Fibrocytes: A Novel Stromal Cells to Regulate Resistance to Anti-Angiogenic Therapy and Cancer Progression

Abstract
:1. Introduction
2. Stromal Cells Are Involved in the Resistance to VEGF Blockade
2.1. Endothelial Cells
2.2. Pericytes
2.3. Tumor-Associated Macrophages
2.4. Myeloid-Derived Suppressor Cells
2.5. Cancer-Associated Fibroblasts
2.6. Other Stromal Cells
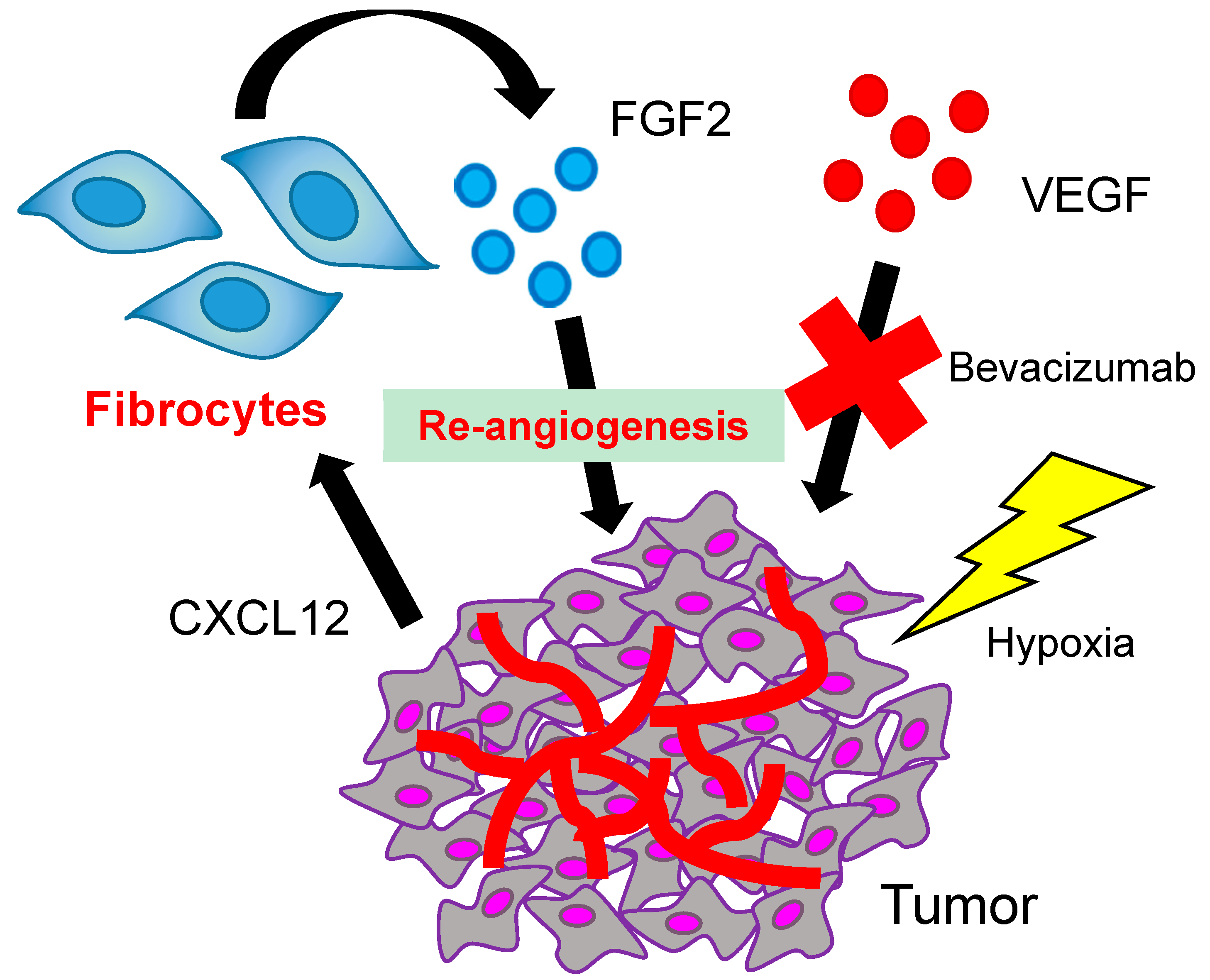
3. The Role of Fibrocytes in Re-Angiogenesis after VEGF Blockade
4. Other Emerging Roles of Fibrocytes in the Tumor Microenvironment
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Lenzi, P.; Bocci, G.; Natale, G. John Hunter and the origin of the term ‘‘angiogenesis”. Angiogenesis 2016, 19, 255–256. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J.; Shing, Y. Control of angiogenesis by heparin and other sulfated polysaccharides. Adv. Exp. Med. Biol. 1992, 313, 355–364. [Google Scholar] [PubMed]
- Folkman, J. Anti-angiogenesis: New concept for therapy of solid tumors. Ann. Surg. 1972, 175, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Griffioen, A.W.; Molema, G. Angiogenesis: Potentials for pharmacologic intervention in the treatment of cancer, cardiovascular diseases, and chronic inflammation. Pharmacol. Rev. 2000, 52, 237–268. [Google Scholar] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angio-genesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; Duda, D.G.; Clark, J.W.; Loeffler, J.S. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat. Clin. Pract. Oncol. 2006, 3, 24–40. [Google Scholar] [CrossRef] [PubMed]
- Sandler, A.; Gray, R.; Perry, M.C.; Brahmer, J.; Schiller, J.H.; Dowlati, A.; Lilenbaum, R.; Johnson, D.H. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N. Engl. J. Med. 2006, 355, 2542–2550. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- Kerbel, R.S. Inhibition of tumor angiogenesis as a strategy to circumvent acquired resistance to anti-cancer therapeutic agents. Bioessays 1991, 13, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Kerbel, R.; Ellis, L.M.; Harris, A.L. Antiangiogenic therapy in oncology: Current status and future directions. Lancet 2016, 388, 518–529. [Google Scholar] [CrossRef]
- Rapisarada, A.; Melillo, G. Overcoming disappointing results with antiangiogenic therapy by targeting hypoxia. Nat. Rev. Clin. Oncol. 2012, 9, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Ebos, J.M.; Kerbel, R.S. Antiangiogenic therapy: Impact on invasion, disease progression, and metastasis. Nat. Rev. Clin. Oncol. 2011, 8, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Montero, A.J.; Vogel, C. Fighting fire with fire: Rekindling the bevacizumab debate. N. Engl. J. Med. 2012, 366, 374–375. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.M.; Hicklin, D.J. Pathways mediating resistance to vascular endothelial growth factor-targeted therapy. Clin. Cancer Res. 2008, 14, 6371–6375. [Google Scholar] [CrossRef] [PubMed]
- Huijbers, E.J.; van Beijnum, J.R.; Thijssen, V.L.; Sabrkhany, S.; Nowak-Sliwinska, P.; Griffioen, A.W. Role of the tumor stroma in resistance to anti-angiogenic therapy. Drug Resist. Updates 2016, 25, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Al-Abd, A.M.; Alamoudi, A.J.; Abdel-Naim, A.B.; Neamatallah, T.A.; Ashour, O.M. Anti-angiogenic agents for the treatment of solid tumors: Potential pathways, therapy and current strategies—A review. J. Adv. Res. 2017, 8, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Thijssen, V.L.; van Beijnum, J.R.; Mayo, K.H.; Griffioen, A.W. Identification of novel drug targets for angiostatic cancer therapy: It takes two to tango. Curr. Pharm. Des. 2007, 13, 3576–3583. [Google Scholar] [CrossRef] [PubMed]
