, Philippe Lieutaud, Stefano Gianni, Sonia Longhi, and Christophe Bignon. 2018. "InSiDDe: A Server for Designing Artificial Disordered Proteins" International Journal of Molecular Sciences 19, no. 1: 91. https://doi.org/10.3390/ijms19010091
APA StyleSchramm, A., Lieutaud, P., Gianni, S., Longhi, S., & Bignon, C. (2018). InSiDDe: A Server for Designing Artificial Disordered Proteins. International Journal of Molecular Sciences, 19(1), 91. https://doi.org/10.3390/ijms19010091