Ubiquitylation at the Fork: Making and Breaking Chains to Complete DNA Replication


Abstract
: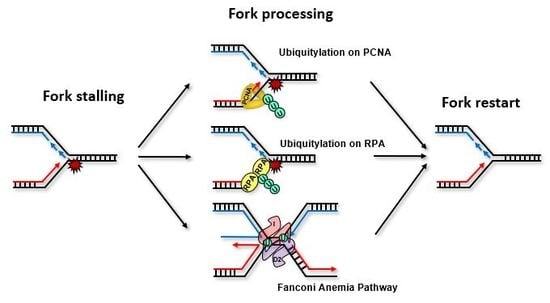
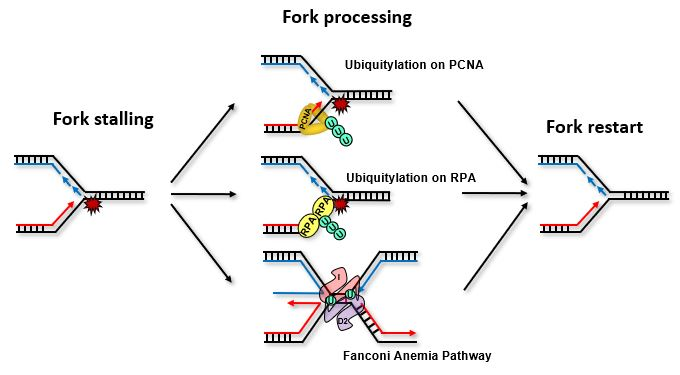{kind=link}
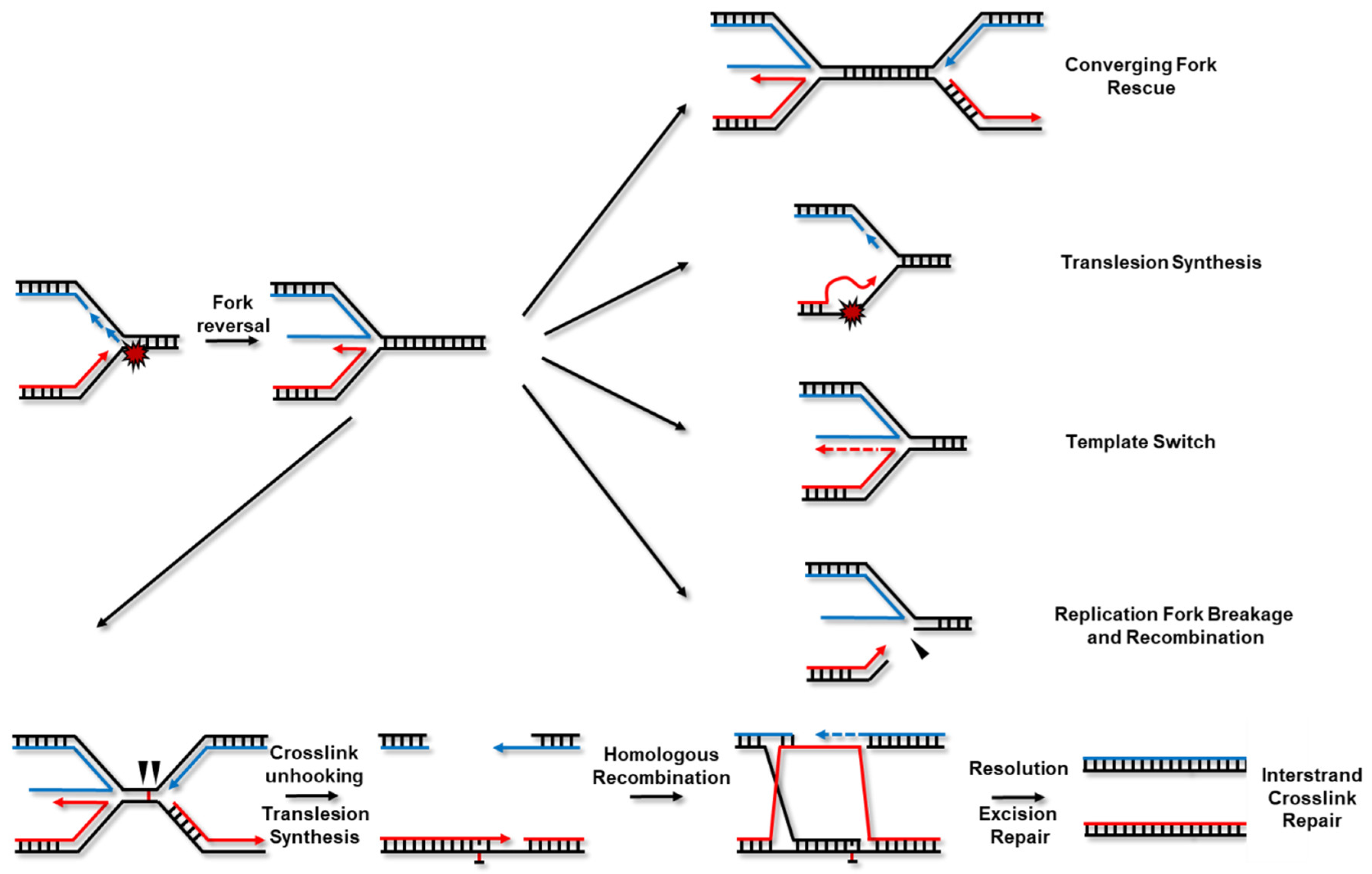{kind=link}
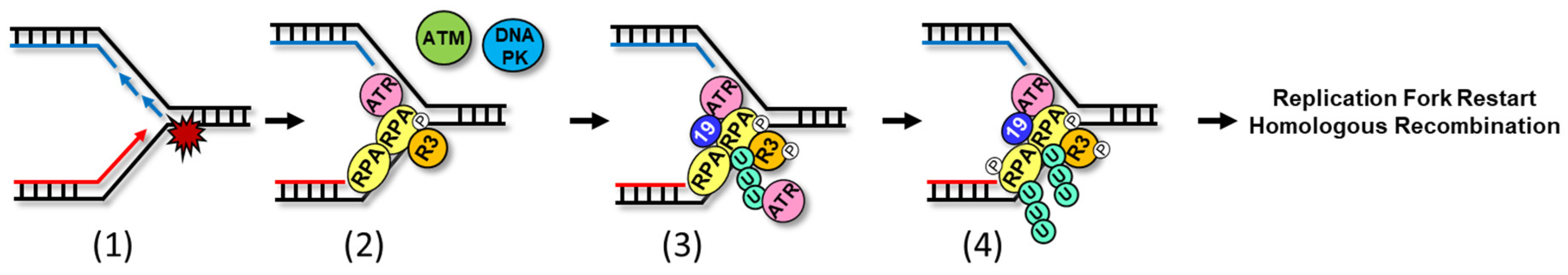{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Ubiquitylation on the RPA-ssDNA Platform
2.1. PRP19, at the Intersection of mRNA Maturation and Genome Maintenance
2.2. RFWD3, a Novel Fanconi Anemia Player on RPA-ssDNA
2.3. Potential Interplay between PRP19 and RFWD3 on RPA-ssDNA
3. Ubiquitylation on the PCNA Platform
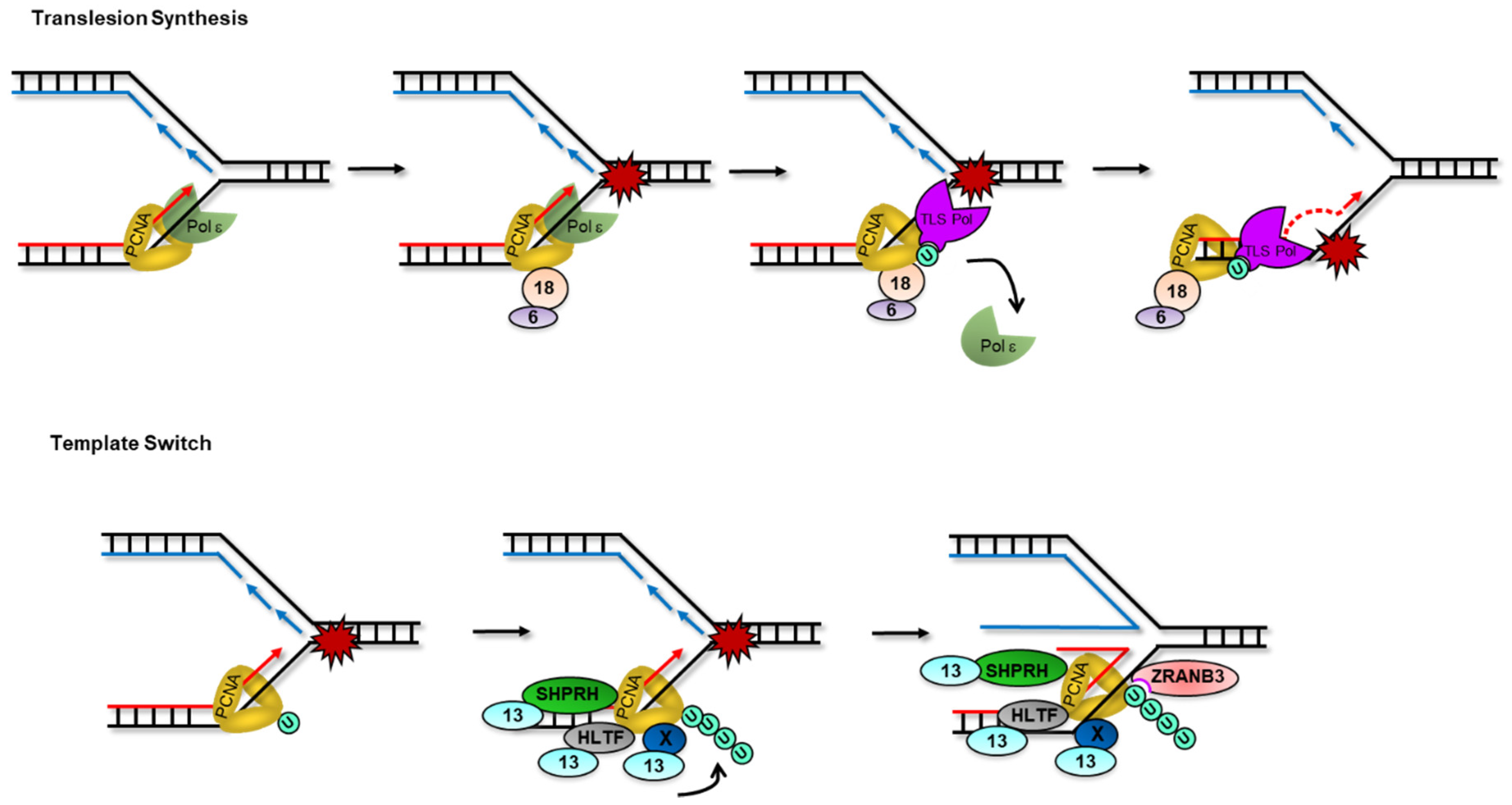
3.1. PCNA Mono-Ubiquitylation and Translesion Synthesis
3.2. PCNA Poly-Ubiquitylation and Template Switching
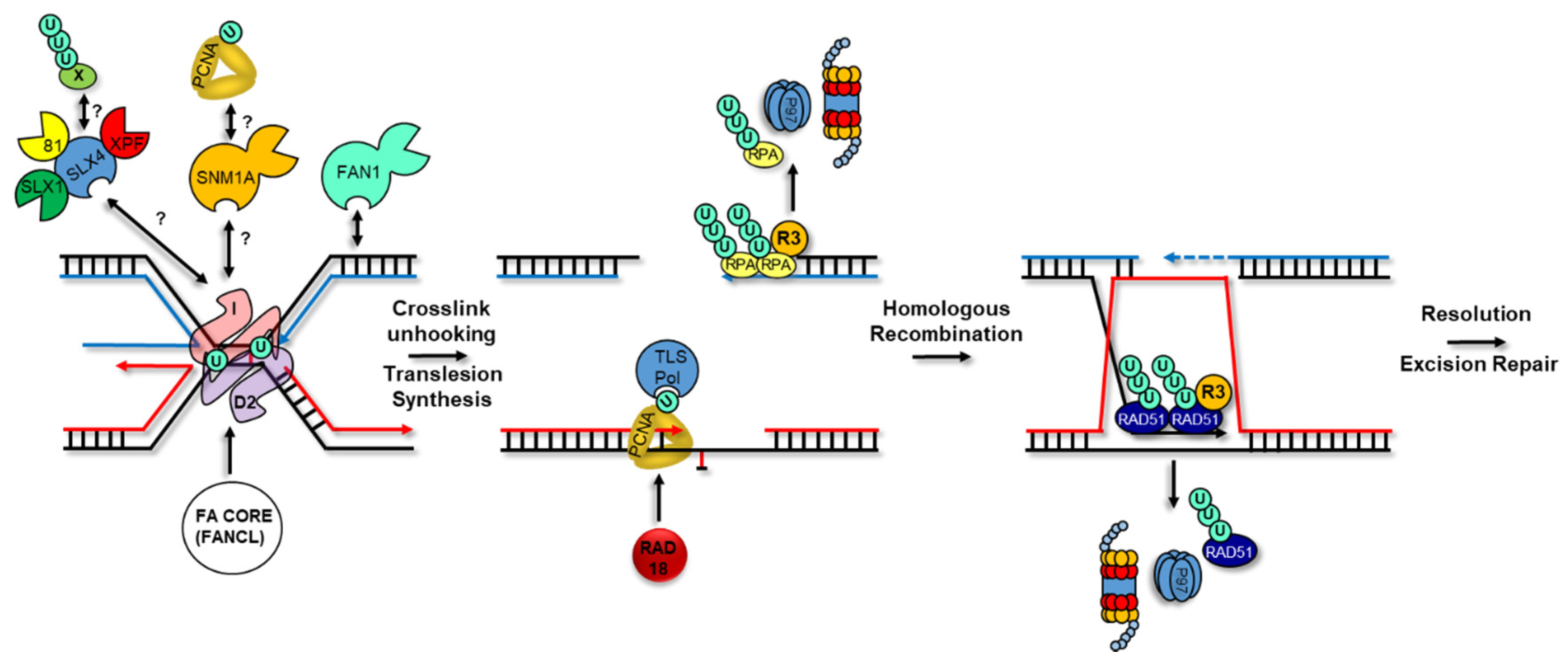
4. Ubiquitylation and the Fanconi Anemia Pathway
5. De-Ubiquitylation and Replication Stress
6. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 4-NQO | 4-Nitroquinoline |
| AAD | ATR-activating domain |
| APH | Aphidicolin |
| BRCT | BRCA1 C Terminus |
| CPD | Cyclobutane thymine-thymine dimer |
| CPT | Camptothecin |
| DCLRE1A | DNA Cross-link Repair 1A |
| DSB | Double-stranded DNA break |
| DUB | Deubiquitylating enzyme |
| ETAA1 | Ewing’s Tumor Associated Antigen 1 |
| FA | Fanconi anemia |
| HIRAN | HIP116 and RAD5 N-terminus |
| HLTF | Helicase-like transcription factor |
| HR | Homologous recombination |
| HU | Hydroxyurea |
| ICL | Inter-strand cross-link |
| IR | Irradiation |
| ISG15 | Interferon-stimulated gene 15 |
| KD | Knockdown |
| KIN | Karyomegalic interstitial nephritis |
| KO | Knockout |
| MEF | Mouse embryonic fibroblast |
| NZF | NPL4 zinf finger |
| OB | Oligonucleotide/oligosaccharide binding |
| PARI | PCNA-associated recombination inhibitor |
| PCNA | Proliferating Cell Nuclear Antigen |
| PIP | PCNA interacting-peptide |
| PTM | Post-translational modification |
| RING | Really Interesting New Gene |
| RPA | Replication Protein A |
| RSR | Replication stress response |
| SHPRH | SNF2 histone-linker PHD-finger RING-finger helicase |
| SIM | SUMO-interacting motif |
| SLD | SUMO-like domain |
| SNM1A | Sensitive to Nitrogen Mustard 1A |
| STUbL | SUMO-targeted ubiquitin ligase |
| SUMO | Small ubiquitin-like modifier |
| TLS | Translesion synthesis |
| TOPBP1 | Topoisomerase II Binding protein 1 |
| TS | Template switch |
| UBM | Ubiquitin-binding motif |
| UBZ | Ubiquitin-binding zinc finger |
| USP1 | Ubiquitin specific protease 1 |
| USP10 | Ubiquitin specific protease 10 |
| VCP | Vasolin-containing protein |
| WRNIP1 | WRN-interacting protein 1 |
| ZRANB3 | Zinc finger RAN-binding domain containing 3 |
| ZUFSP | Zinc finger with UFM1-specific peptidase domain protein |
References
- Bianconi, E.; Piovesan, A.; Facchin, F.; Beraudi, A.; Casadei, R.; Frabetti, F.; Vitale, L.; Pelleri, M.C.; Tassani, S.; Piva, F.; et al. An estimation of the number of cells in the human body. Ann. Hum. Biol. 2013, 40, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K. Causes and consequences of replication stress. Nat. Cell Biol. 2013, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Kotsantis, P.; Petermann, E.; Boulton, S.J. Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling into Place. Cancer Discov. 2018, 1. [Google Scholar] [CrossRef] [PubMed]
- Zellweger, R.; Dalcher, D.; Mutreja, K.; Berti, M.; Schmid, J.A.; Herrador, R.; Vindigni, A.; Lopes, M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 2015, 208, 563–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhuri, A.R.; Hashimoto, Y.; Herrador, R.; Neelsen, K.J.; Fachinetti, D.; Bermejo, R.; Cocito, A.; Costanzo, V.; Lopes, M. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat. Struct. Mol. Biol. 2012, 19, 417–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neelsen, K.J.; Lopes, M. Replication fork reversal in eukaryotes: From dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 2015, 9. [Google Scholar] [CrossRef]
- Atkinson, J.; McGlynn, P. Replication fork reversal and the maintenance of genome stability. Nucleic Acids Res. 2009, 37, 3475–3492. [Google Scholar] [CrossRef] [Green Version]
- Quinet, A.; Lemaçon, D.; Vindigni, A. Replication Fork Reversal: Players and Guardians. Mol. Cell 2017, 68, 830–833. [Google Scholar] [CrossRef]
- Neelsen, K.J.; Chaudhuri, A.R.; Follonier, C.; Herrador, R.; Lopes, M. Visualization and Interpretation of Eukaryotic DNA Replication Intermediates In Vivo by Electron Microscopy. In Functional Analysis of DNA and Chromatin; Stockert, J.C., Espada, J., Blázquez-Castro, A., Eds.; Humana Press: Totowa, NJ, USA, 2014; pp. 177–208. ISBN 978-1-62703-706-8. [Google Scholar]
- Duxin, J.P.; Walter, J.C. What is the DNA repair defect underlying Fanconi anemia? Curr. Opin. Cell Biol. 2015, 37, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Kottemann, M.C.; Smogorzewska, A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 2013, 493, 356–363. [Google Scholar] [CrossRef]
- Che, R.; Zhang, J.; Nepal, M.; Han, B.; Fei, P. Multifaceted Fanconi Anemia Signaling. Trends Genet. 2017, 34, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Kramara, J.; Osia, B.; Malkova, A. Break-induced replication: An unhealthy choice for stress relief? Nat. Struct. Mol. Biol. 2017, 24, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Berti, M.; Vindigni, A. Replication stress: Getting back on track. Nat. Struct. Mol. Biol. 2016, 23, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Wold, M.S. Replication protein A: A heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 1997, 66, 61–92. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.L.; Zou, L. Oligonucleotide/oligosaccharide-binding fold proteins: A growing family of genome guardians. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Tkáč, J.; Xu, G.; Adhikary, H.; Young, J.T.F.; Gallo, D.; Escribano-Díaz, C.; Krietsch, J.; Orthwein, A.; Munro, M.; Sol, W.; et al. HELB Is a Feedback Inhibitor of DNA End Resection. Mol. Cell 2016, 61, 405–418. [Google Scholar] [CrossRef]
- Fanning, E.; Klimovich, V.; Nager, A.R. A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res. 2006, 34, 4126–4137. [Google Scholar] [CrossRef] [Green Version]
- Wan, L.; Lou, J.; Xia, Y.; Su, B.; Liu, T.; Cui, J.; Sun, Y.; Lou, H.; Huang, J. hPrimpol1/CCDC111 is a human DNA primase-polymerase required for the maintenance of genome integrity. EMBO Rep. 2013, 14, 1104–1112. [Google Scholar] [CrossRef] [Green Version]
- Maréchal, A.; Li, J.M.; Ji, X.Y.; Wu, C.S.; Yazinski, S.A.; Nguyen, H.D.; Liu, S.; Jiménez, A.E.; Jin, J.; Zou, L. PRP19 Transforms into a Sensor of RPA-ssDNA after DNA Damage and Drives ATR Activation via a Ubiquitin-Mediated Circuitry. Mol. Cell 2014, 53, 235–246. [Google Scholar] [CrossRef]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Yazinski, S.A.; Zou, L. Functions, Regulation, and Therapeutic Implications of the ATR Checkpoint Pathway. Annu. Rev. Genet. 2016, 50, 155–173. [Google Scholar] [CrossRef] [PubMed]
- Byun, T.S.; Pacek, M.; Yee, M.; Walter, J.C.; Cimprich, K.A. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005, 19, 1040–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, M.; Foiani, M.; Sogo, J.M. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol. Cell 2006, 21, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Shiotani, B.; Zou, L. Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol. Cell 2009, 33, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef]
- Cortez, D.; Guntuku, S.; Qin, J.; Elledge, S.J. ATR and ATRIP: Partners in checkpoint signaling. Science 2001, 294, 1713–1716. [Google Scholar] [CrossRef]
- Kumagai, A.; Lee, J.; Yoo, H.Y.; Dunphy, W.G. TopBP1 activates the ATR-ATRIP complex. Cell 2006, 124, 943–955. [Google Scholar] [CrossRef]
- Lee, Y.C.; Zhou, Q.; Chen, J.; Yuan, J. RPA-Binding Protein ETAA1 Is an ATR Activator Involved in DNA Replication Stress Response. Curr. Biol. 2016, 26, 3257–3268. [Google Scholar] [CrossRef] [Green Version]
- Bass, T.E.; Luzwick, J.W.; Kavanaugh, G.; Carroll, C.; Dungrawala, H.; Glick, G.G.; Feldkamp, M.D.; Putney, R.; Chazin, W.J.; Cortez, D. ETAA1 acts at stalled replication forks to maintain genome integrity. Nat. Cell Biol. 2016, 18, 1185–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haahr, P.; Hoffmann, S.; Tollenaere, M.A.X.; Ho, T.; Toledo, L.I.; Mann, M.; Bekker-Jensen, S.; Räschle, M.; Mailand, N. Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat. Cell Biol. 2016, 18, 1196–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Shiotani, B.; Lahiri, M.; Maréchal, A.; Tse, A.; Leung, C.C.Y.; Glover, J.; Yang, X.H.; Zou, L. ATR Autophosphorylation as a Molecular Switch for Checkpoint Activation. Mol. Cell 2011, 43, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Nam, E.A.; Zhao, R.; Glick, G.G.; Bansbach, C.E.; Friedman, D.B.; Cortez, D. T1989 phosphorylation is a marker of active ataxia telangiectasia-mutated and rad3-related (ATR) kinase. J. Biol. Chem. 2011, 286, 28707–28714. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.A.; Oehler, H.; Voigt, A.; Dalic, D.; Freiwald, A.; Serve, H.; Beli, P. ATR inhibition rewires cellular signaling networks induced by replication stress. Proteomics 2016, 16, 402–416. [Google Scholar] [CrossRef] [PubMed]
- Stokes, M.P.; Rush, J.; Macneill, J.; Ren, J.M.; Sprott, K.; Nardone, J.; Yang, V.; Beausoleil, S.A.; Gygi, S.P.; Livingstone, M.; et al. Profiling of UV-induced ATM/ATR signaling pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 19855–19860. [Google Scholar] [CrossRef] [Green Version]