- Van der Schaft, D.W.; Hillen, F.; Pauwels, P.; Kirschmann, D.A.; Castermans, K.; Egbrink, M.G.; Tran, M.G.; Sciot, R.; Hauben, E.; Hogendoorn, P.C.; et al. Tumor cell plasticity in Ewing sarcoma, an alternative circulatory system stimulated by hypoxia. Cancer Res. 2005, 65, 11520–11528. [Google Scholar] [CrossRef] [PubMed]
- Paulis, Y.W.; Soetekouw, P.M.; Verheul, H.M.; Tjan-Heijnen, V.C.; Griffioen, A.W. Signalling pathways in vasculogenic mimicry. Biochim. Biophys. Acta 2010, 1806, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, J.L.; Kim, J.; Ozawa, T.; Zhang, M.; Westphal, M.; Deen, D.F.; Shuman, M.A. Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia 2000, 2, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Leenders, W.P.; Küsters, B.; Verrijp, K.; Maass, C.; Wesseling, P.; Heerschap, A.; Ruiter, D.; Ryan, A.; de Waal, R. Antiangiogenic therapy of cerebral melanoma metastases results in sustained tumor progression via vessel co-option. Clin. Cancer Res. 2004, 10, 6222–6230. [Google Scholar] [CrossRef] [PubMed]
- Gotink, K.J.; Broxterman, H.J.; Labots, M.; de Haas, R.R.; Dekker, H.; Honeywell, R.J.; Rudek, M.A.; Beerepoot, L.V.; Musters, R.J.; Jansen, G.; et al. Lysosomal sequestration of sunitinib: A novel mechanism of drug resistance. Clin. Cancer Res. 2011, 17, 7337–7346. [Google Scholar] [CrossRef] [PubMed]
- Adar, Y.; Stark, M.; Bram, E.E.; Nowak-Sliwinska, P.; van den Bergh, H.; Szewczyk, G.; Sarna, T.; Skladanowski, A.; Griffioen, A.W.; Assaraf, Y.G. Imidazoacridinone-dependent lysosomal photodestruction: A pharmacological Trojan horse approach to eradicate multidrug-resistant cancers. Cell Death Dis. 2012, 3, e293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomal sequestration of hydrophobic weak base chemotherapeutics triggers lysosomal biogenesis and lysosome-dependent cancer multidrug resistance. Oncotarget 2015, 6, 1143–1156. [Google Scholar] [CrossRef] [PubMed]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomes as mediators of drug resistance in cancer. Drug Resist. Updates 2016, 24, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Nowak-Sliwinska, P.; Weiss, A.; van Beijnum, J.R.; Wong, T.J.; Kilarski, W.W.; Szewczyk, G.; Verheul, H.M.; Sarna, T.; van den Bergh, H.; Griffioen, A.W. Photoactivation of lysosomally sequestered sunitinib after angiostatic treatment causes vascular occlusion and enhances tumor growth inhibition. Cell Death Dis. 2015, 6, e1641. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Van Beijnum, J.R.; Nowak-Sliwinska, P.; Huijbers, E.J.; Thijssen, V.L.; Griffioen, A.W. The great escape: The hallmarks of resistance to antiangiogenic therapy. Pharmacol. Rev. 2015, 67, 441–461. [Google Scholar] [CrossRef] [PubMed]
- Vasudev, N.S.; Reynolds, A.R. Anti-angiogenic therapy for cancer: Current progress, unresolved questions and future directions. Angiogenesis 2014, 17, 471–494. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, K.; Ohga, N.; Hida, Y.; Kawamoto, T.; Sadamoto, Y.; Ishikawa, S.; Maishi, N.; Akino, T.; Kondoh, M.; Matsuda, A.; et al. Tumor endothelial cells acquire drug resistance by MDR1 up-regulation via VEGF signaling in tumor microenvironment. Am. J. Pathol. 2012, 180, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Hu, C.; di Benedetto, M.; Varin, R.; Liu, J.; Wang, L.; Vannier, J.P.; Jin, J.; Janin, A.; Lu, H.; et al. Induction of multiple drug resistance in HMEC-1 endothelial cells after long-term exposure to sunitinib. Oncol. Target Ther. 2014, 7, 2249–2255. [Google Scholar]
- Croci, D.O.; Cerliani, J.P.; Dalotto-Moreno, T.; Méndez-Huergo, S.P.; Mascanfroni, I.D.; Dergan-Dylon, S.; Toscano, M.A.; Caramelo, J.J.; García-Vallejo, J.J.; Ouyang, J.; et al. Glycosylation-dependent lectin-receptor interactions preserve angiogenesis in anti-VEGF refractory tumors. Cell 2014, 156, 744–758. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Sims, D.E. The pericyte—A review. Tissue Cell 1986, 18, 153–174. [Google Scholar] [CrossRef]
- Hellström, M.; Gerhardt, H.; Kalén, M.; Li, X.; Eriksson, U.; Wolburg, H.; Betsholtz, C. Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J. Cell Biol. 2001, 153, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Hellberg, C.; Ostman, A.; Heldin, C.H. PDGF and vessel maturation. Recent Results Cancer Res. 2010, 180, 103–114. [Google Scholar] [PubMed]
- Ribatti, D. Tumor refractoriness to anti-VEGF therapy. Oncotarget 2016, 7, 46668–46677. [Google Scholar] [CrossRef] [PubMed]
- Orlidge, A.; D’Amore, P.A. Inhibition of capillary endothelial cell growth by pericytes and smooth muscle cells. J. Cell Biol. 1987, 105, 1455–1462. [Google Scholar] [CrossRef] [PubMed]
- Bussard, K.M.; Mutkus, L.; Stumpf, K.; Gomez-Manzano, C.; Marini, F.C. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016, 18, 84. [Google Scholar] [CrossRef] [PubMed]
- Cascone, T.; Herynk, M.H.; Xu, L.; Du, Z.; Kadara, H.; Nilsson, M.B.; Oborn, C.J.; Park, Y.Y.; Erez, B.; Jacoby, J.J.; et al. Upregulated stromal EGFR and vascular remodeling in mouse xenograft models of angiogenesis inhibitor-resistant human lung adenocarcinoma. J. Clin. Investig. 2011, 121, 1313–1328. [Google Scholar] [CrossRef] [PubMed]
- Valkovic, T.; Dobrila, F.; Melato, M.; Sasso, F.; Rizzardi, C.; Jonjic, N. Correlation between vascular endothelial growth factor, angiogenesis, and tumor-associated macrophages in invasive ductal breast carcinoma. Virchows Arch. 2002, 440, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Sny-der, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Song, N.; Luo, Y. Role of bone marrow-derived cells in angiogenesis: Focus on macrophages and pericytes. Cancer Microenviron. 2012, 5, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Allavena, P.; Garlanda, C.; Locati, M. Tumor-associated macrophages and the related myeloid-derived suppressor cells as a paradigm of the diversity of macrophage activation. Hum. Immunol. 2009, 70, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Locati, M. Tumor-associated macrophages as a paradigm of macrophage plasticity, diversity, and polarization: Lessons and open questions. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- Hesketh, M.; Sahin, K.B.; West, Z.E.; Murray, R.Z. Macrophage Phenotypes Regulate Scar Formation and Chronic Wound Healing. Int. J. Mol. Sci. 2017, 18, 1545. [Google Scholar] [CrossRef] [PubMed]