- Moldovan, G.L.; Pfander, B.; Jentsch, S. PCNA, the Maestro of the Replication Fork. Cell 2007, 129, 665–679. [Google Scholar] [CrossRef] [Green Version]
- Choe, K.N.; Moldovan, G.L. Forging Ahead through Darkness: PCNA, Still the Principal Conductor at the Replication Fork. Mol. Cell 2017, 65, 380–392. [Google Scholar] [CrossRef] [Green Version]
- Prelich, G.; Tan, C.K.; Kostura, M.; Mathews, M.B.; So, A.G.; Downey, K.M.; Stillman, B. Functional identity of proliferating cell nuclear antigen and a DNA polymerase-δ auxiliary protein. Nature 1987, 326, 517–520. [Google Scholar] [CrossRef]
- O’Donnell, M.; Langston, L.; Stillman, B. Principles and concepts of DNA replication in bacteria, archaea, and eukarya. Cold Spring Harb. Perspect. Biol. 2013, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, E.; Tsurimoto, T. Functions of Multiple Clamp and Clamp-Loader Complexes in Eukaryotic DNA Replication. In DNA Replication: From Old Principles to New Discoveries; Masai, H., Foiani, M., Eds.; Springer: Singapore, 2017; pp. 135–162. ISBN 978-981-10-6955-0. [Google Scholar]
- Mailand, N.; Gibbs-Seymour, I.; Bekker-Jensen, S. Regulation of PCNA-protein interactions for genome stability. Nat. Rev. Mol. Cell Biol. 2013, 14, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Indiani, C.; McInerney, P.; Georgescu, R.; Goodman, M.F.; O’Donnell, M. A sliding-clamp toolbelt binds high- and low-fidelity DNA polymerases simultaneously. Mol. Cell 2005, 19, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Dovrat, D.; Stodola, J.L.; Burgers, P.M.J.; Aharoni, A. Sequential switching of binding partners on PCNA during in vitro Okazaki fragment maturation. Proc. Natl. Acad. Sci. USA 2014, 111, 14118–14123. [Google Scholar] [CrossRef] [PubMed]
- Stodola, J.L.; Burgers, P.M. Resolving individual steps of Okazaki-fragment maturation at a millisecond timescale. Nat. Struct. Mol. Biol. 2016, 23, 402–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Gao, Y. Translesion and Repair DNA Polymerases: Diverse Structure and Mechanism. Annu. Rev. Biochem. 2018, 87, 239–261. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Psakhye, I. DNA damage tolerance. Curr. Opin. Cell Biol. 2016, 40, 137–144. [Google Scholar] [CrossRef]
- Schwab, R.A.; Nieminuszczy, J.; Shah, F.; Langton, J.; Lopez Martinez, D.; Liang, C.C.; Cohn, M.A.; Gibbons, R.J.; Deans, A.J.; Niedzwiedz, W. The Fanconi Anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription. Mol. Cell 2015, 60, 351–361. [Google Scholar] [CrossRef]
- García-Rubio, M.L.; Pérez-Calero, C.; Barroso, S.I.; Tumini, E.; Herrera-Moyano, E.; Rosado, I.V.; Aguilera, A. The Fanconi Anemia Pathway Protects Genome Integrity from R-loops. PLoS Genet. 2015, 11, e1005674. [Google Scholar] [CrossRef] [PubMed]
- Michl, J.; Zimmer, J.; Buffa, F.M.; McDermott, U.; Tarsounas, M. FANCD2 limits replication stress and genome instability in cells lacking BRCA2. Nat. Struct. Mol. Biol. 2016, 23, 755–757. [Google Scholar] [CrossRef] [Green Version]
- Bosch, P.C.; Segura-Bayona, S.; Koole, W.; van Heteren, J.T.; Dewar, J.M.; Tijsterman, M.; Knipscheer, P. FANCJ promotes DNA synthesis through G-quadruplex structures. EMBO J. 2014, 33, 2521–2533. [Google Scholar] [CrossRef] [Green Version]
- Sobinoff, A.P.; Allen, J.A.M.; Neumann, A.A.; Yang, S.F.; Walsh, M.E.; Henson, J.D.; Reddel, R.R.; Pickett, H.A. BLM and SLX 4 play opposing roles in recombination-dependent replication at human telomeres. EMBO J. 2017, 36, 2907–2919. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, A.; Zou, L. RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res. 2015, 25, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Oakley, G.G.; Patrick, S.M. Replication protein A: Directing traffic at the intersection of replication and repair. Front. Biosci. (Landmark Ed.) 2010, 15, 883–900. [Google Scholar] [CrossRef] [PubMed]
- Oakley, G.G.; Tillison, K.; Opiyo, S.A.; Glanzer, J.G.; Horn, J.M.; Patrick, S.M. Physical interaction between replication protein A (RPA) and MRN: Involvement of RPA2 phosphorylation and the N-terminus of RPA1. Biochemistry 2009, 48, 7473–7481. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.K.; Fitzgerald, M.; Ro, T.; Kim, J.H.; Rabinowitsch, A.I.; Chowdhury, D.; Schildkraut, C.L.; Borowiec, J.A. Phosphorylated RPA recruits PALB2 to stalled DNA replication forks to facilitate fork recovery. J. Cell Biol. 2014, 206, 493–507. [Google Scholar] [CrossRef] [Green Version]
- Dubois, J.C.; Yates, M.; Gaudreau-Lapierre, A.; Clément, G.; Cappadocia, L.; Gaudreau, L.; Zou, L.; Maréchal, A. A phosphorylation-and-ubiquitylation circuitry driving ATR activation and homologous recombination. Nucleic Acids Res. 2017, 45, 8859–8872. [Google Scholar] [CrossRef] [PubMed]
- Elia, A.E.H.; Boardman, A.P.; Wang, D.C.; Koren, I.; Gygi, S.P.; Elledge, S.J.; Elia, A.E.H.; Boardman, A.P.; Wang, D.C.; Huttlin, E.L.; et al. Quantitative Proteomic Atlas of Ubiquitination and Acetylation in the DNA Damage Response Resource. Mol. Cell 2015, 59, 867–881. [Google Scholar] [CrossRef] [PubMed]
- Povlsen, L.K.; Beli, P.; Wagner, S.A.; Poulsen, S.L.; Sylvestersen, K.B.; Poulsen, J.W.; Nielsen, M.L.; Bekker-Jensen, S.; Mailand, N.; Choudhary, C. Systems-wide analysis of ubiquitylation dynamics reveals a key role for PAF15 ubiquitylation in DNA-damage bypass. Nat. Cell Biol. 2012, 14, 1089–1098. [Google Scholar] [CrossRef]
- Dungrawala, H.; Rose, K.L.; Bhat, K.P.; Glick, G.G.; Couch, F.B.; Cortez, D.; Dungrawala, H.; Rose, K.L.; Bhat, K.P.; Mohni, K.N.; et al. The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Mol. Cell 2015, 59, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Durocher, D. Regulation of DNA Damage Responses by Ubiquitin and SUMO. Mol. Cell 2013, 49, 795–807. [Google Scholar] [CrossRef] [PubMed]
- García-Rodríguez, N.; Wong, R.P.; Ulrich, H.D. Functions of Ubiquitin and SUMO in DNA Replication and Replication Stress. Front. Genet. 2016, 7, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Schwertman, P.; Bekker-Jensen, S.; Mailand, N. Regulation of DNA double-strand break repair by ubiquitin and ubiquitin-like modifiers. Nat. Rev. Mol. Cell Biol. 2016, 17, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Jentsch, S.; Müller, S. Regulatory Functions of Ubiquitin and SUMO in DNA Repair Pathways. In Conjugation and Deconjugation of Ubiquitin Family Modifiers: Subcellular Biochemistry; Groettrup, M., Ed.; Springer: New York, NY, USA, 2010; pp. 184–194. ISBN 978-1-4419-6676-6. [Google Scholar]
- Elia, A.E.H.; Wang, D.C.; Willis, N.A.; Gygi, S.P.; Scully, R.; Elledge, S.J. RFWD3-Dependent Ubiquitination of RPA Regulates Repair at Stalled Replication Forks. Mol. Cell 2015, 60, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Inano, S.; Sato, K.; Katsuki, Y.; Kobayashi, W.; Tanaka, H.; Nakajima, K.; Nakada, S.; Miyoshi, H.; Knies, K.; Takaori-Kondo, A.; et al. RFWD3-Mediated Ubiquitination Promotes Timely Removal of Both RPA and RAD51 from DNA Damage Sites to Facilitate Homologous Recombination. Mol. Cell 2017, 66, 622–634. [Google Scholar] [CrossRef] [PubMed]
- De Moura, T.R.; Mozaffari-Jovin, S.; Szabó, C.Z.K.; Schmitzová, J.; Dybkov, O.; Cretu, C.; Kachala, M.; Svergun, D.; Urlaub, H.; Lührmann, R.; et al. Prp19/Pso4 Is an Autoinhibited Ubiquitin Ligase Activated by Stepwise Assembly of Three Splicing Factors. Mol. Cell 2018, 69, 979–992. [Google Scholar] [CrossRef] [PubMed]
- Wahl, M.C.; Will, C.L.; Lührmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef]
- Hatakeyama, S.; Yada, M.; Matsumoto, M.; Ishida, N.; Nakayama, K.I. U box proteins as a new family of ubiquitin-protein ligases. J. Biol. Chem. 2001, 276, 33111–33120. [Google Scholar] [CrossRef] [PubMed]
- Ohi, M.D.; Vander Kooi, C.W.; Rosenberg, J.A.; Ren, L.; Hirsch, J.P.; Chazin, W.J.; Walz, T.; Gould, K.L. Structural and Functional Analysis of Essential pre-mRNA Splicing Factor Prp19p. Mol. Cell Biol. 2005, 25, 451–460. [Google Scholar] [CrossRef]
- Fortschegger, K.; Wagner, B.; Voglauer, R.; Katinger, H.; Sibilia, M.; Grillari, J. Early embryonic lethality of mice lacking the essential protein SNEV. Mol. Cell Biol. 2007, 27, 3123–3130. [Google Scholar] [CrossRef]
- Grote, M.; Wolf, E.; Will, C.L.; Lemm, I.; Agafonov, D.E.; Schomburg, A.; Fischle, W.; Urlaub, H.; Lührmann, R. Molecular architecture of the human Prp19/CDC5L complex. Mol. Cell Biol. 2010, 30, 2105–2119. [Google Scholar] [CrossRef] [PubMed]
- Vander Kooi, C.W.; Ren, L.; Xu, P.; Ohi, M.D.; Gould, K.L.; Chazin, W.J. The Prp19 WD40 domain contains a conserved protein interaction region essential for its function. Structure 2010, 18, 584–593. [Google Scholar] [CrossRef]
- Ohi, M.D.; Vander Kooi, C.W.; Rosenberg, J.A.; Chazin, W.J.; Gould, K.L. Structural insights into the U-box, a domain associated with multi-ubiquitination. Nat. Struct. Biol. 2003, 10, 250–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijayraghavan, U.; Company, M.; Abelson, J. Isolation and characterization of pre-mRNA splicing mutants of Saccharomyces cerevisiae. Genes Dev. 1989, 3, 1206–1216. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.C.; Tarn, W.Y.; Tsao, T.Y.; Abelson, J. PRP19: A novel spliceosomal component. Mol. Cell Biol. 1993, 13, 1876–1882. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.-P.; Kao, D.-I.; Tsai, W.-Y.; Cheng, S.-C. The Prp19p-associated complex in spliceosome activation. Science 2003, 302, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Song, E.J.; Werner, S.L.; Neubauer, J.; Stegmeier, F.; Aspden, J.; Rio, D.; Harper, J.W.; Elledge, S.J.; Kirschner, M.W.; Rape, M. The Prp19 complex and the Usp4Sart3 deubiquitinating enzyme control reversible ubiquitination at the spliceosome. Genes Dev. 2010, 24, 1434–1447. [Google Scholar] [CrossRef] [Green Version]