- Edholm, E.S.; Rhoo, K.H.; Robert, J. Evolutionary Aspects of Macrophages Polarization. Results Probl. Cell Differ. 2017, 62, 3–22. [Google Scholar] [PubMed]
- Dirkx, A.E.; Oude Egbrink, M.G.; Wagstaff, J.; Griffioen, A.W. Monocyte/macrophage infiltration in tumors: Modulators of angiogenesis. J. Leukoc. Biol. 2006, 80, 1183–1196. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.; Muthana, M.; Coffelt, S.B.; Lewis, C.E. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, K.A.; Ashton, A.W.; Klein, R.S.; Lenzi, M.L.; Zhu, G.H.; Schwartz, E.L. Mechanisms by which tumor cells and monocytes expressing the angiogenic factor thymidine phosphorylase mediate human endothelial cell migration. Cancer Res. 2002, 63, 527–533. [Google Scholar]
- Lin, E.Y.; Li, J.F.; Gnatovskiy, L.; Deng, Y.; Zhu, L.; Grzesik, D.A.; Qian, H.; Xue, X.N.; Pollard, J.W. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006, 66, 11238–11246. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Brekken, R.; McMahon, G.; Vu, T.H.; Itoh, T.; Tamaki, K.; Tanzawa, K.; Thorpe, P.; Itohara, S.; Werb, Z.; et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2000, 2, 737–744. [Google Scholar] [PubMed]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Huang, S.; Van Arsdall, M.; Tedjarati, S.; McCarty, M.; Wu, W.; Langley, R.; Fidler, I.J. Contributions of stromal metalloproteinase-9 to angiogenesis and growth of human ovarian carcinoma in mice. J. Natl. Cancer Inst. 2002, 94, 1134–1142. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Pucci, F.; Venneri, M.A.; Biziato, D.; Nonis, A.; Moi, D.; Sica, A.; di Serio, C.; Naldini, L.; de Palma, M. A distinguishing gene signature shared by tumor-infiltrating Tie2-expressing monocytes, blood “resident” monocytes, and embryonic macrophages suggests common functions and developmental relationships. Blood 2009, 114, 901–914. [Google Scholar] [CrossRef] [PubMed]
- Lewis, CE.; de Palma, M.; Naldini, L. Tie2-expressing monocytes and tumor angiogenesis: Regulation by hypoxia and angiopoietin-2. Cancer Res. 2007, 67, 8429–8432. [Google Scholar] [CrossRef] [PubMed]
- Gabrusiewicz, K.; Liu, D.; Cortes-Santiago, N.; Hossain, M.B.; Conrad, C.A.; Aldape, K.D.; Fuller, G.N.; Marini, F.C.; Alonso, M.M.; Idoate, M.A.; et al. Anti-vascular endothelial growth factor therapy-induced glioma invasion is associated with accumulation of Tie2-expressing monocytes. Oncotarget 2014, 5, 2208–2220. [Google Scholar] [CrossRef] [PubMed]
- Safarzadeh, E.; Orangi, M.; Mohammadi, H.; Babaie, F.; Baradaran, B. Myeloid-derived suppressor cells: Important contributors to tumor progression and metastasis. J. Cell. Physiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ezernitchi, A.V.; Vaknin, I.; Cohen-Daniel, L.; Levy, O.; Manaster, E.; Halabi, A.; Pikarsky, E.; Shapira, L.; Baniyash, M. TCR zeta down-regulation under chronic inflammation is mediated by myeloid suppressor cells differentially distributed between various lymphatic organs. J. Immunol. 2006, 177, 4763–4772. [Google Scholar] [CrossRef] [PubMed]
- Kanterman, J.; Sade-Feldman, M.; Baniyash, M. New insights into chronic inflammation-induced immunosuppression. Semin. Cancer Biol. 2012, 22, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Filipazzi, P.; Huber, V.; Rivoltini, L. Phenotype, function and clinical implications of myeloid-derived suppressor cells in cancer patients. Cancer Immunol. Immunother. 2012, 61, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ye, T.H.; Han, Y.P.; Song, H.; Zhang, Y.K.; Xia, Y.; Wang, N.Y.; Xiong, Y.; Song, X.J.; Zhu, Y.X.; et al. Reductions in myeloid-derived suppressor cells and lung metastases using AZD4547 treatment of a metastatic murine breast tumor model. Cell. Physiol. Biochem. 2014, 33, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Vences-Catalán, F.; Rajapaksa, R.; Srivastava, M.K.; Marabelle, A.; Kuo, C.C.; Levy, R.; Levy, S. Tetraspanin CD81 promotes tumor growth and metastasis by modulating the functions of T regulatory and myeloid-derived suppressor cells. Cancer Res. 2015, 75, 4517–4526. [Google Scholar] [CrossRef] [PubMed]
- Motallebnezhad, M.; Jadidi-Niaragh, F.; Qamsari, E.S.; Bagheri, S.; Gharibi, T.; Yousefi, M. The immunobiology of myeloid-derived suppressor cells in cancer. Tumour Biol. 2016, 37, 1387–1406. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, F.; Wu, X.; Malik, A.K.; Zhong, C.; Baldwin, M.E.; Schanz, S.; Fuh, G.; Gerber, H.P.; Ferrara, N. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat. Biotechnol. 2007, 25, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, F.; Wu, X.; Zhong, C.; Yu, L.; Liang, X.H.; Yao, J.; Blanchard, D.; Bais, C.; Peale, F.V.; van Bruggen, N.; et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature 2007, 450, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Brandau, S.; Moses, K.; Lang, S. The kinship of neutrophils and granulocytic myeloid-derived suppressor cells in cancer: Cousins, siblings or twins? Semin. Cancer Biol. 2013, 23, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Khaled, Y.S.; Ammori, B.J.; Elkord, E. Myeloid-derived suppressor cells in cancer: Recent progress and prospects. Immunol. Cell Biol. 2013, 91, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Anderberg, C.; Pietras, K. On the origin of cancer-associated fibroblasts. Cell Cycle 2009, 8, 1461–1462. [Google Scholar] [CrossRef] [PubMed]
- Cirri, P.; Chiarugi, P. Cancer associated fibroblasts: The dark side of the coin. Am. J. Cancer Res. 2011, 1, 482–497. [Google Scholar] [PubMed]
- Luo, H.; Tu, G.; Liu, Z.; Liu, M. Cancer-associated fibroblasts: A multifaceted driver of breast cancer progression. Cancer Lett. 2015, 361, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Verdelli, C.; Avagliano, L.; Creo, P.; Guarnieri, V.; Scillitani, A.; Vicentini, L.; Steffano, G.B.; Beretta, E.; Soldati, L.; Costa, E.; et al. Tumour-associated fibroblasts contribute to neoangiogenesis in human parathyroid neoplasia. Endocr. Relat. Cancer 2015, 22, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Crawford, Y.; Ferrara, N. Tumor and stromal pathways mediating refractoriness/resistance to anti-angiogenic therapies. Trends Pharmacol. Sci. 2009, 30, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Kinugasa, Y.; Matsui, T.; Takakura, N. CD44 expressed on cancer-associated fibroblasts is a functional molecule supporting the stemness and drug resistance of malignant cancer cells in the tumor microenvironment. Stem Cells 2014, 32, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.S.; Wu, X.; Zhuang, G.; Ngu, H.; Kasman, I.; Zhang, J.; Vernes, J.M.; Jiang, Z.; Meng, Y.G.; Peale, F.V.; et al. An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nat. Med. 2013, 19, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Verheul, H.M.; Lolkema, M.P.; Qian, D.Z.; Hilkes, Y.H.; Liapi, E.; Akkerman, J.W.; Pili, R.; Voest, E.E. Platelets take up the monoclonal antibody bevacizumab. Clin. Cancer Res. 2007, 13, 5341–5347. [Google Scholar] [CrossRef] [PubMed]