- Grey, M.; Düsterhöft, A.; Henriques, J.A.; Brendel, M. Allelism of PSO4 and PRP19 links pre-mRNA processing with recombination and error-prone DNA repair in Saccharomyces cerevisiae. Nucleic Acids Res. 1996, 24, 4009–4014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Kaur, R.; Lu, X.; Shen, X.; Li, L.; Legerski, R.J. The Pso4 mRNA splicing and DNA repair complex interacts with WRN for processing of DNA interstrand cross-links. J. Biol. Chem. 2005, 280, 40559–40567. [Google Scholar] [CrossRef]
- Voglauer, R.; Chang, M.W.-F.; Dampier, B.; Wieser, M.; Baumann, K.; Sterovsky, T.; Schreiber, M.; Katinger, H.; Grillari, J. SNEV overexpression extends the life span of human endothelial cells. Exp. Cell Res. 2006, 312, 746–759. [Google Scholar] [CrossRef]
- Zhang, N.; Kaur, R.; Akhter, S.; Legerski, R.J. Cdc5L interacts with ATR and is required for the S-phase cell-cycle checkpoint. EMBO Rep. 2009, 10, 1029–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteforte, R.; Beilhack, G.F.; Grausenburger, R.; Mayerhofer, B.; Bittner, R.; Grillari-Voglauer, R.; Sibilia, M.; Dellago, H.; Tschachler, E.; Gruber, F.; et al. SNEVPrp19/PSO4deficiency increases PUVA-induced senescence in mouse skin. Exp. Dermatol. 2016, 25, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Huang, J. The PSO4 Complex Associates with RPA and Modulates the Activation of ATR. J. Biol. Chem. 2014, 289, 6619–6626. [Google Scholar] [CrossRef] [PubMed]
- Onyango, D.O.; Howard, S.M.; Neherin, K.; Yanez, D.A.; Stark, J.M. Tetratricopeptide repeat factor XAB2 mediates the end resection step of homologous recombination. Nucleic Acids Res. 2016, 44, 5702–5716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onyango, D.O.; Lee, G.; Stark, J.M. PRPF8 is important for BRCA1-mediated homologous recombination. Oncotarget 2017, 8, 93319–93337. [Google Scholar] [CrossRef] [PubMed]
- Abbas, M.; Shanmugam, I.; Bsaili, M.; Hromas, R.; Shaheen, M. The role of the human Psoralen 4 (hPso4) complex in replication stress and homologous recombination. J. Biol. Chem. 2014, 289, 14009–14019. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Yucer, N.; Liu, S.; Li, M.; Yi, P.; Mu, J.-J.; Yang, T.; Chu, J.; Jung, S.Y.; O’Malley, B.W.; et al. RFWD3-Mdm2 ubiquitin ligase complex positively regulates p53 stability in response to DNA damage. Proc. Natl. Acad. Sci. USA 2010, 107, 4579–4584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Z.; Chen, J. E3 ligase RFWD3 participates in replication checkpoint control. J. Biol. Chem. 2011, 286, 22308–22313. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Chu, J.; Yucer, N.; Leng, M.; Wang, S.-Y.; Chen, B.P.C.; Hittelman, W.N.; Wang, Y. RING finger and WD repeat domain 3 (RFWD3) associates with replication protein A (RPA) and facilitates RPA-mediated DNA damage response. J. Biol. Chem. 2011, 286, 22314–22322. [Google Scholar] [CrossRef]
- Feeney, L.; Muñoz, I.M.; Lachaud, C.; Toth, R.; Appleton, P.L.; Schindler, D.; Rouse, J. RPA-Mediated Recruitment of the E3 Ligase RFWD3 Is Vital for Interstrand Crosslink Repair and Human Health. Mol. Cell 2017, 66, 610–621. [Google Scholar] [CrossRef]
- Knies, K.; Inano, S.; Ramírez, M.J.; Ishiai, M.; Surrallés, J.; Takata, M.; Schindler, D. Biallelic mutations in the ubiquitin ligase RFWD3 cause Fanconi anemia. J. Clin. Investig. 2017, 127, 3013–3027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binz, S.K.; Sheehan, A.M.; Wold, M.S. Replication protein A phosphorylation and the cellular response to DNA damage. DNA Repair 2004, 3, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Hewings, D.S.; Heideker, J.; Ma, T.P.; AhYoung, A.P.; El Oualid, F.; Amore, A.; Costakes, G.T.; Kirchhofer, D.; Brasher, B.; Pillow, T.; et al. Reactive-site-centric chemoproteomics identifies a distinct class of deubiquitinase enzymes. Nat. Commun. 2018, 9, 1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husnjak, K.; Dikic, I. Ubiquitin-Binding Proteins: Decoders of Ubiquitin-Mediated Cellular Functions. Annu. Rev. Biochem. 2012, 81, 291–322. [Google Scholar] [CrossRef] [PubMed]
- Dou, H.; Huang, C.; Singh, M.; Carpenter, P.B.; Yeh, E.T.H. Regulation of DNA repair through deSUMOylation and SUMOylation of replication protein A complex. Mol. Cell 2010, 39, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Galanty, Y.; Belotserkovskaya, R.; Coates, J.; Jackson, S.P. RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes DNA double-strand break repair. Genes Dev. 2012, 26, 1179–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, S.; Niimi, A.; Lehmann, A.R. Ubiquitination and deubiquitination of PCNA in response to stalling of the replication fork. Cell Cycle 2009, 8, 689–692. [Google Scholar] [CrossRef] [Green Version]
- Niimi, A.; Brown, S.; Sabbioneda, S.; Kannouche, P.L.; Scott, A.; Yasui, A.; Green, C.M.; Lehmann, A.R. Regulation of proliferating cell nuclear antigen ubiquitination in mammalian cells. Proc. Natl. Acad. Sci. USA 2008, 105, 16125–16130. [Google Scholar] [CrossRef] [Green Version]
- Hoege, C.; Pfander, B.; Moldovan, G.L.; Pyrowolakis, G.; Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002, 419, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.L.; Ulrich, H.D. Mechanistic analysis of PCNA poly-ubiquitylation by the ubiquitin protein ligases Rad18 and Rad5. EMBO J. 2009, 28, 3657–3666. [Google Scholar] [CrossRef] [Green Version]
- Masuda, Y.; Suzuki, M.; Kawai, H.; Hishiki, A.; Hashimoto, H.; Masutani, C.; Hishida, T.; Suzuki, F.; Kamiya, K. En bloc transfer of polyubiquitin chains to PCNA in vitro is mediated by two different human E2-E3 pairs. Nucleic Acids Res. 2012, 40, 10394–10407. [Google Scholar] [CrossRef]
- Stelter, P.; Ulrich, H.D. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature 2003, 425, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Tateishi, S.; Kawasuji, M.; Tsurimoto, T.; Inoue, H.; Yamaizumi, M. Rad18 guides polη to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 2004, 23, 3886–3896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blastyák, A.; Pintér, L.; Unk, I.; Prakash, L.; Prakash, S.; Haracska, L. Yeast Rad5 Protein Required for Postreplication Repair Has a DNA Helicase Activity Specific for Replication Fork Regression. Mol. Cell 2007, 28, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Unk, I.; Hajdú, I.; Blastyák, A.; Haracska, L. Role of yeast Rad5 and its human orthologs, HLTF and SHPRH in DNA damage tolerance. DNA Repair 2010, 9, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Gali, H.; Juhasz, S.; Morocz, M.; Hajdu, I.; Fatyol, K.; Szukacsov, V.; Burkovics, P.; Haracska, L. Role of SUMO modification of human PCNA at stalled replication fork. Nucleic Acids Res. 2012, 40, 6049–6059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, J.L.; Bucceri, A.; Davies, A.A.; Heidrich, K.; Windecker, H.; Ulrich, H.D. SUMO modification of PCNA is controlled by DNA. EMBO J. 2008, 27, 2422–2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terai, K.; Abbas, T.; Jazaeri, A.A.; Dutta, A. CRL4Cdt2E3 Ubiquitin Ligase Monoubiquitinates PCNA to Promote Translesion DNA Synthesis. Mol. Cell 2010, 37, 143–149. [Google Scholar] [CrossRef]
- Han, J.; Liu, T.; Huen, M.S.Y.; Hu, L.; Chen, Z.; Huang, J. SIVA1 directs the E3 ubiquitin ligase RAD18 for PCNA monoubiquitination. J. Cell Biol. 2014, 205, 811–827. [Google Scholar] [CrossRef] [Green Version]
- Das-Bradoo, S.; Nguyen, H.D.; Wood, J.L.; Ricke, R.M.; Haworth, J.C.; Bielinsky, A.K. Defects in DNA ligase i trigger PCNA ubiquitylation at Lys 107. Nat. Cell Biol. 2010, 12, 74–79. [Google Scholar] [CrossRef]
- Da Nguyen, H.; Becker, J.; Thu, Y.M.; Costanzo, M.; Koch, E.N.; Smith, S.; Myung, K.; Myers, C.L.; Boone, C.; Bielinsky, A.K. Unligated Okazaki Fragments Induce PCNA Ubiquitination and a Requirement for Rad59-Dependent Replication Fork Progression. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.A.; Huttner, D.; Daigaku, Y.; Chen, S.; Ulrich, H.D. Activation of Ubiquitin-Dependent DNA Damage Bypass Is Mediated by Replication Protein A. Mol. Cell 2008, 29, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, H.D.; Walden, H. Ubiquitin signalling in DNA replication and repair. Nat. Rev. Mol. Cell Biol. 2010, 11, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Boehm, E.M.; Spies, M.; Washington, M.T. PCNA tool belts and polymerase bridges form during translesion synthesis. Nucleic Acids Res. 2016, 44, 8250–8260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, J.G.; Tsaalbi-Shtylik, A.; de Wind, N. Roles of mutagenic translesion synthesis in mammalian genome stability, health and disease. DNA Repair 2015, 29, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Vaisman, A.; Woodgate, R. Translesion DNA polymerases in eukaryotes: What makes them tick? Crit. Rev. Biochem. Mol. Biol. 2017, 52, 274–303. [Google Scholar] [CrossRef] [PubMed]
- Makarova, A.V.; Stodola, J.L.; Burgers, P.M. A four-subunit DNA polymerase ζ complex containing Pol δ accessory subunits is essential for PCNA-mediated mutagenesis. Nucleic Acids Res. 2012, 40, 11618–11626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.E.; Prakash, L.; Prakash, S. Pol31 and Pol32 subunits of yeast DNA polymerase are also essential subunits of DNA polymerase ζ. Proc. Natl. Acad. Sci. USA 2012, 109, 12455–12460. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-S.; Gregory, M.T.; Yang, W. Human Pol ζ purified with accessory subunits is active in translesion DNA synthesis and complements Pol η in cisplatin bypass. Proc. Natl. Acad. Sci. USA 2014, 111, 2954–2959. [Google Scholar] [CrossRef] [PubMed]
- Baranovskiy, A.G.; Lada, A.G.; Siebler, H.M.; Zhang, Y.; Pavlov, Y.I.; Tahirov, T.H. DNA polymerase delta and zeta switch by sharing accessory subunits of DNA polymerase delta. J. Biol. Chem. 2012, 287, 17281–17287. [Google Scholar] [CrossRef]
- Sale, J.E. Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Prakash, S.; Johnson, R.E.; Prakash, L. Eukaryotic Translesion Synthesis DNA Polymerases: Specificity of Structure and Function. Annu. Rev. Biochem. 2005, 74, 317–353. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M.F.; Woodgate, R. Translesion DNA Polymerases. Cold Spring Harb. Perspect. Biol. 2013, 5, a010363. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Aggarwal, A.K.; Rechkoblit, O. Eukaryotic DNA polymerases. Curr. Opin. Struct. Biol. 2018, 53, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.E.; Washington, M.T.; Prakash, S.; Prakash, L. Fidelity of human DNA polymerase η. J. Biol. Chem. 2000, 275, 7447–7450. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.E.; Haracska, L.; Prakash, S.; Prakash, L. Role of DNA Polymerase η in the Bypass of a (6-4) TT Photoproduct. Mol. Cell. Biol. 2001, 21, 1–7. [Google Scholar] [CrossRef]
- Kannouche, P.L.; Wing, J.; Lehmann, A.R. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: A possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell 2004, 14, 491–500. [Google Scholar] [CrossRef]
- Johnson, R.E.; Prakash, S.; Prakash, L. Efficient bypass of a thymine-thymine dimer by yeast DNA polymerase, Polη. Science 1999, 283, 1001–1004. [Google Scholar] [CrossRef]
- Masutani, C.; Kusumoto, R.; Yamada, A.; Dohmae, N.; Yokoi, M.; Yuasa, M.; Araki, M.; Iwai, S.; Takio, K.; Hanaoka, F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature 1999, 399, 700–704. [Google Scholar] [CrossRef]
- Johnson, R.E.; Kondratick, C.M.; Prakash, S.; Prakash, L. hRAD30 mutations in the variant form of xeroderma pigmentosum. Science 1999, 285, 263–265. [Google Scholar] [CrossRef]
- Kannouche, P.; Stary, A. Xeroderma pigmentosum variant and error-prone DNA polymerases. Biochimie 2003, 85, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Vidal, A.E.; Kannouche, P.; Podust, V.N.; Yang, W.; Lehmann, A.R.; Woodgate, R. Proliferating cell nuclear antigen-dependent coordination of the biological functions of human DNA polymerase iota. J. Biol. Chem. 2004, 279, 48360–48368. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.-H.; Acharya, N.; Park, J.; Basu, D.; Prakash, S.; Prakash, L. Identification of two functional PCNA-binding domains in human DNA polymerase κ. Genes Cells 2014, 19, 594–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharya, N.; Yoon, J.-H.; Gali, H.; Unk, I.; Haracska, L.; Johnson, R.E.; Hurwitz, J.; Prakash, L.; Prakash, S. Roles of PCNA-binding and ubiquitin-binding domains in human DNA polymerase in translesion DNA synthesis. Proc. Natl. Acad. Sci. USA 2008, 105, 17724–17729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, Y.; Kanao, R.; Kaji, K.; Ohmori, H.; Hanaoka, F.; Masutani, C. Different types of interaction between PCNA and PIP boxes contribute to distinct cellular functions of Y-family DNA polymerases. Nucleic Acids Res. 2015, 43, 7898–7910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Sonoda, E.; Tang, T.S.; Parker, J.L.; Bielen, A.B.; Takeda, S.; Ulrich, H.D.; Friedberg, E.C. REV1 Protein Interacts with PCNA: Significance of the REV1 BRCT Domain In Vitro and In Vivo. Mol. Cell 2006, 23, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Hibbert, R.G.; Sixma, T.K. Intrinsic flexibility of ubiquitin on proliferating cell nuclear antigen (PCNA) in translesion synthesis. J. Biol. Chem. 2012, 287, 39216–39223. [Google Scholar] [CrossRef] [PubMed]
- Plosky, B.S.; Vidal, A.E.; De Henestrosa, A.R.F.; McLenigan, M.P.; McDonald, J.P.; Mead, S.; Woodgate, R. Controlling the subcellular localization of DNA polymerases ι and η via interactions with ubiquitin. EMBO J. 2006, 25, 2847–2855. [Google Scholar] [CrossRef] [Green Version]
- Bienko, M.; Green, C.M.; Crosetto, N.; Rudolf, F.; Zapart, G.; Coull, B.; Kannouche, P.; Wider, G.; Peter, M.; Lehmann, A.R.; et al. Ubiquitin-binding domains in translesion synthesis polymerases. Science 2005, 310, 1821–1824. [Google Scholar] [CrossRef]
- Hendel, A.; Krijger, P.H.L.; Diamant, N.; Goren, Z.; Langerak, P.; Kim, J.; Reißner, T.; Lee, K.Y.; Geacintov, N.E.; Carell, T.; et al. PCNA ubiquitination is important, but not essential for translesion DNA synthesis in mammalian cells. PLoS Genet. 2011, 7, e1002262. [Google Scholar] [CrossRef] [Green Version]
- Friedberg, E.C.; Lehmann, A.R.; Fuchs, R.P.P. Trading Places: How Do DNA Polymerases Switch during Translesion DNA Synthesis? Mol. Cell 2005, 18, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Fischhaber, P.L.; Luk-Paszyc, M.J.; Masuda, Y.; Zhou, J.; Kamiya, K.; Kisker, C.; Friedberg, E.C. Mouse Rev1 protein interacts with multiple DNA polymerases involved in translesion DNA synthesis. EMBO J. 2003, 22, 6621–6630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohashi, E.; Murakumo, Y.; Kanjo, N.; Akagi, J.I.; Masutani, C.; Hanaoka, F.; Ohmori, H. Interaction of hREV1 with three human Y-family DNA polymerases. Genes Cells 2004, 9, 523–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marians, K.J. Lesion Bypass and the Reactivation of Stalled Replication Forks. Annu. Rev. Biochem. 2018, 87, 217–238. [Google Scholar] [CrossRef]
- Branzei, D. Ubiquitin family modifications and template switching. FEBS Lett. 2011, 585, 2810–2817. [Google Scholar] [CrossRef] [Green Version]
- Ulrich, H.D. Timing and spacing of ubiquitin-dependent DNA damage bypass. FEBS Lett. 2011, 585, 2861–2867. [Google Scholar] [CrossRef] [Green Version]
- Daigaku, Y.; Davies, A.A.; Ulrich, H.D. Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature 2010, 465, 951–955. [Google Scholar] [CrossRef] [Green Version]
- Karras, G.I.; Jentsch, S. The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell 2010, 141, 255–267. [Google Scholar] [CrossRef]
- Shin, S.; Hyun, K.; Kim, J.; Hohng, S. ATP Binding to Rad5 Initiates Replication Fork Reversal by Inducing the Unwinding of the Leading Arm and the Formation of the Holliday Junction. Cell Rep. 2018, 23, 1831–1839. [Google Scholar] [CrossRef]
- Gangavarapu, V.; Haracska, L.; Unk, I.; Johnson, R.E.; Prakash, S.; Prakash, L. Mms2-Ubc13-Dependent and -Independent Roles of Rad5 Ubiquitin Ligase in Postreplication Repair and Translesion DNA Synthesis in Saccharomyces cerevisiae. Mol. Cell. Biol. 2006, 26, 7783–7790. [Google Scholar] [CrossRef] [Green Version]
- Gangavarapu, V.; Prakash, S.; Prakash, L. Requirement of RAD52 Group Genes for Postreplication Repair of UV-Damaged DNA in Saccharomyces cerevisiae. Mol. Cell. Biol. 2007, 27, 7758–7764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz-Bazán, M.Á.; Gallo-Fernández, M.; Saugar, I.; Jiménez-Martín, A.; Vázquez, M.V.; Tercero, J.A. Rad5 Plays a Major Role in the Cellular Response to DNA Damage during Chromosome Replication. Cell Rep. 2014, 23, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Vanoli, F.; Foiani, M. SUMOylation regulates Rad18-mediated template switch. Nature 2008, 456, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Sollier, J.; Liberi, G.; Zhao, X.; Maeda, D.; Seki, M.; Enomoto, T.; Ohta, K.; Foiani, M. Ubc9- and Mms21-Mediated Sumoylation Counteracts Recombinogenic Events at Damaged Replication Forks. Cell 2006, 127, 509–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberi, G.; Maffioletti, G.; Lucca, C.; Chiolo, I.; Baryshnikova, A.; Cotta-ramusino, C.; Lopes, M.; Pellicioli, A.; Haber, J.E.; Foiani, M. Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev. 2005, 19, 339–350. [Google Scholar] [CrossRef] [Green Version]
- Pfander, B.; Moldovan, G.L.; Sacher, M.; Hoege, C.; Jentsch, S. SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature 2005, 436, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Papouli, E.; Chen, S.; Davies, A.A.; Huttner, D.; Krejci, L.; Sung, P.; Ulrich, H.D. Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol. Cell 2005, 19, 123–133. [Google Scholar] [CrossRef]
- Armstrong, A.A.; Mohideen, F.; Lima, C.D. Recognition of SUMO-modified PCNA requires tandem receptor motifs in Srs2. Nature 2012, 483, 59–65. [Google Scholar] [CrossRef]
- Veaute, X.; Jeusset, J.; Soustelle, C.; Kowalczykowski, S.C.; Le Cam, E.; Fahre, F. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature 2003, 423, 309–312. [Google Scholar] [CrossRef]
- Krejci, L.; Van Komen, S.; Li, Y.; Villemain, J.; Reddy, M.S.; Klein, H.; Ellenberger, T.; Sung, P. DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature 2003, 423, 305–309. [Google Scholar] [CrossRef]