- Sabrkhany, S.; Griffioen, A.W.; Verheul, H.M.; Heemskerk, J.W.; Oude Egbrink, M.G.; Kuijpers, M.J. Sunitinib is taken up by platelets and inhibits their function. Angiogenesis 2014, 17, 771. [Google Scholar]
- Nozawa, H.; Chiu, C.; Hanahan, D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 12493–12498. [Google Scholar] [CrossRef] [PubMed]
- Laurent, J.; Touvrey, C.; Botta, F.; Kuonen, F.; Ruegg, C. Emerging paradigms and questions on pro-angiogenic bone marrow-derived myelomonocytic cells. Int. J. Dev. Biol. 2011, 55, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.P.; Rothenberg, M.E. Eosinophils and cancer. Cancer Immunol. Res. 2014, 2, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Coffelt, S.B.; Lewis, C.E.; Naldini, L.; Brown, J.M.; Ferrara, N.; de Palma, M. Elusive identities and overlapping phenotypes of proangiogenic myeloid cells in tumors. Am. J. Pathol. 2010, 176, 1564–1576. [Google Scholar] [CrossRef] [PubMed]
- Crivellato, E.; Nico, B.; Ribatti, D. Mast cells and tumour angiogenesis: New insight from experimental carcinogenesis. Cancer Lett. 2008, 269, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, A.; Elia, A.R.; Cappello, P.; Puppo, M.; Vanni, C.; Fardin, P.; Eva, A.; Munroe, D.; Wu, X.; Giovarelli, M.; et al. Transcriptome of hypoxic immature dendritic cells: Modulation of chemokine/receptor expression. Mol. Cancer Res. 2008, 6, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, A.; Goto, H.; Saijo, A.; Trung, V.T.; Aono, Y.; Ogino, H.; Kuramoto, T.; Tabata, S.; Uehara, H.; Izumi, K.; et al. Fibrocyte-like cells mediate acquired resistance to anti-angiogenic therapy with bevacizumab. Nat. Commun. 2015, 6, 8792. [Google Scholar] [CrossRef] [PubMed]
- Bucala, R.; Spiegel, L.A.; Chesney, J.; Hogan, M.; Cerami, A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol. Med. 1994, 1, 71–81. [Google Scholar] [PubMed]
- Reilkoff, R.A.; Bucala, R.; Herzog, E.L. Fibrocytes: Emerging effector cells in chronic inflammation. Nat. Rev. Immunol. 2011, 11, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Pilling, D.; Fan, T.; Huang, D.; Kaul, B.; Gomer, R.H. Identification of markers that distinguish monocyte-derived fibrocytes from monocytes, macrophages, and fibroblasts. PLoS ONE 2009, 4, e7475. [Google Scholar] [CrossRef] [PubMed]
- Hartlapp, I.; Abe, R.; Saeed, R.W.; Peng, T.; Voelter, W.; Bucala, R.; Metz, C.N. Fibrocytes induce an angiogenic phenotype in cultured endothelial cells and promote angiogenesis in vivo. FASEB J. 2001, 15, 2215–2224. [Google Scholar] [CrossRef] [PubMed]
- Bellini, A.; Mattoli, S. The role of the fibrocyte, a bone marrow-derived mesenchymal progenitor, in reactive and reparative fibroses. Lab. Investig. 2007, 87, 858–870. [Google Scholar] [CrossRef] [PubMed]
- Chesney, J.; Metz, C.; Stavitsky, A.B.; Bacher, M.; Bucala, R. Regulated production of type I collagen and inflammatory cytokines by peripheral blood fibrocytes. J. Immunol. 1998, 160, 419–425. [Google Scholar] [PubMed]