- Kolesar, P.; Sarangi, P.; Altmannova, V.; Zhao, X.; Krejci, L. Dual roles of the SUMO-interacting motif in the regulation of Srs2 sumoylation. Nucleic Acids Res. 2012, 40, 7831–7843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parnas, O.; Zipin-Roitman, A.; Pfander, B.; Liefshitz, B.; Mazor, Y.; Ben-Aroya, S.; Jentsch, S.; Kupiec, M. Elg1, an alternative subunit of the RFC clamp loader, preferentially interacts with SUMOylated PCNA. EMBO J. 2010, 29, 2611–2622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, T.; Nishimura, K.; Kanemaki, M.T.; Donaldson, A.D. The Elg1 Replication Factor C-like Complex Functions in PCNA Unloading during DNA Replication. Mol. Cell 2013, 50, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Katou, Y.; Nakato, R.; Shirahige, K.; Donaldson, A.D. Replication-Coupled PCNA Unloading by the Elg1 Complex Occurs Genome-wide and Requires Okazaki Fragment Ligation. Cell Rep. 2015, 12, 774–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.Y.; Fu, H.; Aladjem, M.I.; Myung, K. ATAD5 regulates the lifespan of DNA replication factories by modulating PCNA level on the chromatin. J. Cell Biol. 2013, 200, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.L.; Ulrich, H.D. A SUMO-interacting motif activates budding yeast ubiquitin ligase Rad18 towards SUMO-modified PCNA. Nucleic Acids Res. 2012, 40, 11380–11388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urulangodi, M.; Sebesta, M.; Menolfi, D.; Szakal, B.; Sollier, J.; Sisakova, A.; Krejci, L.; Branzei, D. Local regulation of the Srs2 helicase by the SUMO-like domain protein Esc2 promotes recombination at sites of stalled replication. 2015, 29, 2067–2080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saponaro, M.; Callahan, D.; Zheng, X.; Krejci, L.; Haber, J.E.; Klein, H.L.; Liberi, G. Cdk1 targets Srs2 to complete synthesis-dependent strand annealing and to promote recombinational repair. PLoS Genet. 2010, 6, e1000858. [Google Scholar] [CrossRef]
- Wu, L.; Hickson, I.O. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature 2003, 426, 870–874. [Google Scholar] [CrossRef]
- Sebesta, M.; Urulangodi, M.; Stefanovie, B.; Szakal, B.; Pacesa, M.; Lisby, M.; Branzei, D.; Krejci, L. Esc2 promotes Mus81 complex-activity via its SUMO-like and DNA binding domains. Nucleic Acids Res. 2017, 45, 215–230. [Google Scholar] [CrossRef]
- Szakal, B.; Branzei, D. Premature Cdk1/Cdc5/Mus81 pathway activation induces aberrant replication and deleterious crossover. EMBO J. 2013, 32, 1155–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashton, T.M.; Mankouri, H.W.; Heidenblut, A.; McHugh, P.J.; Hickson, I.D. Pathways for Holliday Junction Processing during Homologous Recombination in Saccharomyces cerevisiae. Mol. Cell. Biol. 2011, 31, 1921–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saugar, I.; Parker, J.L.; Zhao, S.; Ulrich, H.D. The genome maintenance factor Mgs1 is targeted to sites of replication stress by ubiquitylated PCNA. Nucleic Acids Res. 2012, 40, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Hishida, T.; Ohno, T. Saccharomyces cerevisiae MGS1 is essential in strains deficient in the RAD6 -dependent DNA damage tolerance pathway. EMBO J. 2002, 21, 2019–2029. [Google Scholar] [CrossRef]
- Hishida, T.; Iwasaki, H.; Ohno, T.; Morishita, T.; Shinagawa, H. A yeast gene, MGS1, encoding a DNA-dependent AAA+ ATPase is required to maintain genome stability. Proc. Natl. Acad. Sci. USA 2001, 98, 8283–8289. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Gan, W.; Su, S.; Hauenstein, A.V.; Fu, T.; Brasher, B.; Schwerdtfeger, C.; Liang, A.C.; Xu, M.; Wei, W. K63-linked polyubiquitin chains bind to DNA to facilitate DNA damage repair. Sci. Signal. 2018, 11, eaar8133. [Google Scholar] [CrossRef] [Green Version]
- Motegi, A.; Liaw, H.-J.; Lee, K.-Y.; Roest, H.P.; Maas, A.; Wu, X.; Moinova, H.; Markowitz, S.D.; Ding, H.; Hoeijmakers, J.H.J.; et al. Polyubiquitination of proliferating cell nuclear antigen by HLTF and SHPRH prevents genomic instability from stalled replication forks. Proc. Natl. Acad. Sci. USA 2008, 105, 12411–12416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unk, I.; Hajdu, I.; Fatyol, K.; Hurwitz, J.; Yoon, J.-H.; Prakash, L.; Prakash, S.; Haracska, L. Human HLTF functions as a ubiquitin ligase for proliferating cell nuclear antigen polyubiquitination. Proc. Natl. Acad. Sci. USA 2008, 105, 3768–3773. [Google Scholar] [CrossRef] [PubMed]
- Unk, I.; Hajdú, I.; Fátyol, K.; Szakál, B.; Blastyák, A.; Bermudez, V.; Hurwitz, J.; Prakash, L.; Prakash, S.; Haracska, L. Human SHPRH is a ubiquitin ligase for Mms2-Ubc13-dependent polyubiquitylation of proliferating cell nuclear antigen. Proc. Natl. Acad. Sci. USA 2006, 103, 18107–18112. [Google Scholar] [CrossRef] [Green Version]
- Burkovics, P.; Sebesta, M.; Balogh, D.; Haracska, L.; Krejci, L. Strand invasion by HLTF as a mechanism for template switch in fork rescue. Nucleic Acids Res. 2014, 42, 1711–1720. [Google Scholar] [CrossRef] [PubMed]
- Motegi, A.; Sood, R.; Moinova, H.; Markowitz, S.D.; Liu, P.P.; Myung, K. Human SHPRH suppresses genomic instability through proliferating cell nuclear antigen polyubiquitination. J. Cell Biol. 2006, 175, 703–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krijger, P.H.L.; Lee, K.Y.; Wit, N.; van den Berk, P.C.M.; Wu, X.; Roest, H.P.; Maas, A.; Ding, H.; Hoeijmakers, J.H.J.; Myung, K.; et al. HLTF and SHPRH are not essential for PCNA polyubiquitination, survival and somatic hypermutation: Existence of an alternative E3 ligase. DNA Repair 2011, 10, 438–444. [Google Scholar] [CrossRef] [Green Version]
- Blastyak, A.; Hajdu, I.; Unk, I.; Haracska, L. Role of Double-Stranded DNA Translocase Activity of Human HLTF in Replication of Damaged DNA. Mol. Cell. Biol. 2010, 30, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Achar, Y.J.; Balogh, D.; Haracska, L. Coordinated protein and DNA remodeling by human HLTF on stalled replication fork. Proc. Natl. Acad. Sci. USA 2011, 108, 14073–14078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kile, A.C.; Chavez, D.A.; Bacal, J.; Eldirany, S.; Korzhnev, D.M.; Bezsonova, I.; Eichman, B.F.; Cimprich, K.A. HLTF’s Ancient HIRAN Domain Binds 3′ DNA Ends to Drive Replication Fork Reversal. Mol. Cell 2015, 58, 1090–1100. [Google Scholar] [CrossRef]
- Hishiki, A.; Hara, K.; Ikegaya, Y.; Yokoyama, H.; Shimizu, T.; Sato, M.; Hashimoto, H. Structure of a novel DNA-binding domain of Helicase-like Transcription Factor (HLTF) and its functional implication in DNA damage tolerance. J. Biol. Chem. 2015, 290, 13215–13223. [Google Scholar] [CrossRef] [PubMed]
- Achar, Y.J.; Balogh, D.; Neculai, D.; Juhasz, S.; Morocz, M.; Gali, H.; Dhe-Paganon, S.; Venclovas, Č.; Haracska, L. Human HLTF mediates postreplication repair by its HIRAN domain-dependent replication fork remodelling. Nucleic Acids Res. 2015, 43, 10277–10291. [Google Scholar] [CrossRef] [PubMed]
- Chavez, D.A.; Greer, B.H.; Eichman, B.F. The HIRAN domain of helicase-like transcription factor positions the DNA translocase motor to drive efficient DNA fork regression. J. Biol. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.R.; Zeman, M.K.; Chen, J.Y.; Yee, M.C.; Cimprich, K.A. SHPRH and HLTF Act in a Damage-Specific Manner to Coordinate Different Forms of Postreplication Repair and Prevent Mutagenesis. Mol. Cell 2011, 42, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Coulon, S.; Ramasubramanyan, S.; Alies, C.; Philippin, G.; Lehmann, A.; Fuchs, R.P. Rad8Rad5/Mms2-Ubc13 ubiquitin ligase complex controls translesion synthesis in fission yeast. EMBO J. 2010, 29, 2048–2058. [Google Scholar] [CrossRef] [PubMed]
- Vujanovic, M.; Krietsch, J.; Raso, M.C.; Terraneo, N.; Zellweger, R.; Schmid, J.A.; Taglialatela, A.; Huang, J.W.; Holland, C.L.; Zwicky, K.; et al. Replication Fork Slowing and Reversal upon DNA Damage Require PCNA Polyubiquitination and ZRANB3 DNA Translocase Activity. Mol. Cell 2017, 67, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Nimonkar, A.V.; Hu, Y.; Hajdu, I.; Achar, Y.J.; Izhar, L.; Petit, S.A.; Adamson, B.; Yoon, J.C.; Kowalczykowski, S.C.; et al. Polyubiquitinated PCNA Recruits the ZRANB3 Translocase to Maintain Genomic Integrity after Replication Stress. Mol. Cell 2012, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Taglialatela, A.; Alvarez, S.; Leuzzi, G.; Sannino, V.; Ranjha, L.; Huang, J.W.; Madubata, C.; Anand, R.; Levy, B.; Rabadan, R.; et al. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol. Cell 2017, 68, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Ghosal, G.; Chen, J. The HARP-like Domain-Containing Protein AH2/ZRANB3 Binds to PCNA and Participates in Cellular Response to Replication Stress. Mol. Cell 2012, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Weston, R.; Peeters, H.; Ahel, D. ZRANB3 is a structure-specific ATP-dependent endonuclease involved in replication stress response. Genes Dev. 2012, 26, 1558–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebesta, M.; Cooper, C.D.O.; Ariza, A.; Carnie, C.J.; Ahel, D. Structural insights into the function of ZRANB3 in replication stress response. Nat. Commun. 2017, 8, 15847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badu-Nkansah, A.; Mason, A.C.; Eichman, B.F.; Cortez, D. Identification of a substrate recognition domain in the replication stress response protein zinc finger ran-binding domain-containing protein 3 (ZRANB3). J. Biol. Chem. 2016, 291, 8251–8257. [Google Scholar] [CrossRef]
- Kolinjivadi, A.M.; Sannino, V.; De Antoni, A.; Zadorozhny, K.; Kilkenny, M.; Técher, H.; Baldi, G.; Shen, R.; Ciccia, A.; Pellegrini, L.; et al. Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol. Cell 2017, 67, 867–881. [Google Scholar] [CrossRef]
- Crosetto, N.; Bienko, M.; Hibbert, R.G.; Perica, T.; Ambrogio, C.; Kensche, T.; Hofmann, K.; Sixma, T.K.; Dikic, I. Human Wrnip1 is localized in replication factories in a ubiquitin-binding zinc finger-dependent manner. J. Biol. Chem. 2008, 283, 35173–35185. [Google Scholar] [CrossRef]
- Bish, R.A.; Myers, M.P. Werner helicase-interacting protein 1 binds polyubiquitin via its zinc finger domain. J. Biol. Chem. 2007, 282, 23184–23193. [Google Scholar] [CrossRef]
- Nomura, H.; Yoshimura, A.; Edo, T.; Kanno, S.I.; Tada, S.; Seki, M.; Yasui, A.; Enomoto, T. WRNIP1 accumulates at laser light irradiated sites rapidly via its ubiquitin-binding zinc finger domain and independently from its ATPase domain. Biochem. Biophys. Res. Commun. 2012, 417, 1145–1150. [Google Scholar] [CrossRef]
- Kanu, N.; Zhang, T.; Burrell, R.A.; Chakraborty, A.; Cronshaw, J.; Costa, C.D.; Grönroos, E.; Pemberton, H.N.; Anderton, E.; Gonzalez, L.; et al. RAD18, WRNIP1 and ATMIN promote ATM signalling in response to replication stress. Oncogene 2016, 35, 4009–4019. [Google Scholar] [CrossRef]
- Yoshimura, A.; Seki, M.; Kanamori, M.; Tateishi, S.; Tsurimoto, T.; Tada, S.; Enomoto, T. Physical and functional interaction between WRNIP1 and RAD18. Genes Genet. Syst. 2009, 84, 171–178. [Google Scholar] [CrossRef] [Green Version]
- Leuzzi, G.; Marabitti, V.; Pichierri, P.; Franchitto, A. WRNIP1 protects stalled forks from degradation and promotes fork restart after replication stress. EMBO J. 2016, 35, 1437–1451. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Seki, M.; Inoue, E.; Yoshimura, A.; Kusa, Y.; Tada, S.; Enomoto, T. Vertebrate WRNIP1 and BLM are required for efficient maintenance of genome stability. Genes Genet. Syst. 2008, 83, 95–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moldovan, G.-L.; Dejsuphong, D.; Petalcorin, M.I.R.; Hofmann, K.; Takeda, S.; Boulton, S.J.; D’Andrea, A.D. Inhibition of homologous recombination by the PCNA-interacting protein PARI. Mol. Cell 2012, 45, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Burkovics, P.; Dome, L.; Juhasz, S.; Altmannova, V.; Sebesta, M.; Pacesa, M.; Fugger, K.; Sorensen, C.S.; Lee, Y.W.T.; Haracska, L.; et al. The PCNA-associated protein PARI negatively regulates homologous recombination via the inhibition of DNA repair synthesis. Nucleic Acids Res. 2016, 44, 3176–3189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreassen, P.R.; D’Andrea, A.D.; Taniguchi, T. ATR couples FANCD2 monoubiquitination to the DNA-damage responsey. Genes Dev. 2004, 18, 1958–1963. [Google Scholar] [CrossRef] [PubMed]
- Ishiai, M.; Kitao, H.; Smogorzewska, A.; Tomida, J.; Kinomura, A.; Uchida, E.; Saberi, A.; Kinoshita, E.; Kinoshita-Kikuta, E.; Koike, T.; et al. FANCI phosphorylation functions as a molecular switch to turn on the Fanconi anemia pathway. Nat. Struct. Mol. Biol. 2008, 15, 1138–1146. [Google Scholar] [CrossRef] [PubMed]
- Knipscheer, P.; Räschle, M.; Smogorzewska, A.; Enoiu, M.; Ho, T.V.; Schärer, O.D.; Elledge, S.J.; Walter, J.C. The Fanconi Anemia Pathway Promotes Replication-Dependent DNA Interstrand Cross-Link Repair. Science 2009, 2, 1698–1701. [Google Scholar] [CrossRef] [PubMed]
- Meetei, A.R.; de Winter, J.P.; Medhurst, A.L.; Wallisch, M.; Waisfisz, Q.; van de Vrugt, H.J.; Oostra, A.B.; Yan, Z.; Ling, C.; Bishop, C.E.; et al. A novel ubiquitin ligase is deficient in Fanconi anemia. Nat. Genet. 2003, 35, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Machida, Y.J.; Machida, Y.; Chen, Y.; Gurtan, A.M.; Kupfer, G.M.; D’Andrea, A.D.; Dutta, A. UBE2T Is the E2 in the Fanconi Anemia Pathway and Undergoes Negative Autoregulation. Mol. Cell 2006, 23, 589–596. [Google Scholar] [CrossRef]
- Smogorzewska, A.; Matsuoka, S.; Vinciguerra, P.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Ballif, B.A.; Gygi, S.P.; Hofmann, K.; D’Andrea, A.D.; et al. Identification of the FANCI Protein, a Monoubiquitinated FANCD2 Paralog Required for DNA Repair. Cell 2007, 129, 289–301. [Google Scholar] [CrossRef] [Green Version]
- Alpi, A.F.; Pace, P.E.; Babu, M.M.; Patel, K.J. Mechanistic Insight into Site-Restricted Monoubiquitination of FANCD2 by Ube2t, FANCL, and FANCI. Mol. Cell 2008, 32, 767–777. [Google Scholar] [CrossRef]
- Yan, Z.; Guo, R.; Paramasivam, M.; Shen, W.; Ling, C.; Fox, D.; Wang, Y.; Oostra, A.B.; Kuehl, J.; Lee, D.Y.; et al. A Ubiquitin-Binding Protein, FAAP20, Links RNF8-Mediated Ubiquitination to the Fanconi Anemia DNA Repair Network. Mol. Cell 2012, 47, 61–75. [Google Scholar] [CrossRef]
- Ali, A.M.; Pradhan, A.; Singh, T.R.; Du, C.; Li, J.; Wahengbam, K.; Grassman, E.; Auerbach, A.D.; Pang, Q.; Meetei, A.R. FAAP20: A novel ubiquitin-binding FA nuclear core-complex protein required for functional integrity of the FA-BRCA DNA repair pathway. Blood 2012, 119, 3285–3294. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.W.C.; Wang, Y.; Fong, K.W.; Huen, M.S.Y.; Li, L.; Chen, J. Fanconi anemia (FA) binding protein FAAP20 stabilizes FA complementation group A (FANCA) and participates in interstrand cross-link repair. Proc. Natl. Acad. Sci. USA 2012, 109, 4491–4496. [Google Scholar] [CrossRef] [Green Version]
- Stoepker, C.; Hain, K.; Schuster, B.; Hilhorst-Hofstee, Y.; Rooimans, M.A.; Steltenpool, J.; Oostra, A.B.; Eirich, K.; Korthof, E.T.; Nieuwint, A.W.M.; et al. SLX4, a coordinator of structure-specific endonucleases, is mutated in a new Fanconi anemia subtype. Nat. Genet. 2011, 43, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Svendsen, J.M.; Smogorzewska, A.; Sowa, M.E.; O’Connell, B.C.; Gygi, S.P.; Elledge, S.J.; Harper, J.W. Mammalian BTBD12/SLX4 Assembles A Holliday Junction Resolvase and Is Required for DNA Repair. Cell 2009, 138, 63–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, S.L.; Bergstralh, D.T.; Kohl, K.P.; LaRocque, J.R.; Moore, C.B.; Sekelsky, J. Drosophila MUS312 and the Vertebrate Ortholog BTBD12 Interact with DNA Structure-Specific Endonucleases in DNA Repair and Recombination. Mol. Cell 2009, 35, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Fekairi, S.; Scaglione, S.; Chahwan, C.; Taylor, E.R.; Tissier, A.; Coulon, S.; Dong, M.Q.; Ruse, C.; Yates, J.R.; Russell, P.; et al. Human SLX4 Is a Holliday Junction Resolvase Subunit that Binds Multiple DNA Repair/Recombination Endonucleases. Cell 2009, 138, 78–89. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, I.M.; Hain, K.; Déclais, A.C.; Gardiner, M.; Toh, G.W.; Sanchez-Pulido, L.; Heuckmann, J.M.; Toth, R.; Macartney, T.; Eppink, B.; et al. Coordination of Structure-Specific Nucleases by Human SLX4/BTBD12 Is Required for DNA Repair. Mol. Cell 2009, 35, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Crossan, G.P.; Van Der Weyden, L.; Rosado, I.V.; Langevin, F.; Gaillard, P.H.L.; McIntyre, R.E.; Gallagher, F.; Kettunen, M.I.; Lewis, D.Y.; Brindle, K.; et al. Disruption of mouse Slx4, a regulator of structure-specific nucleases, phenocopies Fanconi anemia. Nat. Genet. 2011, 43, 147–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Lach, F.P.; Desetty, R.; Hanenberg, H.; Auerbach, A.D.; Smogorzewska, A. Mutations of the SLX4 gene in Fanconi anemia. Nat. Genet. 2011, 43, 142–146. [Google Scholar] [CrossRef]
- Kashiyama, K.; Nakazawa, Y.; Pilz, D.T.; Guo, C.; Shimada, M.; Sasaki, K.; Fawcett, H.; Wing, J.F.; Lewin, S.O.; Carr, L.; et al. Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes Cockayne syndrome, xeroderma pigmentosum, and Fanconi anemia. Am. J. Hum. Genet. 2013, 92, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Bogliolo, M.; Schuster, B.; Stoepker, C.; Derkunt, B.; Su, Y.; Raams, A.; Trujillo, J.P.; Minguillón, J.; Ramírez, M.J.; Pujol, R.; et al. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am. J. Hum. Genet. 2013, 92, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Spitz, G.S.; Veturi, U.; Lach, F.P.; Auerbach, A.D.; Smogorzewska, A. Regulation of multiple DNA repair pathways by the Fanconi anemia protein SLX4. Blood 2013, 121, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Bhagwat, N.; Olsen, A.L.; Wang, A.T.; Hanada, K.; Stuckert, P.; Kanaar, R.; D’Andrea, A.; Niedernhofer, L.J.; McHugh, P.J. XPF-ERCC1 Participates in the Fanconi Anemia Pathway of Cross-Link Repair. Mol. Cell. Biol. 2009, 29, 6427–6437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douwel, D.K.; Boonen, R.A.; Long, D.T.; Szypowska, A.A.; Räschle, M.; Walter, J.C.; Knipscheer, P. XPF-ERCC1 Acts in Unhooking DNA Interstrand Crosslinks in Cooperation with FANCD2 and FANCP/SLX4. Mol. Cell 2014, 54, 460–471. [Google Scholar] [CrossRef]
- Castor, D.; Nair, N.; Déclais, A.C.; Lachaud, C.; Toth, R.; Macartney, T.J.; Lilley, D.M.J.; Arthur, J.S.C.; Rouse, J. Cooperative control of holliday junction resolution and DNA Repair by the SLX1 and MUS81-EME1 nucleases. Mol. Cell 2013, 52, 221–233. [Google Scholar] [CrossRef]
- Nowotny, M.; Gaur, V. Structure and mechanism of nucleases regulated by SLX4. Curr. Opin. Struct. Biol. 2016, 36, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.N.; Kobayashi, S.; Tsuda, M.; Kurumizaka, H.; Takata, M.; Kono, K.; Jiricny, J.; Takeda, S.; Hirota, K. Involvement of SLX4 in interstrand cross-link repair is regulated by the Fanconi anemia pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 6492–6496. [Google Scholar] [CrossRef] [Green Version]