- Gomperts, B.N.; Strieter, R.M. Fibrocytes in lung disease. J. Leukoc. Biol. 2007, 82, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Yeager, M.E.; Nguyen, C.M.; Belchenko, D.D.; Colvin, K.L.; Takatsuki, S.; Ivy, D.D.; Stenmark, K.R. Circulating fibrocytes are increased in children and young adults with pulmonary hypertension. Eur. Respir. J. 2012, 39, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Huang, C.D.; Lin, H.C.; Lee, K.Y.; Lin, S.M.; Liu, C.Y.; Huang, H.; Ko, Y.S.; Chung, K.F.; Kuo, H.P. Increased circulating fibrocytes in asthma with chronic airflow obstruction. Am. J. Respir. Crit. Care Med. 2008, 178, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Medbury, H.J.; Tarran, S.L.; Guiffre, A.K.; Williams, M.M.; Lam, T.H.; Vicaretti, M.; Fletcher, J.P. Monocytes contribute to the atherosclerotic cap by transformation into fibrocytes. Int. Angiol. 2008, 27, 114–123. [Google Scholar] [CrossRef]
- Mathai, S.K.; Gulati, M.; Peng, X.; Russell, T.R.; Shaw, A.C.; Rubinowitz, A.N.; Murray, L.A.; Siner, J.M.; Antin-Ozerkis, D.E.; Montgomery, R.R.; et al. Circulating monocytes from systemic sclerosis patients with interstitial lung disease show an enhanced profibrotic phenotype. Lab. Investig. 2010, 90, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Galligan, C.L.; Fish, E.N. Circulating fibrocytes contribute to the pathogenesis of collagen antibody-induced arthritis. Arthritis Rheum. 2012, 64, 3583–3593. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Maric, I.; DiPrima, M.J.; Khan, J.; Orentas, R.J.; Kaplan, R.N.; Mackall, C.L. Fibrocytes represent a novel MDSC subset circulating in patients with metastatic cancer. Blood 2013, 122, 1105–1113. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yano, S.; Ogino, H.; Wang, W.; Uehara, H.; Nishioka, Y.; Sone, S. The therapeutic efficacy of anti-vascular endothelial growth factor antibody, bevacizumab, and pemetrexed against orthotopically implanted human pleural mesothelioma cells in severe combined immunodeficient mice. Clin. Cancer Res. 2007, 13, 5918–5925. [Google Scholar] [CrossRef] [PubMed]
- Ikuta, K.; Yano, S.; Trung, V.T.; Hanibuchi, M.; Goto, H.; Li, Q.; Wang, W.; Yamada, T.; Ogino, H.; Kakiuchi, S.; et al. E7080, a multi-tyrosine kinase inhibitor, suppresses the progression of malignant pleural mesothelioma with different proangiogenic cytokine production profiles. Clin. Cancer Res. 2009, 15, 7229–7237. [Google Scholar] [CrossRef] [PubMed]
- Welford, A.F.; Biziato, D.; Coffelt, S.B.; Nucera, S.; Fisher, M.; Pucci, F.; Di Serio, C.; Naldini, L.; de Palma, M.; Tozer, G.M.; et al. TIE2-expressing macrophages limit the therapeutic efficacy of the vascular-disrupting agent combretastatin A4 phosphate in mice. J. Clin. Investig. 2011, 121, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Ceradini, D.J.; Kulkarni, A.R.; Callaghan, M.J.; Tepper, O.M.; Bastidas, N.; Kleinman, M.E.; Capla, J.M.; Galiano, R.D.; Levine, J.P.; Gurtner, G.C. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 2004, 10, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Shinohara, S.; Hayashi, S.; Morizumi, S.; Abe, S.; Okazaki, H.; Chen, Y.; Goto, H.; Aono, Y.; Ogawa, H.; et al. Anti-fibrotic efficacy of nintedanib in pulmonary fibrosis via the inhibition of fibrocyte activity. Respir. Res. 2017, 18, 172. [Google Scholar] [CrossRef] [PubMed]
- Saijo, A.; Goto, H. Bone marrow-derived fibrocytes promote stem cell-like properties of lung cancer cells. in preparation.