- Lachaud, C.; Castor, D.; Hain, K.; Muñoz, I.; Wilson, J.; MacArtney, T.J.; Schindler, D.; Rouse, J. Distinct functional roles for the two SLX4 ubiquitin-binding UBZ domains mutated in Fanconi anemia. J. Cell Sci. 2014, 127, 2811–2817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, J.; Garner, E.; Hallet, A.; Nguyen, H.D.; Rickman, K.A.; Gill, G.; Smogorzewska, A.; Zou, L. Noncovalent Interactions with SUMO and Ubiquitin Orchestrate Distinct Functions of the SLX4 Complex in Genome Maintenance. Mol. Cell 2015, 57, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Guervilly, J.H.; Takedachi, A.; Naim, V.; Scaglione, S.; Chawhan, C.; Lovera, Y.; Despras, E.; Kuraoka, I.; Kannouche, P.; Rosselli, F.; et al. The SLX4 complex is a SUMO E3 ligase that impacts on replication stress outcome and genome stability. Mol. Cell 2015, 57, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Yoshikiyo, K.; Kratz, K.; Hirota, K.; Nishihara, K.; Takata, M.; Kurumizaka, H.; Horimoto, S.; Takeda, S.; Jiricny, J. KIAA1018/FAN1 nuclease protects cells against genomic instability induced by interstrand cross-linking agents. Proc. Natl. Acad. Sci. USA 2010, 107, 21553–21557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smogorzewska, A.; Desetty, R.; Saito, T.T.; Schlabach, M.; Lach, F.P.; Sowa, M.E.; Clark, A.B.; Kunkel, T.A.; Harper, J.W.; Colaiácovo, M.P.; et al. A Genetic Screen Identifies FAN1, a Fanconi Anemia-Associated Nuclease Necessary for DNA Interstrand Crosslink Repair. Mol. Cell 2010, 39, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Ghosal, G.; Yuan, J.; Chen, J.; Huang, J. FAN1 acts with FANCI-FANCD2 to promote DNA interstrand cross-link repair. Science 2010, 329, 693–696. [Google Scholar] [CrossRef]
- MacKay, C.; Déclais, A.C.; Lundin, C.; Agostinho, A.; Deans, A.J.; MacArtney, T.J.; Hofmann, K.; Gartner, A.; West, S.C.; Helleday, T.; et al. Identification of KIAA1018/FAN1, a DNA Repair Nuclease Recruited to DNA Damage by Monoubiquitinated FANCD2. Cell 2010, 142, 65–76. [Google Scholar] [CrossRef]
- Lachaud, C.; Slean, M.; Marchesi, F.; Lock, C.; Odell, E.; Castor, D.; Toth, R.; Rouse, J. Karyomegalic interstitial nephritis and DNA damage-induced polyploidy in Fan1 nuclease-defective knock-in mice. Genes Dev. 2016, 30, 639–644. [Google Scholar] [CrossRef] [Green Version]
- Thongthip, S.; Bellani, M.; Gregg, S.Q.; Sridhar, S.; Conti, B.A.; Chen, Y.; Seidman, M.M.; Smogorzewska, A. Fan1 deficiency results in DNA interstrand cross-link repair defects, enhanced tissue karyomegaly, and organ dysfunction. Genes Dev. 2016, 30, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, J.P.; Mina, L.B.; Pujol, R.; Bogliolo, M.; Andrieux, J.; Holder, M.; Schuster, B.; Schindler, D.; Surrallés, J.; Dc, W.; et al. On the role of FAN1 in Fanconi anemia. Blood 2013, 120, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Otto, E.A.; Cluckey, A.; Airik, R.; Hurd, T.W.; Chaki, M.; Diaz, K.; Lach, F.P.; Bennett, G.R.; Gee, H.Y.; et al. FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat. Genet. 2012, 44, 910–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seguí, N.; Mina, L.B.; Lázaro, C.; Sanz-Pamplona, R.; Pons, T.; Navarro, M.; Bellido, F.; López-Doriga, A.; Valdés-Mas, R.; Pineda, M.; et al. Germline Mutations in FAN1 Cause Hereditary Colorectal Cancer by Impairing DNA Repair. Gastroenterology 2015, 149, 563–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Airik, R.; Schueler, M.; Airik, M.; Cho, J.; Porath, J.D.; Mukherjee, E.; Sims-Lucas, S.; Hildebrandt, F. A FANCD2/FANCI-Associated Nuclease 1-Knockout Model Develops Karyomegalic Interstitial Nephritis. J. Am. Soc. Nephrol. 2016, 27, 3552–3559. [Google Scholar] [CrossRef]
- Lachaud, C.; Moreno, A.; Marchesi, F.; Toth, R.; Blow, J.J.; Rouse, J. Ubiquitinated Fancd2 recruits Fan1 to stalled replication forks to prevent genome instability. Science 2016, 5634, 1–8. [Google Scholar] [CrossRef]
- Lossaint, G.; Larroque, M.; Ribeyre, C.; Bec, N.; Larroque, C.; Décaillet, C.; Gari, K.; Constantinou, A. FANCD2 Binds MCM Proteins and Controls Replisome Function upon Activation of S Phase Checkpoint Signaling. Mol. Cell 2013, 51, 678–690. [Google Scholar] [CrossRef]
- Chaudhury, I.; Stroik, D.R.; Sobeck, A. FANCD2-Controlled Chromatin Access of the Fanconi-Associated Nuclease FAN1 Is Crucial for the Recovery of Stalled Replication Forks. Mol. Cell. Biol. 2014, 34, 3939–3954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porro, A.; Berti, M.; Pizzolato, J.; Bologna, S.; Kaden, S.; Saxer, A.; Ma, Y.; Nagasawa, K.; Sartori, A.A.; Jiricny, J. FAN1 interaction with ubiquitylated PCNA alleviates replication stress and preserves genomic integrity independently of BRCA2. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Pizzolato, J.; Mukherjee, S.; Schaerer, O.D.; Jiricny, J. FANCD2-associated nuclease 1, but not exonuclease 1 or flap endonuclease 1, is able to unhook DNA interstrand cross-links in vitro. J. Biol. Chem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Persky, N.S.; Yoo, B.; Ouerfelli, O.; Smogorzewska, A.; Elledge, S.J.; Pavletich, N.P. DNA repair. Mechanism of DNA interstrand cross-link processing by repair nuclease FAN1. Science 2014, 346, 1127–1130. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.T.; Sengerová, B.; Cattell, E.; Inagawa, T.; Hartley, J.M.; Kiakos, K.; Burgess-Brown, N.A.; Swift, L.P.; Enzlin, J.H.; Schofield, C.J.; et al. Human SNM1A and XPF—ERCC1 collaborate to initiate DNA interstrand cross-link repair. Genes Dev. 2011, 25, 1859–1870. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Akhter, S.; Zhang, X.; Legerski, R. The multifunctional SNM1 gene family: Not just nucleases. Future Oncol. 2010, 6, 1015–1029. [Google Scholar] [CrossRef]
- Ishiai, M.; Kimura, M.; Namikoshi, K.; Yamazoe, M.; Yamamoto, K.; Arakawa, H.; Agematsu, K.; Matsushita, N.; Takeda, S.; Buerstedde, J.-M.; et al. DNA Cross-Link Repair Protein SNM1A Interacts with PIAS1 in Nuclear Focus Formation. Mol. Cell. Biol. 2004, 24, 10733–10741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dronkert, M.L.G.; de Wit, J.; Boeve, M.; Vasconcelos, M.L.; van Steeg, H.; Tan, T.L.R.; Hoeijmakers, J.H.J.; Kanaar, R. Disruption of Mouse SNM1 Causes Increased Sensitivity to the DNA Interstrand Cross-Linking Agent Mitomycin C. Mol. Cell. Biol. 2000, 20, 4553–4561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazrati, A.; Ramis-Castelltort, M.; Sarkar, S.; Barber, L.J.; Schofield, C.J.; Hartley, J.A.; McHugh, P.J. Human SNM1A suppresses the DNA repair defects of yeast pso2 mutants. DNA Repair 2008, 7, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Fontebasso, Y.; Etheridge, T.J.; Oliver, A.W.; Murray, J.M.; Carr, A.M. The conserved Fanconi anemia nuclease Fan1 and the SUMO E3 ligase Pli1 act in two novel Pso2-independent pathways of DNA interstrand crosslink repair in yeast. DNA Repair 2013, 12, 1011–1023. [Google Scholar] [CrossRef]
- Hemphill, A.W.; Bruun, D.; Thrun, L.; Akkari, Y.; Torimaru, Y.; Hejna, K.; Jakobs, P.M.; Hejna, J.; Jones, S.; Olson, S.B.; et al. Mammalian SNM1 is required for genome stability. Mol. Genet. Metab. 2008, 94, 38–45. [Google Scholar] [CrossRef] [Green Version]
- Abdullah, U.B.; McGouran, J.F.; Brolih, S.; Ptchelkine, D.; El-Sagheer, A.H.; Brown, T.; McHugh, P.J. RPA activates the XPF-ERCC1 endonuclease to initiate processing of DNA interstrand crosslinks. EMBO J. 2017, e201796664. [Google Scholar] [CrossRef]
- Yang, K.; Moldovan, G.L.; D’Andrea, A.D. RAD18-dependent recruitment of SNM1A to DNA repair complexes by a ubiquitin-binding zinc finger. J. Biol. Chem. 2010, 285, 19085–19091. [Google Scholar] [CrossRef]
- Niedzwiedz, W.; Mosedale, G.; Johnson, M.; Ong, C.Y.; Pace, P.; Patel, K.J. The Fanconi anaemia gene FANCC promotes homologous recombination and error-prone DNA repair. Mol. Cell 2004, 15, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Simpson, L.J.; Sale, J.E. Rev1 is essential for DNA damage tolerance and non-templated immunoglobulin gene mutation in a vertebrate cell line. EMBO J. 2003, 22, 1654–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, T.; Sonoda, E.; Yoshimura, M.; Kawano, Y.; Saya, H.; Kohzaki, M. Multiple Roles of Vertebrate REV Genes in DNA Repair and Recombination. Mol. Cell. Biol. 2005, 25, 6103–6111. [Google Scholar] [CrossRef] [PubMed]
- Räschle, M.; Knipsheer, P.; Enoiu, M.; Angelov, T.; Sun, J.; Griffith, J.D.; Ellenberger, T.E.; Schärer, O.D.; Walter, J.C. Mechanism of Replication-Coupled DNA Interstrand Crosslink Repair. Cell 2008, 134, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Budzowska, M.; Graham, T.G.; Sobeck, A.; Waga, S.; Walter, J.C. Regulation of the Rev1-pol complex during bypass of a DNA interstrand cross-link. EMBO J. 2015, 34, 1971–1985. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Tang, T.-S.; Bienko, M.; Parker, J.L.; Bielen, A.B.; Sonoda, E.; Takeda, S.; Ulrich, H.D.; Dikic, I.; Friedberg, E.C. Ubiquitin-Binding Motifs in REV1 Protein Are Required for Its Role in the Tolerance of DNA Damage. Mol. Cell. Biol. 2006, 26, 8892–8900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bluteau, D.; Masliah-Planchon, J.; Clairmont, C.; Rousseau, A.; Ceccaldi, R.; D’Enghien, C.D.; Bluteau, O.; Cuccuini, W.; Gachet, S.; De Latour, R.P.; et al. Biallelic inactivation of REV7 is associated with Fanconi anemia. J. Clin. Investig. 2016, 126, 3580–3584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomida, J.; Takata, K.I.; Lange, S.S.; Schibler, A.C.; Yousefzadeh, M.J.; Bhetawal, S.; Dent, S.Y.R.; Wood, R.D. REV7 is essential for DNA damage tolerance via two REV3L binding sites in mammalian DNA polymerase ζ. Nucleic Acids Res. 2015, 43, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Somyajit, K.; Narita, T.; Maskey, E.; Stanlie, A.; Kremer, M.; Typas, D.; Lammers, M.; Mailand, N.; Nussenzweig, A.; et al. DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell 2018, 173, 972–988. [Google Scholar] [CrossRef]
- Dev, H.; Chiang, T.W.W.; Lescale, C.; de Krijger, I.; Martin, A.G.; Pilger, D.; Coates, J.; Sczaniecka-Clift, M.; Wei, W.; Ostermaier, M.; et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol. 2018, 20, 954–965. [Google Scholar] [CrossRef]
- Mirman, Z.; Lottersberger, F.; Takai, H.; Kibe, T.; Gong, Y.; Takai, K.; Bianchi, A.; Zimmermann, M.; Durocher, D.; de Lange, T. 53BP1–RIF1–shieldin counteracts DSB resection through CST- and Polα-dependent fill-in. Nature 2018, 560, 112–116. [Google Scholar] [CrossRef]
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Álvarez-Quilón, A.; Moatti, N.; Zimmermann, M.; et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Tomida, J.; Takata, K.; Bhetawal, S.; Person, M.D.; Chao, H.; Tang, D.G.; Wood, R.D. FAM35A associates with REV7 and modulates DNA damage responses of normal and BRCA1-defective cells. EMBO J. 2018, e99543. [Google Scholar] [CrossRef] [PubMed]
- Findlay, S.; Heath, J.; Luo, V.M.; Malina, A.; Morin, T.; Coulombe, Y.; Djerir, B.; Li, Z.; Samiei, A.; Simo-Cheyou, E.; et al. FAM35A co-operates with REV7 to coordinate DNA double-strand break repair pathway choice. EMBO J. 2018, e100158. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Chapman, J.R.; Brandsma, I.; Yuan, J.; Mistrik, M.; Bouwman, P.; Bartkova, J.; Gogola, E.; Warmerdam, D.; Barazas, M.; et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 2015, 521, 541–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boersma, V.; Moatti, N.; Segura-Bayona, S.; Peuscher, M.H.; van der Torre, J.; Wevers, B.A.; Orthwein, A.; Durocher, D.; Jacobs, J.J.L. MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5′ end resection. Nature 2015, 521, 537–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kee, Y.; Huang, T.T. Role of Deubiquitinating Enzymes in DNA Repair. Mol. Cell. Biol. 2016, 36, 524–544. [Google Scholar] [CrossRef] [PubMed]
- Jacq, X.; Kemp, M.; Martin, N.M.B.; Jackson, S.P. Deubiquitylating Enzymes and DNA Damage Response Pathways. Cell Biochem. Biophys. 2013, 67, 25–43. [Google Scholar] [CrossRef]
- Nijman, S.M.B.; Huang, T.T.; Dirac, A.M.G.; Brummelkamp, T.R.; Kerkhoven, R.M.; D’Andrea, A.D.; Bernards, R. The deubiquitinating enzyme USP1 regulates the fanconi anemia pathway. Mol. Cell 2005, 17, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Parmar, K.; Huang, M.; Weinstock, D.M.; Ruit, C.A.; Kutok, J.L.; D’Andrea, A.D. Inactivation of Murine Usp1 Results in Genomic Instability and a Fanconi Anemia Phenotype. Dev. Cell 2009, 16, 314–320. [Google Scholar] [CrossRef] [Green Version]
- Oestergaard, V.H.; Langevin, F.; Kuiken, H.J.; Pace, P.; Niedzwiedz, W.; Simpson, L.J.; Ohzeki, M.; Takata, M.; Sale, J.E.; Patel, K.J. Deubiquitination of FANCD2 Is Required for DNA Crosslink Repair. Mol. Cell 2007, 28, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Kim, J.M.; Primack, B.; Weinstock, D.M.; Moreau, L.A.; Parmar, K.; D’Andrea, A.D. Inactivation of Uaf1 Causes Defective Homologous Recombination and Early Embryonic Lethality in Mice. Mol. Cell. Biol. 2013, 33, 4360–4370. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Moldovan, G.L.; Vinciguerra, P.; Murai, J.; Takeda, S.; D’Andrea, A.D. Regulation of the Fanconi anemia pathway by a SUMO-like delivery network. Genes Dev. 2011, 25, 1847–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Twest, S.; Murphy, V.J.; Hodson, C.; Tan, W.; Swuec, P.; O’Rourke, J.J.; Heierhorst, J.; Crismani, W.; Deans, A.J. Mechanism of Ubiquitination and Deubiquitination in the Fanconi Anemia Pathway. Mol. Cell 2017, 65, 247–259. [Google Scholar]
- Huang, T.T.; Nijman, S.M.B.; Mirchandani, K.D.; Galardy, P.J.; Cohn, M.A.; Haas, W.; Gygi, S.P.; Ploegh, H.L.; Bernards, R.; D’Andrea, A.D. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat. Cell Biol. 2006, 8, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Yang, S.W.; Yu, K.R.; Ka, S.H.; Lee, S.W.; Seol, J.H.; Jeon, Y.J.; Chung, C.H. Modification of PCNA by ISG15 Plays a Crucial Role in Termination of Error-Prone Translesion DNA Synthesis. Mol. Cell 2014, 54, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Haahr, P.; Borgermann, N.; Guo, X.; Typas, D.; Achuthankutty, D.; Hoffman, S.; Shearer, R.; Sixma, T.; Mailand, N. ZUFSP deubiquitylates K63-linked polyubiquitin chains to promote genome stability. Mol. Cell 2018, 70, 165–174. [Google Scholar] [CrossRef]
- Kwasna, D.; Rehman, S.A.A.; Natarajan, J.; Matthews, S.; Madden, R.; De Cesare, V.; Weidlich, S.; Virdee, S.; Ahel, I.; Gibbs Seymour, I.; et al. Discovery and characterization of ZUFSP, a novel DUB class important for genome stability. Mol. Cell 2018, 70, 150–164. [Google Scholar] [CrossRef]
- Hermanns, T.; Pichlo, C.; Woiwode, I.; Klopffleisch, K.; Witting, K.F.; Ovaa, H.; Baumann, U.; Hofmann, K. A family of unconventional deubiquitinases with modular chain specificity determinants. Nat. Commun. 2018, 9, 799. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Paudyal, S.C.; Chin, R.I.; You, Z. PCNA promotes processive DNA end resection by Exo1. Nucleic Acids Res. 2013, 41, 9325–9338. [Google Scholar] [CrossRef] [Green Version]
- Soo Lee, N.; Jin Chung, H.; Kim, H.-J.; Yun Lee, S.; Ji, J.-H.; Seo, Y.; Hun Han, S.; Choi, M.; Yun, M.; Lee, S.-G.; et al. TRAIP/RNF206 is required for recruitment of RAP80 to sites of DNA damage. Nat. Commun. 2016, 7, 10463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harley, M.E.; Murina, O.; Leitch, A.; Higgs, M.R.; Bicknell, L.S.; Yigit, G.; Blackford, A.N.; Zlatanou, A.; Mackenzie, K.J.; Reddy, K.; et al. TRAIP promotes DNA damage response during genome replication and is mutated in primordial dwarfism. Nat. Genet. 2016, 48, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Guo, Y.; Huang, J.; Deng, Y.; Zang, J.; Huen, M.S. TRAIP regulates replication fork recovery and progression via PCNA. Cell Discov. 2016, 2, 16016. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.; Smedegaard, S.; Nakamura, K.; Mortuza, G.B.; Räschle, M.; de Opakua, A.I.; Oka, Y.; Feng, Y.; Blanco, F.J.; Mann, M.; et al. TRAIP is a PCNA-binding ubiquitin ligase that protects genome stability after replication stress. J. Cell Biol. 2015. [Google Scholar] [CrossRef]
- Baranes-Bachar, K.; Levy-Barda, A.; Oehler, J.; Reid, D.A.; Soria-Bretones, I.; Voss, T.C.; Chung, D.; Park, Y.; Liu, C.; Yoon, J.-B.; et al. The Ubiquitin E3/E4 Ligase UBE4A Adjusts Protein Ubiquitylation and Accumulation at Sites of DNA Damage, Facilitating Double-Strand Break Repair. Mol. Cell 2018, 69, 866–878. [Google Scholar] [CrossRef] [PubMed]
- Forment, J.V.; O’Connor, M.J. Targeting the replication stress response in cancer. Pharmacol. Ther. 2018, 188, 155–167. [Google Scholar] [CrossRef]
- Glanzer, J.G.; Liu, S.; Wang, L.; Mosel, A.; Peng, A.; Oakley, G.G. RPA Inhibition increases Replication Stress and Suppresses Tumor Growth. Cancer Res. 2014, 74, 5165–5172. [Google Scholar] [CrossRef] [PubMed]
- Feldkamp, M.D.; Frank, A.O.; Kennedy, J.P.; Patrone, J.D.; Vangamudi, B.; Waterson, A.G.; Fesik, S.W.; Chazin, W.J. Surface Reengineering of RPA70N Enables Cocrystallization with an Inhibitor of the Replication Protein A Interaction Motif of ATR Interacting Protein. Biochemistry 2013, 52, 6515–6524. [Google Scholar] [CrossRef]
- Shuck, S.C.; Turchi, J.J. Targeted inhibition of RPA reveals cytotoxic activity, synergy with chemotherapeutic DNA damaging agents and insight into cellular function. Cancer Res. 2010, 70, 3189–3198. [Google Scholar] [CrossRef]
- Smith, S.J.; Gu, L.; Phipps, E.A.; Dobrolecki, L.E.; Mabrey, K.S.; Gulley, P.; Dillehay, K.L.; Dong, Z.; Fields, G.B.; Chen, Y.-R.; et al. A Peptide Mimicking a Region in Proliferating Cell Nuclear Antigen Specific to Key Protein Interactions Is Cytotoxic to Breast Cancer. Mol. Pharmacol. 2015, 87, 263–276. [Google Scholar] [CrossRef]
- Inoue, A.; Kikuchi, S.; Hishiki, A.; Shao, Y.; Heath, R.; Evison, B.J.; Actis, M.; Canman, C.E.; Hashimoto, H.; Fujii, N. A Small Molecule Inhibitor of Monoubiquitinated Proliferating Cell Nuclear Antigen (PCNA) inhibits Repair of Interstrand DNA Crosslink, enhances DNA Double-strand Break, and sensitizes Cancer Cells to Cisplatin. J. Biol. Chem. 2014, 289, 7109–7120. [Google Scholar] [CrossRef] [PubMed]
- Chirnomas, D. Chemosensitization to cisplatin by inhibitors of the Fanconi anemia/BRCA pathway. Mol. Cancer Ther. 2006, 5, 952–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, C.-H.; Wang, Y.; Chen, P.; Jiang, Q.; Lan, T.; Li, M.-Y.; Su, J.-Y.; Wu, Y.; Li, J. Suppression of the FA pathway combined with CHK1 inhibitor hypersensitize lung cancer cells to gemcitabine. Sci. Rep. 2017, 7, 15031. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yates, M.; Maréchal, A. Ubiquitylation at the Fork: Making and Breaking Chains to Complete DNA Replication. Int. J. Mol. Sci. 2018, 19, 2909. https://doi.org/10.3390/ijms19102909
Yates M, Maréchal A. Ubiquitylation at the Fork: Making and Breaking Chains to Complete DNA Replication. International Journal of Molecular Sciences. 2018; 19(10):2909. https://doi.org/10.3390/ijms19102909
Chicago/Turabian StyleYates, Maïlyn, and Alexandre Maréchal. 2018. "Ubiquitylation at the Fork: Making and Breaking Chains to Complete DNA Replication" International Journal of Molecular Sciences 19, no. 10: 2909. https://doi.org/10.3390/ijms19102909
APA StyleYates, M., & Maréchal, A. (2018). Ubiquitylation at the Fork: Making and Breaking Chains to Complete DNA Replication. International Journal of Molecular Sciences, 19(10), 2909. https://doi.org/10.3390/ijms19102909