- Willett, C.G.; Duda, D.G.; di Tomaso, E.; Boucher, Y.; Ancukiewicz, M.; Sahani, D.V.; Lahdenranta, J.; Chung, D.C.; Fischman, A.J.; Lauwers, G.Y.; et al. Efficacy, safety, and biomarkers of neoadjuvant bevacizumab, radiation therapy, and fluorouracil in rectal cancer: A multidisciplinary phase II study. J. Clin. Oncol. 2009, 27, 3020–3026. [Google Scholar] [CrossRef] [PubMed]
- Dowlati, A.; Gray, R.; Sandler, A.B.; Schiller, J.H.; Johnson, D.H. Cell adhesion molecules, vascular endothelial growth factor, and basic fibroblast growth factor in patients with non-small cell lung cancer treated with chemotherapy with or without bevacizumab: An Eastern Cooperative Oncology Group study. Clin. Cancer Res. 2008, 14, 1407–1412. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Hoff, P.M.; Morris, J.S.; Wolff, R.A.; Eng, C.; Glover, K.Y.; Adinin, R.; Overman, M.J.; Valero, V.; Wen, S.; et al. Phase II trial of infusional fluorouracil, irinotecan, and bevacizumab for metastatic colorectal cancer: Efficacy and circulating angiogenic biomarkers associated with therapeutic resistance. J. Clin. Oncol. 2010, 28, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Baar, J.; Silverman, P.; Lyons, J.; Fu, P.; Abdul-Karim, F.; Ziats, N.; Wasman, J.; Hartman, P.; Jesberger, J.; Dumadag, L.; et al. A vasculature-targeting regimen of preoperative docetaxel with or without bevacizumab for locally advanced breast cancer: Impact on angiogenic biomarkers. Clin. Cancer Res. 2009, 15, 3583–3590. [Google Scholar] [CrossRef] [PubMed]
- Boige, V.; Malka, D.; Bourredjem, A.; Dromain, C.; Baey, C.; Jacques, N.; Pignon, J.P.; Vimond, N.; Bouvet-Forteau, N.; de Baere, T.; et al. Efficacy, safety, and biomarkers of single-agent bevacizumab therapy in patients with advanced hepatocellular carcinoma. Oncologist 2012, 17, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Loupakis, F.; Cremolini, C.; Fioravanti, A.; Orlandi, P.; Salvatore, L.; Masi, G.; di Desidero, T.; Canu, B.; Schirripa, M.; Frumento, P.; et al. Pharmacodynamic and pharmacogenetic angiogenesis-related markers of first-line FOLFOXIRI plus bevacizumab schedule in metastatic colorectal cancer. Br. J. Cancer 2011, 104, 1262–1269. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Tumor Cell-Mediated Mechanisms | Stromal Cells Involved | Cells Possibly Involved |
|---|---|---|
| Growth factor redundancy Vascular mimicry Vessel co-option Vessel intussusception Intracellular drug sequestration Induction of stemness Endothelial cell differentiation Pericyte differentiation | Endothelial cells (including progenitor cells) TAMs (including TEMs) MDSCs CAFs Pericytes Platelets Lymphoid cells Fibrocytes | TANs Eosinophils Mast cells Dendritic cells |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goto, H.; Nishioka, Y. Fibrocytes: A Novel Stromal Cells to Regulate Resistance to Anti-Angiogenic Therapy and Cancer Progression. Int. J. Mol. Sci. 2018, 19, 98. https://doi.org/10.3390/ijms19010098
Goto H, Nishioka Y. Fibrocytes: A Novel Stromal Cells to Regulate Resistance to Anti-Angiogenic Therapy and Cancer Progression. International Journal of Molecular Sciences. 2018; 19(1):98. https://doi.org/10.3390/ijms19010098
Chicago/Turabian StyleGoto, Hisatsugu, and Yasuhiko Nishioka. 2018. "Fibrocytes: A Novel Stromal Cells to Regulate Resistance to Anti-Angiogenic Therapy and Cancer Progression" International Journal of Molecular Sciences 19, no. 1: 98. https://doi.org/10.3390/ijms19010098
APA StyleGoto, H., & Nishioka, Y. (2018). Fibrocytes: A Novel Stromal Cells to Regulate Resistance to Anti-Angiogenic Therapy and Cancer Progression. International Journal of Molecular Sciences, 19(1), 98. https://doi.org/10.3390/ijms19010098